
Selective hydrogenation of amides and imides over heterogeneous Pt-based catalysts†
Ruiyang
Qu
 ,
Shuxin
Mao
,
Jana
Weiß
,
Vita A.
Kondratenko
,
Evgenii V.
Kondratenko
,
Stephan
Bartling
,
Haifeng
Qi
,
Annette-Enrica
Surkus
,
Kathrin
Junge
* and
Matthias
Beller
*
,
Shuxin
Mao
,
Jana
Weiß
,
Vita A.
Kondratenko
,
Evgenii V.
Kondratenko
,
Stephan
Bartling
,
Haifeng
Qi
,
Annette-Enrica
Surkus
,
Kathrin
Junge
* and
Matthias
Beller
*
Leibniz-Institut für Katalyse e.V., Albert-Einstein-Straße 29a, 18059 Rostock, Germany. E-mail: Kathrin.Junge@catalysis.de; Matthias.Beller@catalysis.de
First published on 4th December 2023
Abstract
The hydrogenation of amides is an important but highly challenging reaction, which usually is applied under drastic conditions (temperature >160 °C). Here, we report a heterogeneous Pt–MoOx/TiO2 catalyst for amide and imide hydrogenation under milder conditions. The catalytic reactivity is proposed to originate from the synergistic effect between surface active species. Pt promotes formation of partially reduced MoOx that is responsible for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O activation, and MoOx-modified Pt can more efficiently generate surface species from gas-phase H2. This catalyst system enables the selective hydrogenation of several 2° and 3° amides as well as imides.
O activation, and MoOx-modified Pt can more efficiently generate surface species from gas-phase H2. This catalyst system enables the selective hydrogenation of several 2° and 3° amides as well as imides.
1. Introduction
Amines constitute one of the most important classes of organic compounds. The majority of industrially applied amines are used for life science applications, specifically agrochemicals and pharmaceuticals. For the preparation of these products constantly new amine building blocks are needed. Among the various synthetic methodologies used for their preparation, reduction of amides is regarded as one of the most desired methodologies to produce amines since a plethora of starting materials is conveniently available.1 However, this transformation also remains one of the most difficult and challenging reactions due to the low electrophilicity of CO bond in amides,2 which is less susceptible for nucleophilic attack. Thus, traditionally stoichiometric amounts (in reality in excess) of metal hydride reagents like LiAlH4 or NaBH4 are used for amide reduction, which inherently limit industrial-scale syntheses due to their price and safety aspects.3 In addition, considerable quantity of waste metal salts is generated during such processes. Obviously, catalytic reduction of amides to amines using molecular hydrogen offers a green solution since water is ideally the only by-product. Hence, the development of efficient and general catalysts for the hydrogenation of amides continues to be the research interest for many academic and industrial chemists.
In early studies, the hydrogenation of model amides, e.g., N-octanamide, acetanilide and N,N-dimethyldodecanamide, has been performed at rather harsh reaction conditions, i.e., at 200–350 °C and 200–370 bar H2,4 using Ni-,5 Cu-6 and Re-based catalysts.7 In recent years, there is a growing interest in developing new catalysts for amide hydrogenation working at milder reaction conditions and significant progress has been made. From 2010, Whyman and co-workers developed a series of bimetallic materials (Rh/Mo,8 Ru/Mo,9 Rh/Re,10 and Ru/Re10 which are able to perform amide hydrogenation at milder conditions (130–160 °C and 50–100 bar H2). Later on, Breit and co-workers11 reported bimetallic Pd–Re/graphite and Pt–Re/graphite catalysts working at 120–160 °C and 5–70 bar H2. Similarly, an effective Pt–Re/TiO2 catalyst was reported by Thompson12 and Cole-Hamilton13 as well as their co-workers. A solvent-free hydrogenation process was also achieved by Shimizu and co-workers14 using Lewis acidic Nb2O5 supported Pt catalyst at 180 °C and 50 bar H2. In 2017, an interesting discovery was made by Kaneda and co-workers.15 They developed a Pt–V2O5 catalyst supported on hydroxyapatite (HAP) and obtained good yields of the corresponding amines. However, the toxicity of vanadium oxide limits its application potential. More recently, Wang and co-workers16 developed a bifunctional RuW/SiO2 catalyst which afforded the hydrogenation of primary amides at 160 °C and 50 bar H2. It is worth noting that molecularly-defined catalysts have been reported to be active for amide hydrogenations, too.1,17 However, these homogeneous systems have several drawbacks from a practical point of view, like difficulties in catalyst recycling, requirement of acidic or basic additives, and complexity in ligand synthesis, restricting their application. In conclusion, the development of efficient and environmentally benign catalysts which allow for a practical and selective hydrogenation of amides is still highly desired and demanding.
Herein, we report a step towards this goal utilizing a Pt–MoOx/TiO2 heterogeneous catalyst, which permits the hydrogenation of several 2° and 3° amides as well as imides at comparably mild conditions (100–150 °C and 50 bar H2). This bifunctional catalyst material was rationally designed to allow for both dissociation of H2 and activation of CO bond in an efficient manner. The synergistic effect of the key components Pt and MoOx is revealed using various characterization techniques.
