
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A general and expedient amination of alcohols catalysed by a single-site (NN)Co(II)-bidentate complex under solventless conditions†
Rohit
Kumar
a,
Ankit Kumar
Srivastava
a,
Palaniyappan
Nagarasu
b,
Vedichi
Madhu
 *b and
Ekambaram
Balaraman
*a
*b and
Ekambaram
Balaraman
*a
aDepartment of Chemistry, Indian Institute of Science Education and Research (IISER), Tirupati – 517507, India. E-mail: eb.raman@iisertirupati.ac.in
bDepartment of Applied Chemistry, Karunya Institute of Technology and Science (Deemed to be University), Coimbatore – 641114, Tamil Nadu, India. E-mail: madhu@karunya.edu
First published on 22nd November 2023
Abstract
Here we designed and synthesized a NN–CoII bidentate complex and efficiently used it for general and expedient amination of alcohols under benign, solventless conditions. Both primary (including unactivated aliphatic) alcohols and sterically hindered secondary alcohols exhibited very good reactivity and provided diverse amines with good substrate scope (88 examples; up to 95% yields) and excellent functional group tolerance (methoxy, thiomethoxy, phenoxy, trifluoromethyl, amino, alcoholic and halides including bromo and iodo groups). Furthermore, a sequential bis-N-alkylation of diamines was also demonstrated. It was observed that the pyrazole moiety in the ligand backbone plays a crucial role in the amination reaction. Very interestingly, the reusability of the present homogeneous cobalt catalyst was successfully demonstrated.
Introduction
Amines are fundamentally important compounds and play a vital role in the chemical and biological sciences.1,2 They are industrially significant commodity chemicals, as well as versatile building blocks to produce fine and specialty chemicals. In particular, N-alkylated amine derivatives have widespread applications in agrochemicals, pharmaceuticals, lubricants, organic dyes, corrosion inhibitors, surfactants, and polymer industries.3–5 In view of this, numerous synthetic routes have been developed for C–N bond formation reactions to N-alkylated amines.6–12 Conventionally, this process involves nucleophilic substitution with organic halides,13 hydroamination,12,14,15 and reductive amination of aldehydes or carboxylic acids.16–19 However, these methods have been curtailed due to the need for harsh reaction conditions, utilization of toxic and harmful reagents, and preactivated starting materials, leading to the generation of a large amount of inorganic waste, resulting in poor selectivity and low yield, thereby limiting their applicability. Thus, the development of new catalytic systems for the sustainable and affordable benign synthesis of N-alkylated amines is highly demanding and challenging.The borrowing hydrogen (BH) and the hydrogen auto transfer (HA) amination strategies are powerful approaches to access N-alkylated amine derivatives, starting from simple and abundantly available alcohols as alkylating agents.20–25 The C–N bond formation reactions via the BH/HA approach are superior from the step- and atom-economic point of view, as they integrate transfer hydrogenation by circumventing the direct use of hydrogen gas with other (in situ) intermediate reactions to selectively yield the desired compounds. Thus, BH/HA catalysis offers several advantages over traditional methods, as this tandem process replaces the use of hazardous and mutagenic alkylating agents by activating the alcohol moiety, resulting in expedient production of N-alkylated amines with water as the sole by-product. There are seminal reports on N-alkylation amines or amination of alcohols via the BH/HA strategy, mostly catalysed by rare noble-metals.26–38 Of late, the development of sustainable, earth-abundant, and non-precious transition-metal-based catalytic systems (Cu, Ni, Co, Fe, and Mn) are becoming more appealing for the replacement of rare element-based catalytic chemical production.39–41
In recent years, there is considerable growing evidence that molecular cobalt complexes can be potential catalysts for (de)hydrogenation and related reactions.40,42,43 However, there are very limited reports on cobalt-catalysed selective amine alkylation reactions44–54 and BH C–C and C–N bond forming reactions.55–59 Most of the molecular Co-complexes employed for catalytic N-alkylation of amines with alcohols are based on tridentate PNP or PNN ligand systems (Scheme 1). In 2015, Kempe and co-workers reported a PN5P ligand with triazine backbone stabilized Co(II)–PNP pincer complex employed for the efficient N-alkylation of both aromatic and aliphatic amines with alcohols.54 Kirchner46 developed a molecular cobalt(II) complex stabilized by an anionic PCP ligand based on the 1,3-diaminobenzene scaffold for the alkylation of amines by primary alcohols. A base-free N-alkylation of anilines catalysed by aliphatic PNP–Co(II) complex was reported by Zhang and co-workers.47
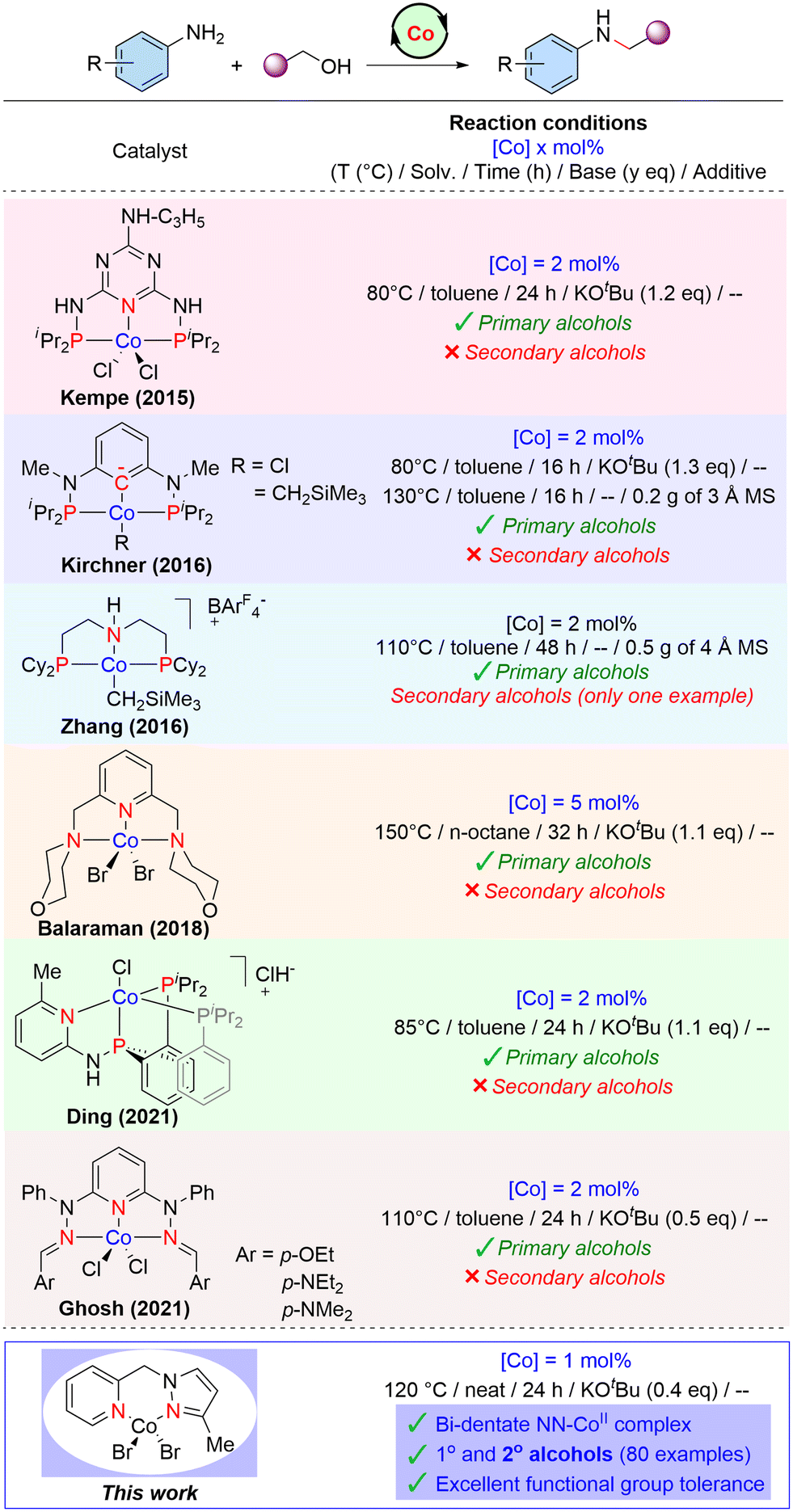 | ||
| Scheme 1 Molecular Co(II)-complexes for amine alkylation. | ||
Recently, the research groups of Ding48 and Ghosh50 independently reported cobalt-catalysed N-alkylation of amines based on tridentate PNP and NNN systems, respectively. Özdemir described the alkylation of anilines by activated benzyl alcohols catalysed by an alkyl benzimidazole–cobalt(II) complex.52 However, this reaction is unselective and produced a mixture of N-alkylated amines and imines. Our group also developed a NNN–Co(II) complex for the N-alkylation of amines using primary alcohols.51 The prior examples of molecular cobalt(II) complexes employed for the N-alkylation reaction are mainly based on tridentate-supported ligand systems, and applicable only to primary alcohols. Indeed, the application of a secondary alcohol for the N-alkylation reaction is scarcely explored due to their lower reactivity compared to the primary ones. Here, we have established a bench-stable, phosphine-free Co-complex supported by an NNPyMe (L1) bidentate ligand, which has been proven to be an efficient and versatile precatalyst for the expedient N-alkylation of amines under benign conditions. Both primary (including unactivated aliphatic) alcohols and sterically hindered secondary alcohols exhibited excellent reactivity with broad substrate scope under the present Co-catalysed conditions.