2. Experimental
2.1 Catalyst preparation
The catalysts were prepared by a wet-impregnation method.18 In a typical synthesis procedure, a desired amount of aqueous H2PtCl6 solution (7.5 mg Pt per mL) and ammonium molybdate tetrahydrate was first dissolved in 16 mL deionized water at 60 °C with vigorous stirring at 800 rpm for 15 min, followed by the addition of a desired amount of TiO2 support. The metal loading of Pt is set as 5 wt% and the molar ratio of Mo/Pt is 1. The mixture was stirred for 16 h with reflux. Then, the mixture was cooled to room temperature and water was removed in vacuo using a rotatory evaporator. The solid sample was dried in an oven at 110 °C over night, after which it was thoroughly grounded to fine powders. Finally, the as-prepared samples were calcined in static air at 450 °C for 4 h (heating rate: 10 °C min−1) in a muffle furnace. Before reaction, the samples were reduced in 5% H2/Ar at 250 °C for 1 h (heating rate: 5 °C min−1) in a flow-tube furnace. The preparation of other catalysts followed the similar procedure, except different metal precursors were used. For the preparation of Pt–VOx/TiO2, Pt(acac)2 and V(acac)3 were dissolved in acetone.2.2 Activity tests
The catalyst (2 mol% of the corresponding metal), substrate (0.5 mmol) and solvent (2 mL) were added in a 4 mL vial with magnetic stirring bar and septum cap. Then, a needle was inserted in the septum, allowing H2 to enter the vial. The vials (up to eight) were set in an alloy plate and placed in a 300 mL steel Parr autoclave. The autoclave was flushed with H2 for 3 times at 10 bar and finally pressurized to the desired value. Then, it was placed into an aluminum block and heated to the desired temperature. When the reaction is completed, the autoclave was quickly cooled down to room temperature with an ice bath and vented. Finally, the samples were removed from the autoclave with the addition of internal standard (n-hexadecane, 50 μL) and ethyl acetate to the crude mixture, followed by filtration to separate the solid catalysts using a celite pad. The organic layer was analyzed at the GC-FID (Agilent 7890A).2.3 Characterizations
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2:Ar = 1:1:1 mixture were performed at the same temperature. The overall pulse size in these experiments was between 9 × 1014–1.1 × 1015 molecules per pulse. Under such conditions, gas-phase reactions are strongly suppressed, and heterogeneously catalyzed reaction steps were studied.
:H2:Ar = 1:1:1 mixture were performed at the same temperature. The overall pulse size in these experiments was between 9 × 1014–1.1 × 1015 molecules per pulse. Under such conditions, gas-phase reactions are strongly suppressed, and heterogeneously catalyzed reaction steps were studied.
The feed mixture was prepared using D2 (2.8, CK Special Gases Limited), H2 (Air Liquide, 5.0) and Ar (Air Liquide, 5.0) without additional purification. Transient responses of the feed components and the reaction products were monitored at the reactor outlet using a quadrupole mass spectrometer (HAL RD 301 Hidden Analytical). The following m/z signals were recorded 4 (D2), 3.0 (HD), 2.0 (H2) and 40 (Ar). Ar was used as an inert standard. For each m/z, pulses were repeated 10 times and averaged to improve the signal-to-noise ratio. For a proper comparison of the appearance order and the shape of transient responses of substances differing in their molecular weights like D2 and H2, the data were transformed to a dimensionless form.19
3. Results and discussion
3.1 Reaction optimization
The general design strategy for the development of a more active amide hydrogenation catalyst is based on the idea of a bifunctional catalyst which combines specific metal sites for H2 dissociation and an oxophilic promoter to adsorb and activate the carbonyl group. Following this concept, we initially screened diverse heterogeneous catalysts with different metal species (Table 1, entries 1–4), Lewis acidic promoters (Table 1, entries 1, 5–8) and supports (Table 1, entries 1, 9–11) for the hydrogenation of 4-acetylmorpholine to 4-ethylmorpholine as a model reaction. Here, Pt–MoOx/TiO2 catalyst showed a promising yield (80%) under comparably mild reaction conditions (120 °C, 50 bar H2 and 16 h), outperforming other catalyst compositions, including previously developed as well as commercially available system (Table 1, entry 8). This may be because of the stronger Lewis acidic property of MoOx and TiO2 which could activate CO group of amide more efficiently.14 Simply increasing the reaction time to 24 h resulted in an excellent yield (92%) of the desired product (Table 1, entry 12 and Fig. S1†). Stability test was performed at ca. 50% conversion, as shown in Fig. S2.† A slight decrease of activity was observed after each run. Inductively coupled plasma optical emission spectrometry (ICP-OES) test showed a slight Pt leaching after reaction (Table S2†), which could be the major reason for the decrease of activity. Although decreasing the reaction temperature or pressure led to a decline in the product yield (Table 1, entries 1, 13, 15–17), still 86% of N-ethyl morpholine was achieved at 100 °C (Table 1, entry 14). Notably, solvent screening showed that the best product yield was observed using the green solvent ethanol (Table S1†), which displayed in previous investigations with Pd/Re catalysts only a negligible activity.11 To clarify this difference, we made a comparison using ethanol with different purities (>99.9% HPLC grade vs. 96% HPLC grade, Table S1,† entries 1 and 2). Only 15% yield of 4-ethylmorpholine was obtained using 96% ethanol as solvent. In addition, when 4% of water was added to the >99.9% ethanol a similar yield (22%) was detected (Table S1,† entry 3). These results clearly suggest that the presence of water in the reaction mixture plays a vital role in determining the reactivity. This is not surprising as water is one of the products for C–O cleavage reaction, the existence of which would shift the reaction equilibrium, inhibiting the reaction. In addition, the formed water (together with the amine product) would likely occupy a certain amount of catalytic active sites due to its incomplete desorption, resulting in a decreased reaction rate. This can be evidenced by the time-on-line plot (Table S1†), in which the reaction proceeds rapidly in the initial period, followed by being slowed down with increasing hours.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | T/°C | P/bar | t/h | Y/% |
| T: temperature; P: H2 pressure; t: reaction time; Y: yield. Reaction conditions: 4-acetylmorpholine (0.5 mmol), [M] catalyst (2 mol%), EtOH (2 mL). Yield was determined by GC-FID using n-hexadecane as an internal standard. | |||||
| 1 | Pt–MoOx/TiO2 | 120 | 50 | 16 | 80 |
| 2 | Pd–MoOx/TiO2 | 120 | 50 | 16 | 2 |
| 3 | Ru–MoOx/TiO2 | 120 | 50 | 16 | 11 |
| 4 | Ni–MoOx/TiO2 | 120 | 50 | 16 | 0 |
| 5 | Pt–WOx/TiO2 | 120 | 50 | 16 | 15 |
| 6 | Pt–NbOx/TiO2 | 120 | 50 | 16 | 8 |
| 7 | Pt–VOx/TiO2 | 120 | 50 | 16 | 31 |
| 8 | Pt–Re/TiO2 | 120 | 50 | 16 | 57 |
| 9 | Pt–MoOx/ZrO2 | 120 | 50 | 16 | 2 |
| 10 | Pt–MoOx/Al2O3 | 120 | 50 | 16 | 2 |
| 11 | Pt–MoOx/C | 120 | 50 | 16 | 1 |
| 12 | Pt–MoOx/TiO2 | 120 | 50 | 24 | 92 |
| 13 | Pt–MoOx/TiO2 | 100 | 50 | 16 | 37 |
| 14 | Pt–MoOx/TiO2 | 100 | 50 | 48 | 86 |
| 15 | Pt–MoOx/TiO2 | 80 | 50 | 16 | 13 |
| 16 | Pt–MoOx/TiO2 | 120 | 30 | 16 | 38 |
| 17 | Pt–MoOx/TiO2 | 120 | 10 | 16 | 6 |
| 18 | Pt/TiO2 | 120 | 50 | 16 | 8 |
| 19 | MoOx/TiO2 | 120 | 50 | 16 | 0 |
| 20 | TiO2 | 120 | 50 | 16 | 0 |
| 21 | Pt/TiO2 + MoOx/TiO2 | 120 | 50 | 16 | 9 |
| 22 | Blank | 120 | 50 | 16 | 0 |
To understand the contribution of the individual components of the most active catalysts, Pt/TiO2, MoOx/TiO2 and TiO2 materials were also prepared and tested showing negligible activity (Table 1, entries 18–20). In addition, the physical mixture of Pt/TiO2 and MoOx/TiO2 only showed a comparable yield with that of Pt/TiO2 (Table 1, entry 21). These results, compared with the activity of the bifunctional Pt–MoOx/TiO2 catalyst, clearly indicate that a synergistic effect exists between Pt and MoOx, which will be further discussed in the following section. As expected, no activity was observed without catalyst (Table 1, entry 21).