The NNPyMe–Co(II) bi-dentate complex is easy to synthesize and simple to activate. This can be synthesized quantitatively on a gram scale and is an air-stable crystalline material for several months (both in solid and solution states). The reaction of a phosphine-free NNPyMe bi-dentate ligand (L1) with anhydrous CoBr2 in acetonitrile solvent at room temperature under ambient conditions resulted in the corresponding Co-complex in 90% isolated yield (Scheme 2). In a similar manner, the (NN)Mor–CoBr2 complex was also synthesized (see the ESI†).
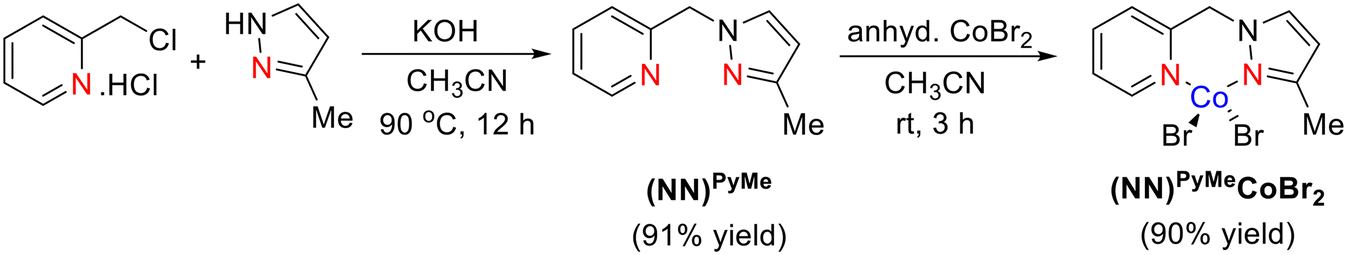 | ||
| Scheme 2 Synthesis of the bi-dentate (NN)PyMeCoBr2 bidentate complex. | ||
The NNPyMe–CoBr2 complex was fully characterized using various analytical (elemental analysis, high-resolution mass spectrometry) and spectroscopic techniques such as IR, UV-vis, and 1H NMR. The magnetic behavior of (NN)PyMeCoBr2 was confirmed using the Evans method, suggesting a magnetic moment of 1.88 BM at the CoII-center (S = 1/2) (see the ESI†). The complex crystallized in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . The molecular structure of complex I was confirmed by a single-crystal X-ray diffraction study, as shown in Fig. 1. The X-ray diffraction analysis reveals that the Co(II) center in Co_L1 displayed a four coordinated, distorted tetrahedral structure with two nitrogen donor atoms of the ligand (L1) and two bromide ions. The bond angle of N(1)–Co(1)–N(2), N(2)–Co(1)–Br(2), N(1)–Co(1)–Br(2), N(2)–Co(1)–Br(1) and N(1)–Co(1)–Br(1) are found to be 92.28(17)°, 110.72(13)°, 113.33(12), 116.66(12) and 115.51(12), respectively.
. The molecular structure of complex I was confirmed by a single-crystal X-ray diffraction study, as shown in Fig. 1. The X-ray diffraction analysis reveals that the Co(II) center in Co_L1 displayed a four coordinated, distorted tetrahedral structure with two nitrogen donor atoms of the ligand (L1) and two bromide ions. The bond angle of N(1)–Co(1)–N(2), N(2)–Co(1)–Br(2), N(1)–Co(1)–Br(2), N(2)–Co(1)–Br(1) and N(1)–Co(1)–Br(1) are found to be 92.28(17)°, 110.72(13)°, 113.33(12), 116.66(12) and 115.51(12), respectively.