3.2 Catalyst characterizations
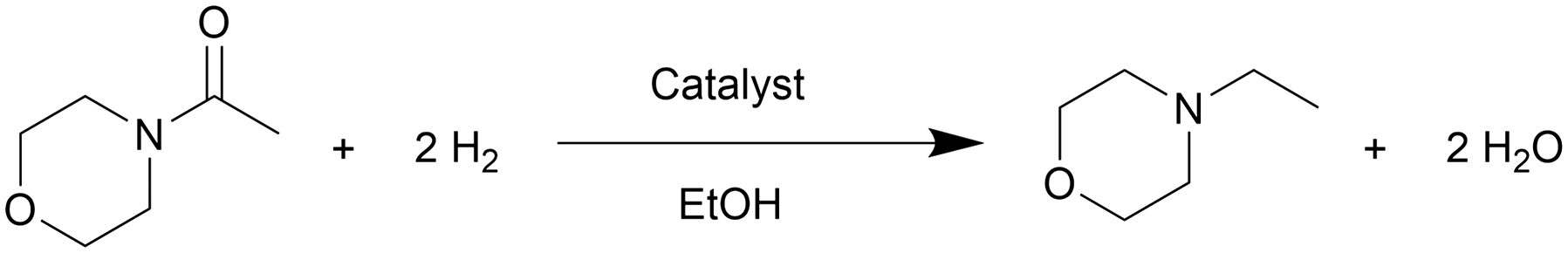
X-ray Photoelectron Spectroscopy (XPS) measurements of the Pt–MoOx/TiO2 catalyst with the MoOx/TiO2 were performed to determine the surface valence state of Mo, as shown in Fig. 1. The peak deconvolution of both materials revealed two doublets for each peak. The doublet with Mo 3d5/2 at 231.9 eV and Mo 3d3/2 at 235.0 eV can be assigned to Mo5+.20 The other peaks including Mo 3d3/2 around 236.2 eV and Mo 3d5/2 at 233.0 eV are characteristic peaks for Mo6+.20,21 Additionally, we calculated the ratio of Mo5+ and Mo6+ in both samples. As shown in Fig. 1, a higher Mo5+ ratio (27 at%) was observed over Pt–MoOx/TiO2 compared to MoOx/TiO2 (23 at%).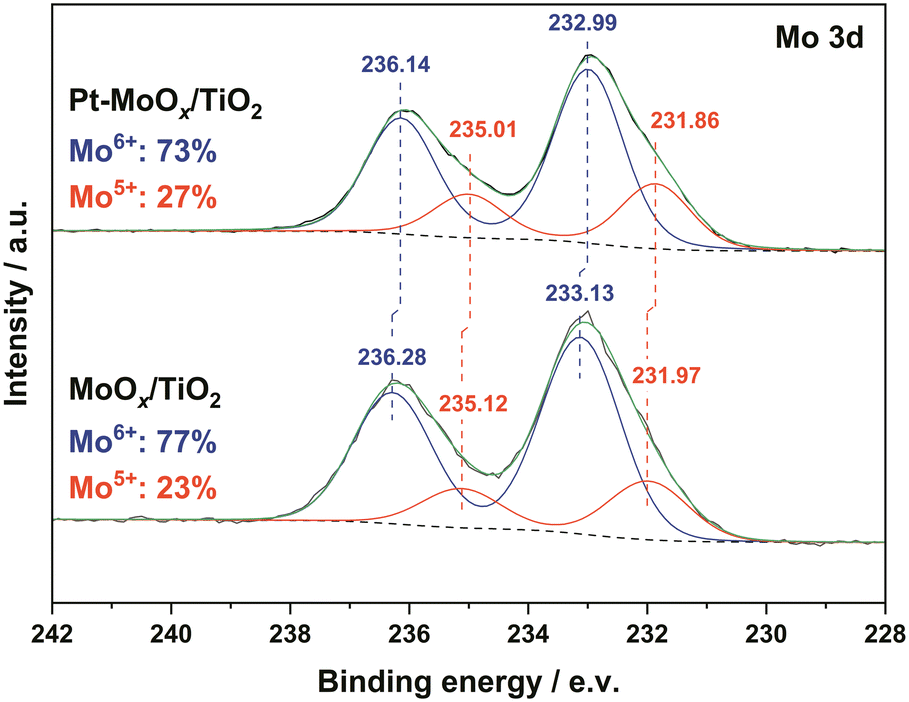 | ||
| Fig. 1 XPS spectra of the Mo 3d region of Pt–MoOx/TiO2 (upper graph) and MoOx/TiO2 catalysts (lower graph). | ||
These results suggest that the existence of Pt nanoparticles could reduce their neighboring Mo species to a certain extend. In other reported bifunctional catalysts (Ru–WOx/SiO216 and Pt–V2O5/HAP15), the low-coordinated W5+ and V3+ were proposed to activate the CO bond in amides. Hence, we would naturally consider that in our case, these coordinately unsaturated Mo species might also contribute to the adsorption and activation of carbonyl group in the amide. To verify the above-mentioned effect of Mo on CO activation, we carried out the adsorption of 4-acetylmorpholine over four samples (TiO2, MoOx/TiO2, Pt/TiO2, Pt–MoOx/TiO2) using in situ diffuse reflectance infrared Fourier transform spectroscopy (in situ DRIFTS), as shown in Fig. 2. The peaks at 1589–1596 cm−1 are ascribed to the stretching vibration of CO group (νCO) in the amide. The bands at 2841 cm−1 and 2931 cm−1 are attributed to the symmetric and asymmetric stretching vibration of cyclic –CH2–, respectively. The ones at 2855 and 2960–2972 cm−1 can be assigned to the symmetric and asymmetric stretching vibration of aliphatic –CH3, respectively.22 A red shift of νCO over Pt–MoOx/TiO2 (1589 cm−1) is observed compared with the peaks of the other three samples (1596 cm−1), suggesting that CO moiety is activated most by Pt–MoOx/TiO2. This result confirms our previous hypothesis, i.e., the Mo species reduced by its neighboring Pt nanoparticles contributes to the activation of carbonyl group.
 | ||
| Fig. 2 In situ DRIFTS for the adsorption of 4-acetylmorpholine over TiO2, MoOx/TiO2, Pt/TiO2, Pt–MoOx/TiO2 at 120 °C. | ||
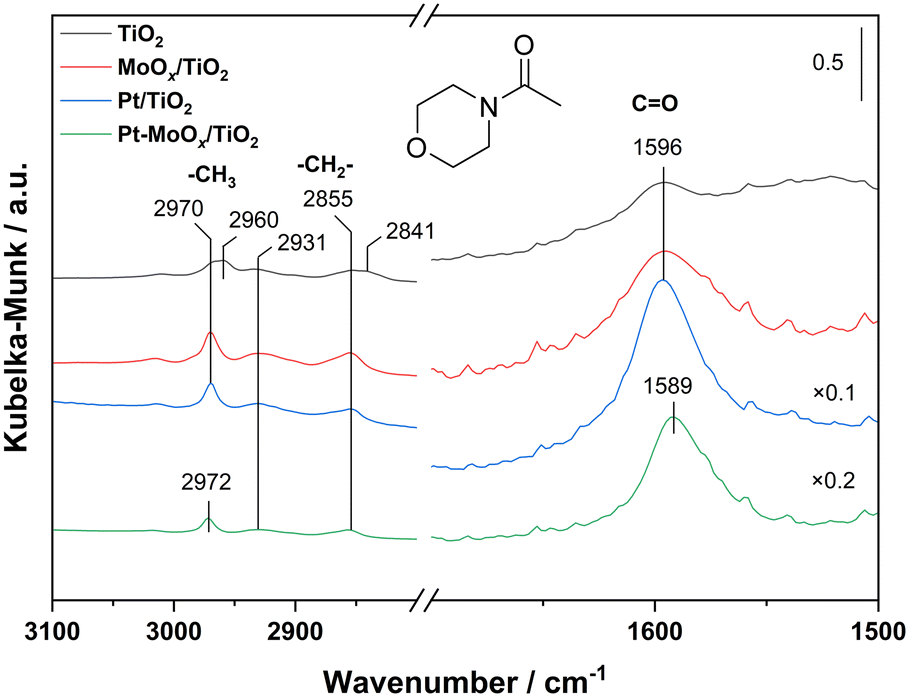
Besides CO activation, the ability of the catalysts to activate gas-phase H2, i.e., to provide surface hydrogen species, is also an important factor determining their overall performance in the hydrogenation of 4-acetylmorpholine. To check if the presence of MoOx is decisive for this catalyst property, we carried out H/D exchange tests with Pt–MoOx/TiO2 and Pt/TiO2 under transient conditions in the TAP reactor.19,23 As evidenced by the formation of HD (Fig. 3), both catalysts are able to break H–H and D–D bonds and to form new H–D bonds. These reactions occur exclusively on the surface of catalysts. Although H2 and D2 are partially converted into HD, there is an intersection between their responses and the response of Ar. According to the theory of the TAP reactor,19 such intersection is a fingerprint of reversible adsorption of the reactants. Thus, the presence of MoOx does not influence the mechanism of hydrogen activation. However, it influences the rate of H/D exchange. To illustrate this, we calculated the difference (Δtmax) between the dimensionless time of the maximal dimensionless flow of Ar (this time characterizes simple diffusion only) and that of HD (this time represents both diffusion and HD formation). The Δtmax values for Pt/TiO2 and Pt–MoOx/TiO2 are 0.74 and 0.60, respectively. Thus, the MoOx promoter enhances the rate of H/D exchange. This conclusion is also supported by the amount of HD formed in these experiments. The HD yield determined for Pt/TiO2 is 35%, while in the presence of MoOx it increases to 49%.
 | ||
| Fig. 3 Dimensionless responses of Ar, D2, H2 and HD recorded after pulsing of a D2:H2:Ar = 1:1:1 mixture in the TAP reactor over (a) Pt/TiO2 and (b) Pt–MoOx/TiO2 at 120 °C. | ||
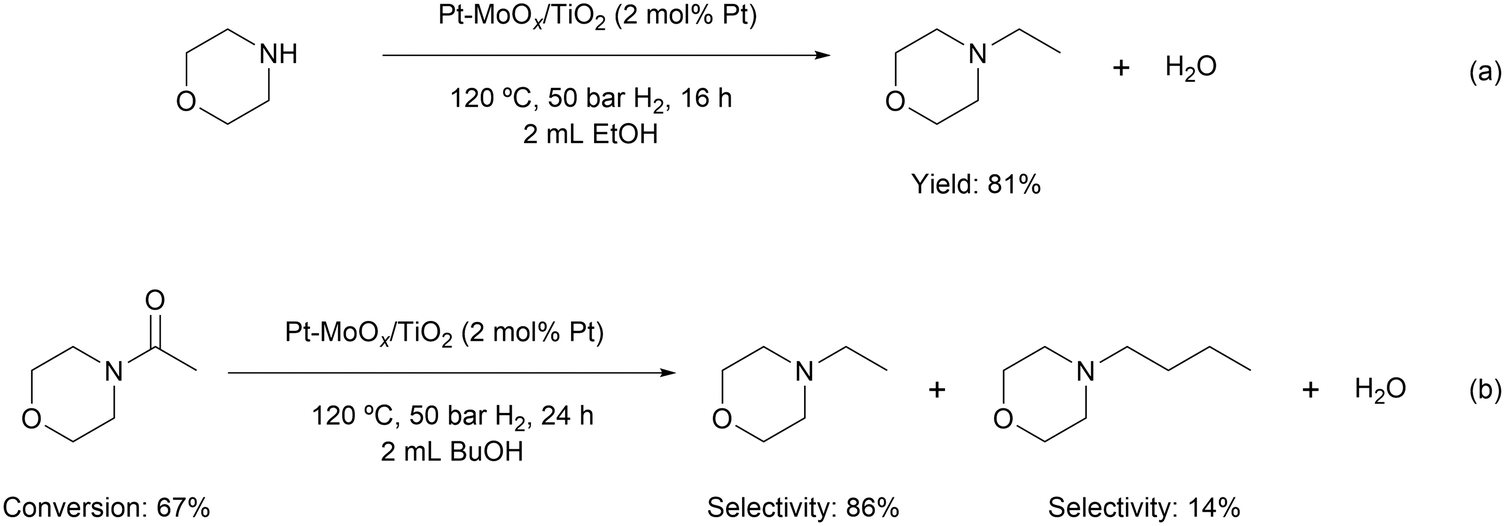
To probe the reaction process, we carried out in situ DRIFTS measurements for the hydrogenation of 4-acetylmorpholine over Pt–MoOx/TiO2. The substrate was carried by He and introduced into the in situ cell, being adsorbed on the catalyst surface. Then, the gas was switched to H2 for the hydrogenation reaction. The spectra were recorded as a function of time. As shown in Fig. 4, a peak around 2040 cm−1, corresponding to Pt–H bond,22 immediately appeared after the dosage of H2. This is easy to understand as Pt is very active for H2 adsorption and dissociation. Meanwhile, the peak intensity of νCO at 1589 cm−1 quickly decreased, as also described in the inset of Fig. 4. This result indicates that the carbonyl group of the amide, which was activated by the coordinatively unsaturated Mo species, could readily react with H dissociated on Pt site. In addition, a control experiment was carried out using morpholine and ethanol solvent at 120 °C and 81% yield of N-ethylmorpholine was obtained (eqn (a)). This result suggests that the Pt–MoOx/TiO2 catalyst is also active and selective for alcohol amination reactions. It explains the high product selectivity when using ethanol as solvent. As shown in Table S1,† the reaction could also proceed with other solvents like i-PrOH, ethers, cyclohexane, and toluene. While the selectivity is not as high as that in ethanol, since C–N cleavage competitively took place which gives morpholine as the byproduct. In ethanol, it is also possible that C–N cleavage occurred. But the formed morpholine would readily react with ethanol to give N-ethylmorpholine. This result clearly shows that the desired secondary amine can be formed either directly or in a two-step process involving initial hydrogenolysis, followed by subsequent hydrogen borrowing alkylation. To understand which reaction pathway is dominating in the presence of our catalyst, the hydrogenation of N-acetylmorpholine was performed in 1-butanol as solvent (eqn (b)). The ratio of N-(n-butyl)morpholine and N-ethylmorpholine is indicative for the ratio of the two- or one-step hydrogenation process. As shown in eqn (b), 86% N-ethylmorpholine and 14% N-(n-butyl)morpholine are observed in the product mixture, suggesting that the one-step hydrodeoxygenation process dominates in the reaction pathway. Based on the above results, we proposed the reaction pathway as shown in Fig. 5.