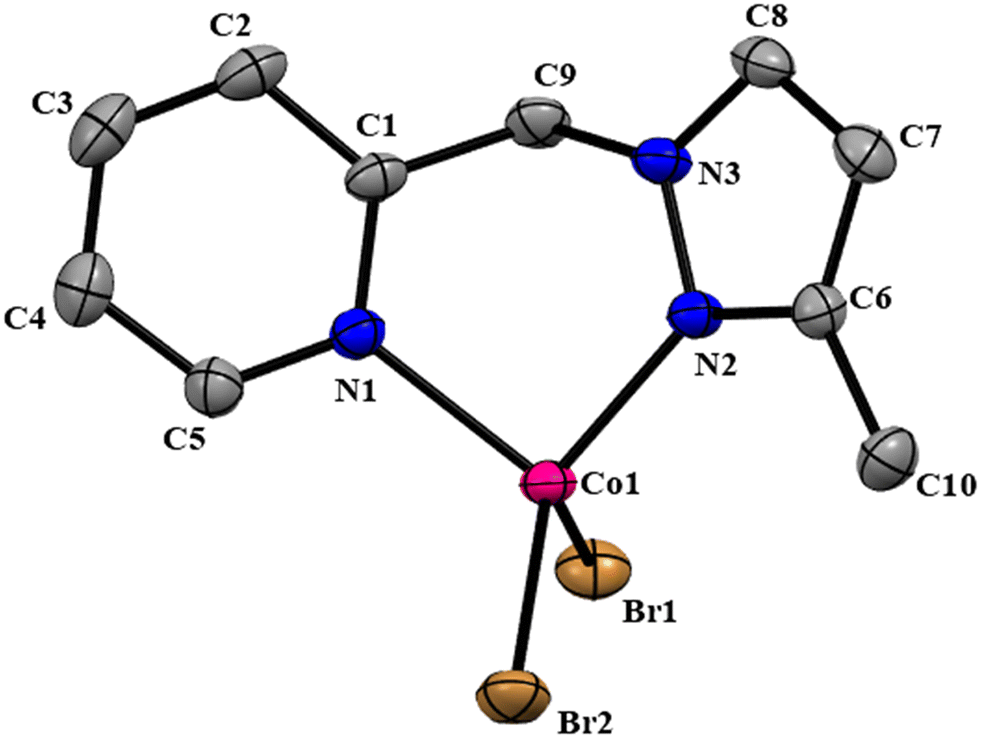 | ||
| Fig. 1 ORTEP diagram of the (NN)PyMeCoBr2 complex with 50% probability ellipsoids. Hydrogen atoms are omitted for clarity. CCDC no: 2264033. | ||
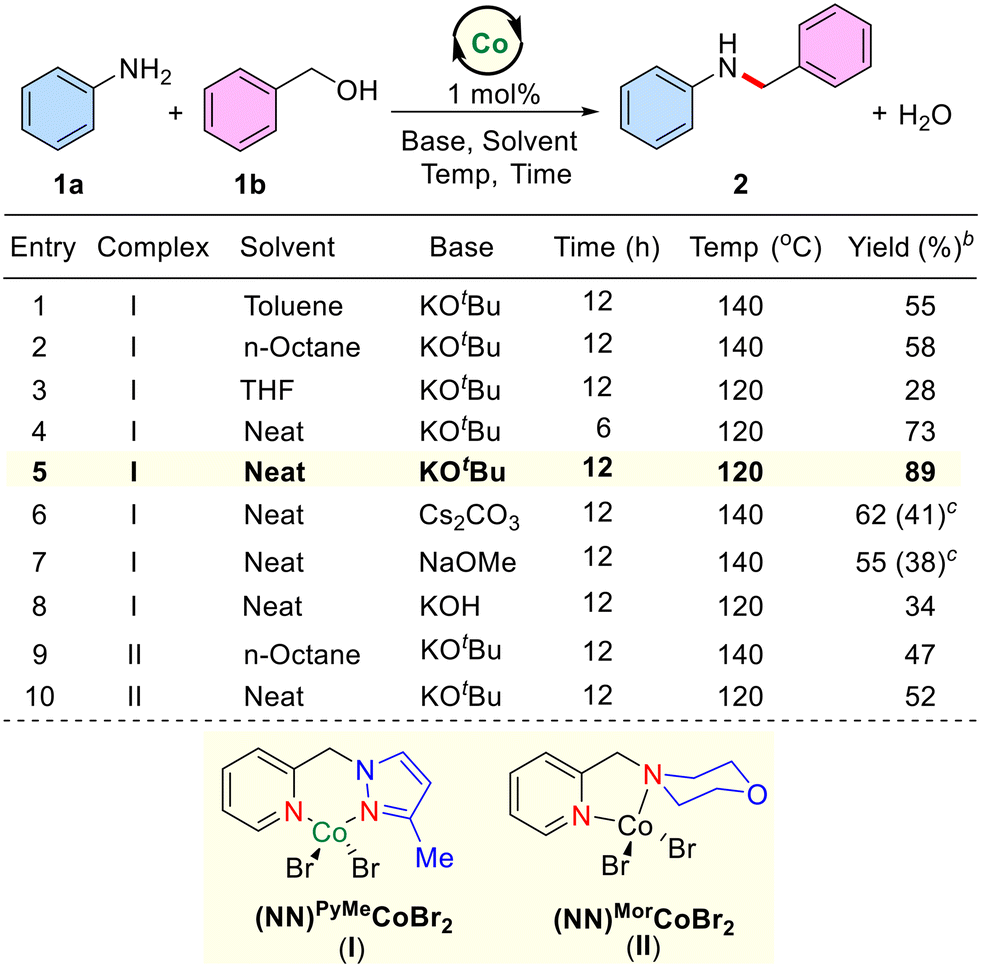
Intrigued by literature precedents, we commenced to explore the potential catalytic activity of the newly synthesized phosphine ligand-free NN–Co(II) bidentate complex as a precatalyst for the selective amine alkylation reaction using alcohols as alkylating agents. For our initial studies, unsubstituted aniline (1a) and benzyl alcohol (1b) were chosen as the benchmark substrates. Several reaction parameters, including solvent, reaction temperature, and base were examined systematically by using these substrates (Table 1 and see the ESI,† for optimization studies). Thus, in a typical experiment, the reaction of aniline (1a), benzyl alcohol (1b), a catalytic amount of (NN)PyMe cobalt complex (I; 1 mol%) and KOtBu (40 mol%) in toluene at 140 °C (silicon oil-bath temperature) after 12 h afforded the corresponding amine alkylated product N-benzylaniline (2) in 55% yield. Indeed, continuing the reaction for 24 h at the same temperature didn't improve the yield of the product. Under similar reaction conditions, n-octane and THF gave 58% and 28% yields, respectively. Interestingly, the effective N-alkylation of 1a with 1b proceeded in 73% yield under solventless (neat) conditions at 120 °C after 6 h. Encouraged by this result, we performed the same reaction for 12 h and yielded product 2 in an 89% isolated yield. Screening of bases such as Cs2CO3, NaOMe, and KOH under solvent-free conditions gave unsatisfactory results. Notably, in the absence of catalyst or base, no formation of product was observed. Employing a low catalytic amount of a Co-complex (0.05 mol%), the N-alkylation reaction