 | ||
| Fig. 4 In situ DRIFTS for the hydrogenation of 4-acetylmorpholine over Pt–MoOx/TiO2 at 120 °C. Inset: integrated area of the bands at 1589 cm−1vs. time. | ||
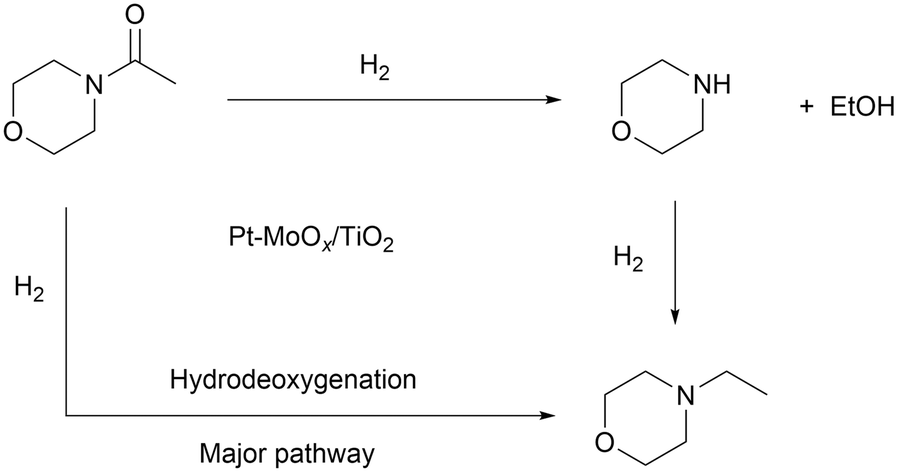 | ||
| Fig. 5 Proposed mechanism for the hydrogenation of 4-acetylmorpholine over Pt–MoOx/TiO2. | ||
3.3 Substrate scope
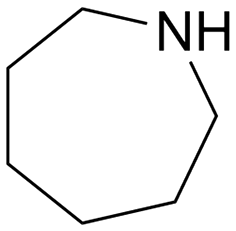
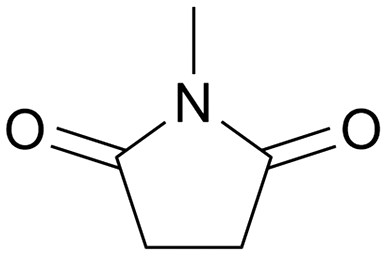
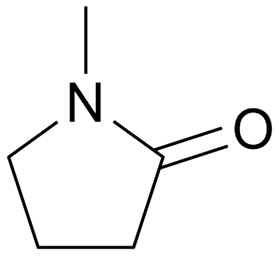
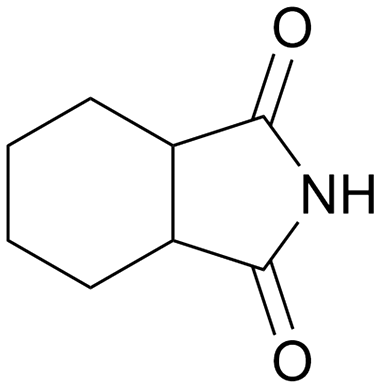
With an optimized amide hydrogenation catalyst and suitable reaction conditions in hand, we then investigated the reactivity of selected other substrates utilizing this Pt–MoOx/TiO2 catalyst (Table 2). Moderate to good yields were observed for the hydrogenation of tertiary amides at 120–150 °C (Table 2, entries 1–8). In case of a strongly electron-withdrawing group (–CF3) – presented next to the CO bond, C–N cleavage readily took place (Table 2, entry 6). With respect to the hydrogenation of a secondary amide (ε-caprolactam), 66% yield of the secondary amine (azepan, Table 2, entry 10) was obtained using 1,4-dioxane as solvent to prevent further alcohol amination. Applying a primary amide, no desired product was observed with this Pt–MoOx/TiO2 catalyst (Table S2,† entry 1), similar with the results reported using Pt-based catalysts.15 Interestingly, this catalyst also works well for the selective hydrogenation of imides (Table 2, entries 11–13) and esters (Table S2,† entry 4), giving >90% yield of corresponding amides and 87% of benzyl alcohol, respectively.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | Product | Temperature/°C | Time/h | Selectivity/% | Yield/% |
| Reaction conditions: substrate: 0.5 mmol, [Pt] catalyst (2 mol%), H2 (50 bar), EtOH (2 mL). Yield was determined by GC-FID using n-hexadecane as an internal standard.a 1,4-Dioxane as solvent.b C–N cleavage mainly accounts for the byproducts. | ||||||