also worked efficiently, and product 2 was isolated in 76% yield (see the ESI†). Notably, under similar reaction conditions, a (NN)Mor–CoBr2 complex (II) derived from the 4-(5yridine-2-ylmethyl)morpholine as the ligand did not perform better than the (NN)PyMe cobalt complex (I). It was observed that the pyrazole moiety in the ligand backbone plays a crucial role in the amination reactions. Indeed, the pyrazole ligand is an excellent π-acceptor, which stabilizes low-valent cobalt species and also provides extra stability to the Co(II)-complex due to its kinetically inert nature.
Having identified the optimized reaction conditions (1 mol% of complex I, 40 mol% of KOtBu under solventless conditions), we investigated the N-alkylation of anilines with various benzyl alcohols catalysed by the NNPyMe–cobalt bidentate complex for the generality of this reaction as well as to extend the substrate scope. It is interesting to see that the NMR, GC, and GC–MS analyses of the crude reaction mixture showed no formation of N,N′-dialkylated products under our standard conditions.
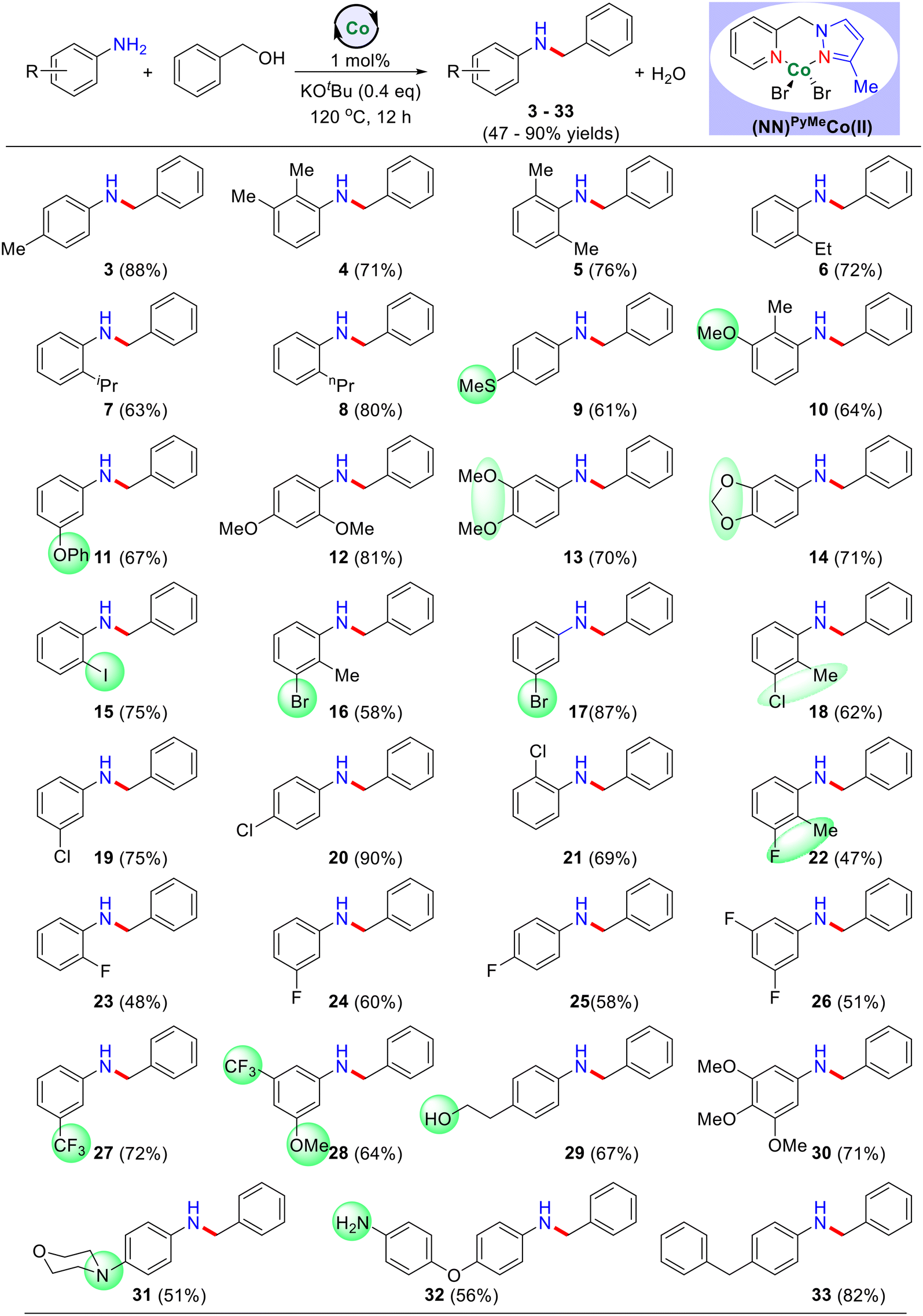
Initially, we explored the selective N-alkylation of benzyl alcohols 1b with various substituted aniline derivatives (Table 2). Regardless of the positions (para and meta) of the electron-releasing and the electron-withdrawing substituents on anilines, the reaction proceeded smoothly under the present cobalt-catalysis and yielded the corresponding N-alkylated products in good to excellent yields. Thus, electron-donating groups (–SMe, –OMe and –benzyl) at the para position of aniline afforded the corresponding N-alkylated products (9, 11, and 33) in good yields up to 82%. Similarly, the electron-withdrawing substituents (–F and –CF3) on aniline also yielded the desired N-alkylated products in very good yields (products 