| 1 |
|
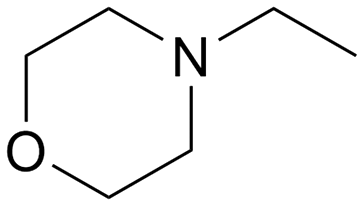
|
120 | 24 | 99 | 92 |
| 2 |
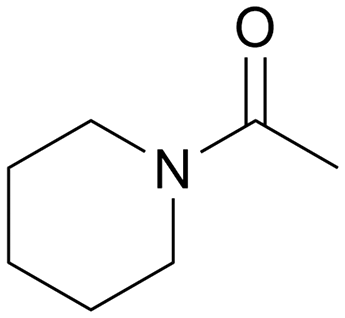
|
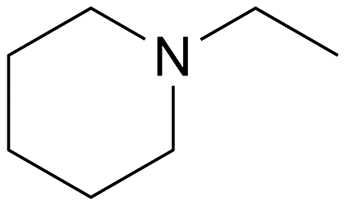
|
120 | 24 | 99 | 35 |
| 3 | 140 | 48 | 99 | 54 | ||
| 4 |
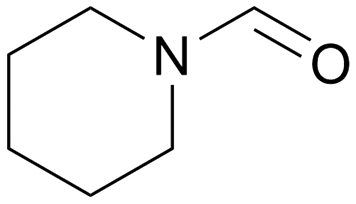
|
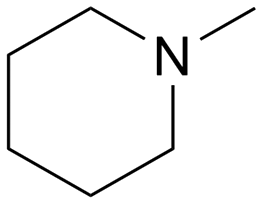
|
120 | 24 | 98 | 56a |
| 5 | 150 | 48 | 97 | 97a | ||
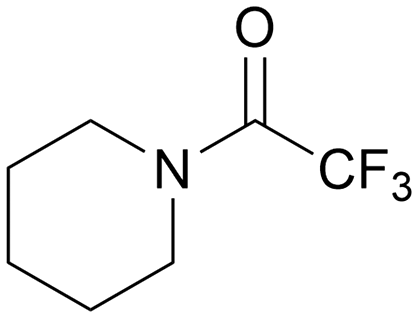
| 6 |
|
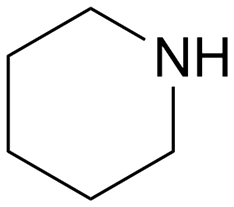
|
120 | 24 | 99 | 98a |
| 7 |
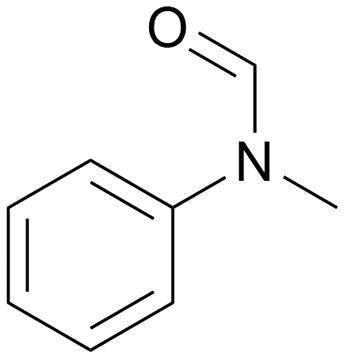
|
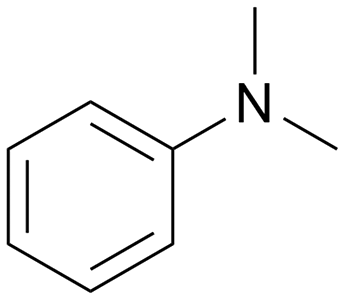
|
120 | 24 | 60b | 53 |
| 8 |
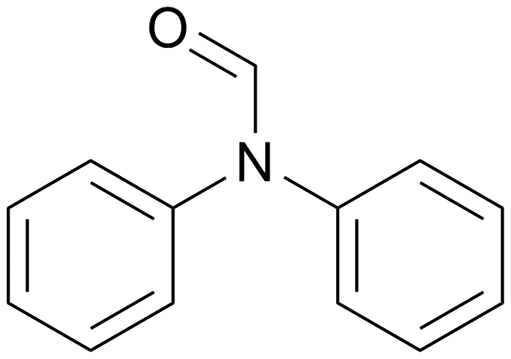
|
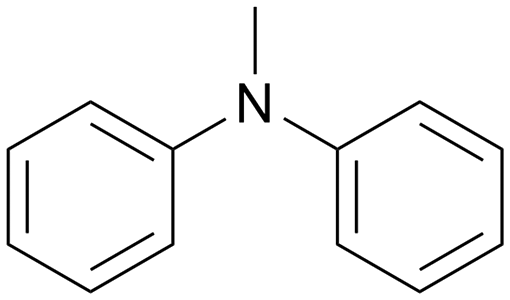
|
120 | 24 | 75b | 74 |
| 9 |
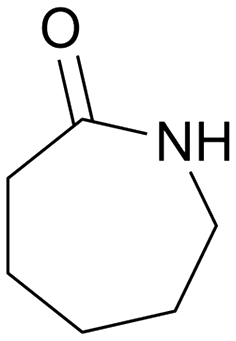
|
|
120 | 24 | 70b | 37a |
| 10 | 140 | 48 | 67b | 66a | ||
| 11 |
|
|
120 | 24 | 98 | 96 |
| 12 |
|
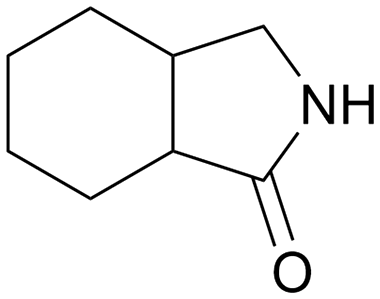
|
120 | 24 | 90 | 75a |
| 13 | 140 | 24 | 95 | 90a | ||

4. Conclusions
We presented a Pt–MoOx/TiO2 catalyst for the hydrogenation of amides to amines. Compared to most known catalysts for this challenging transformation, this novel material allows for selective reductions under comparably mild conditions. Characterizations revealed the synergistic effect between Pt and MoOx. In more detail, the latter species which is reduced by neighboring Pt nanoparticles, is responsible for amide adsorption and activation of its CO bond. On the other side, modifying Pt by MoOx enhances the capability of the former to dissociate H2. The bifunction of Pt and MoOx contributes to the good catalytic performances at a temperature of 120 °C. Control experiments revealed a one/two-step hydrogenation pathway as the main mechanism. Apart from amide reductions, this catalyst system also offers interesting possibilities for the green and sustainable alkylation of amines by alcohols.