22–28; up to 72% yields). Interestingly, anilines possessing the ortho substituents, such as alkyl (methyl, ethyl, isopropyl, n-propyl), methoxy and halide (–F, –Cl, –I) substituents were very well tolerated, and gave the corresponding products in moderate to good yields (products 3–8, 10, 12, 16, and 21–23; 47–81% isolated yields). These reaction conditions were also compatible with methyl ether and methylene dioxy groups and afforded the corresponding products in good yields (products 13 in 70% and 14 in 71% yields). Challenging substituents such as bromide and iodides also worked very well under the optimized reaction conditions and gave the expected N-alkylated products with hydrodehalogenation (products 15 in 75%, 16 in 58%, and 17 in 87% yields). Next, we investigated the effect of the withdrawing groups like –F, –CF3, –OCF3 and observed a moderate yield of the product. Notably, a trace amount of product was only observed in the case of the –NO2 and –COOH groups. It may be due to the formation of other side reactions like hydrogenation of nitro compounds and esterification of carboxylic acid, respectively. It was also observed that highly electron-rich substrates showed excellent reactivity with good product yield, while electron-withdrawing groups had lower catalytic activity. Importantly, substrates bearing an amino group (Table 2, products 31–32) were well-tolerated with this method and provided 51–56% yields. The reaction of aniline with benzyl alcohols containing an alcoholic group under standard reaction conditions led to the N-alkylated product (29) in 67% isolated yield (product). Notably, the alcoholic motif in 29 remains intact and indeed, there was no formation of self-coupled product. This is evidence that activated benzyl alcohols showed excellent reactivity compared to unactivated alcohols.
| a Reaction conditions: aniline derivatives (0.55 mmol), benzyl alcohol (0.5 mmol), catalyst I (1 mol%), and KOtBu (0.2 mmol) heated at 120 °C (silicon oil-bath temperature) for 12 h under an argon atmosphere. The yields of isolated products is in parentheses. |
|---|
|
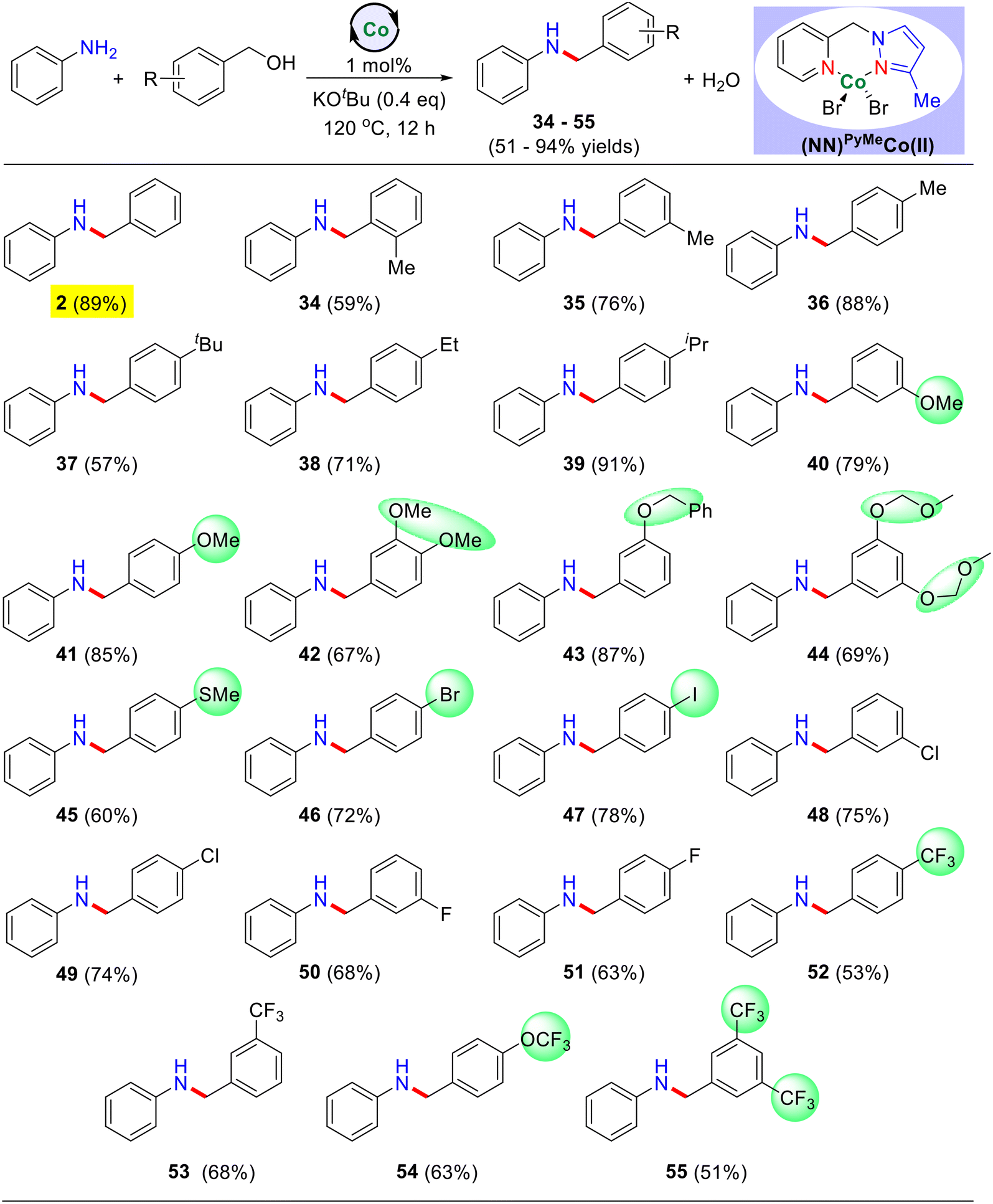
Next, we have investigated a variety of substituted benzyl alcohols for the N-alkylation reaction using unsubstituted aniline as the benchmark substrate (Table 3). Notably, electron-donating substituents (−Me, –OMe, –SMe, –iPr, –tBu, −Et, and others) at the meta and para positions of the benzyl alcohol substrate gave the corresponding N-alkylated products in good to excellent yields ranging from 57–91% (Table 3, products 34–45). Additionally, meta-substituted dimethoxy, phenoxy, and 3,5-bismethoxymethoxy groups also yielded the desired products in good yield (Table 3, products 42–44). Similarly, electron-withdrawing groups such as –F, –Cl, –CF3, and –OCF3 were well-tolerated and resulted in the corresponding N-alkylated products in moderate yields ranging from 51–68% (Table 3, products 50–55). Furthermore, para-substituted benzyl alcohols bearing –I and –Br were also successfully employed, yielding the desired N-alkylated products in good yield (Table 3, products 46–47).