Author contributions
R. Q. conceived the idea and designed the experiments; R. Q. prepared the catalysts; R. Q., S. M. and H. Q. performed the experiments and analyzed the data; J. W. performed in situ DRIFTS measurements; V. K., and E. K. were responsible for TAP experiments; S. B. carried out XPS experiments; A.-E. S. worked on the heat treatment of the catalysts; K. J. and M. B. supervised the research activities and supported the project with funding acquisition; R. Q., K. J. and M. B. co-wrote the paper; all the authors contributed to editing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the staff in the analytical department of the Leibniz Institute for Catalysis for their excellent service. R. Q. and H. Q. gratefully acknowledges the financial support from the Alexander von Humboldt Foundation (CHN 1212814 HFST-P and CHN 1220532 HFST-P).Notes and references
- J. R. Cabrero-Antonino, R. Adam, V. Papa and M. Beller, Nat. Commun., 2020, 11, 3893 CrossRef CAS PubMed.
- T. Leischner, L. A. Suarez, A. Spannenberg, K. Junge, A. Nova and M. Beller, Chem. Sci., 2019, 10, 10566–10576 RSC.
- V. Papa, J. R. Cabrero-Antonino, E. Alberico, A. Spanneberg, K. Junge, H. Junge and M. Beller, Chem. Sci., 2017, 8, 3576–3585 RSC.
- A. M. Smith and R. Whyman, Chem. Rev., 2014, 114, 5477–5510 CrossRef CAS PubMed.
- A. Guyer, A. Bieler and G. Gerliczy, Helv. Chim. Acta, 1955, 38, 1649–1654 CrossRef CAS.
- H. J. Schneider, H. Adkins and S. M. McElvain, J. Am. Chem. Soc., 1952, 74, 4287–4290 CrossRef CAS.
- H. S. Broadbent and W. J. Bartley, J. Org. Chem., 1963, 28, 2345–2347 CrossRef CAS.
- G. Beamson, A. J. Papworth, C. Philipps, A. M. Smith and R. Whyman, J. Catal., 2010, 269, 93–102 CrossRef CAS.
- G. Beamson, A. J. Papworth, C. Philipps, A. M. Smith and R. Whyman, Adv. Synth. Catal., 2010, 352, 869–883 CrossRef CAS.
- G. Beamson, A. J. Papworth, C. Philipps, A. M. Smith and R. Whyman, J. Catal., 2011, 278, 228–238 CrossRef CAS.
- M. Stein and B. Breit, Angew. Chem., Int. Ed., 2013, 52, 2231–2234 CrossRef CAS PubMed.
- R. Burch, C. Paun, X. M. Cao, P. Crawford, P. Goodrich, C. Hardacre, P. Hu, L. McLaughlin, J. Sá and J. M. Thompson, J. Catal., 2011, 283, 89–97 CrossRef CAS.
- J. Coetzee, H. G. Manyar, C. Hardacre and D. J. Cole-Hamilton, ChemCatChem, 2013, 5, 2843–2847 CrossRef CAS.
- K.-i. Shimizu, W. Onodera, A. S. Touchy, S. M. A. H. Siddiki, T. Toyao and K. Kon, ChemistrySelect, 2016, 1, 736–740 CrossRef CAS.
- T. Mitsudome, K. Miyagawa, Z. Maeno, T. Mizugaki, K. Jitsukawa, J. Yamasaki, Y. Kitagawa and K. Kaneda, Angew. Chem., Int. Ed., 2017, 56, 9381–9385 CrossRef CAS PubMed.
- Y. Zhang, L. Li, F. Liu, H. Qi, L. Zhang, W. Guan, Y. Liu, A. Wang and T. Zhang, ACS Catal., 2022, 12, 6302–6312 CrossRef CAS.
- S. Werkmeister, K. Junge and M. Beller, Org. Process Res. Dev., 2014, 18, 289–302 CrossRef CAS.
- M. Macino, A. J. Barnes, S. M. Althahban, R. Qu, E. K. Gibson, D. J. Morgan, S. J. Freakley, N. Dimitratos, C. J. Kiely, X. Gao, A. M. Beale, D. Bethell, Q. He, M. Sankar and G. J. Hutchings, Nat. Catal., 2019, 2, 873–881 CrossRef CAS.
- J. T. Gleaves, G. S. Yablonskii, P. Phanawadee and Y. Schuurman, Appl. Catal., A, 1997, 160, 55–88 CrossRef CAS.
- J. Baltrusaitis, B. Mendoza-Sanchez, V. Fernandez, R. Veenstra, N. Dukstiene, A. Roberts and N. Fairley, Appl. Surf. Sci., 2015, 326, 151–161 CrossRef CAS.
- J. Chang, T. Danuthai, S. Dewiyanti, C. Wang and A. Borgna, ChemCatChem, 2013, 5, 3041–3049 CrossRef CAS.
- G. Socrates, Infrared and Raman Characteristic Group Frequencies: Tables and Charts, John Wiley and Sons, Chichester, 3rd edn, 2001 Search PubMed.
- K. Morgan, N. Maguire, R. Fushimi, J. T. Gleaves, A. Goguet, M. P. Harold, E. V. Kondratenko, U. Menon, Y. Schuurman and G. S. Yablonsky, Catal. Sci. Technol., 2017, 7, 2416–2439 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cy01274c |
| This journal is © The Royal Society of Chemistry 2024 |