| a Reaction conditions: aniline (0.5 mmol), benzyl alcohol derivatives (0.55 mmol), catalyst I (1 mol%), and KOtBu (0.2 mmol) heated at 120 °C (silicon oil-bath temperature) for 12 h under an argon atmosphere. Yield of isolated products are in parentheses. |
|---|
|
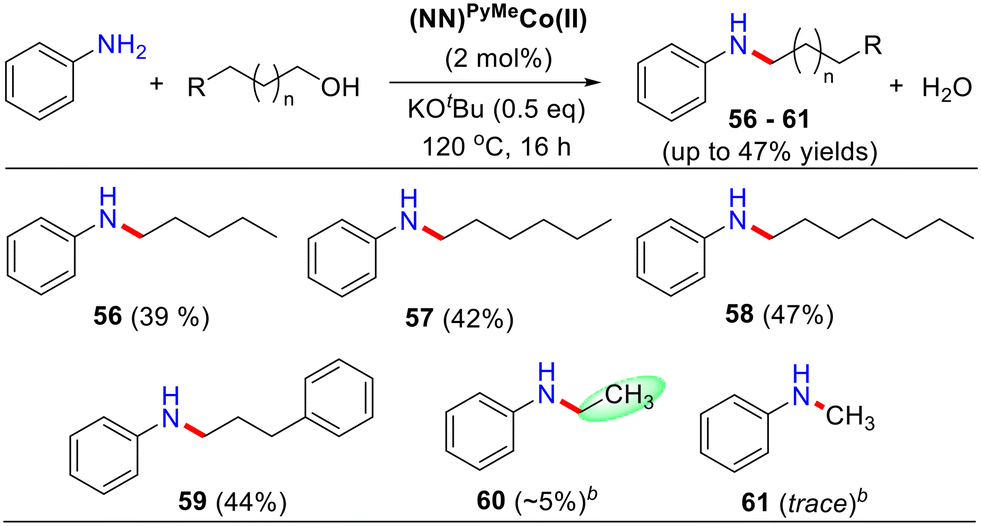
To expand the substrate scope, we examined a comprehensive list of activated primary alcohols (benzyl alcohol analogs) and some inactivated aliphatic alcohols for selective N-alkylation of aniline (Table 4). Initially, the homologous series of linear pentanol to heptanol were dehydrogenated for alkylation with aniline, resulting in low yields of 39–47% (Table 4, products 56–58). With an increase in the –CH2– units in linear alcohols, there was a slight increase in the yield of N-alkylated products. 3-Phenylpropanol also yielded a moderate yield of 47% (Table 4, 59). However, the dehydrogenative coupling of ethanol and methanol was not compatible with this method.
Motivated by the remarkable catalytic activity demonstrated in the N-alkylation of aniline with primary (aromatic and aliphatic) alcohols, we aimed to investigate the reactivity of the present phosphine-free (NN)PyMeCOBr2 bidentate complex in the N-alkylation with sterically demanding secondary alcohols. Indeed, the N-alkylation of amines using secondary alcohols as alkylating agents is very challenging. This is due to the steric hindrance of secondary alcohols and the difficulty in dehydrogenating their corresponding ketones, followed by hydrogenation of the tetrasubstituted imine intermediate. Notably, the present bidentate pyridine–pyrazole (NN)–Co(II) complex has been proven to be an efficient and versatile precatalyst for the N-alkylation of amines with both primary alcohols and sterically hindered secondary alcohols. However, the present bidentate ligand has less steric crowding around the metal than the tridentate pincer ligand, which is often used for N-alkylation reactions. Our ligand has a pyrazole–pyridine unit with an angle of about 120 Å, which makes it easier for bulky secondary alcohols like biphenylmethanol to reach the metal. The metal can then catalyze the alcohol dehydrogenation and the imine hydrogenation. Also, the pyrazole ligand is a good π-acceptor, which stabilizes low-valent cobalt species and improves the kinetic stability of the Co(II)-complex. Thus, through a systematic exploration of various secondary alcohols, benzhydrol emerged as the optimal candidate, delivering 55% of the desired N-alkylated product under neat reaction conditions. However, due to its solid nature and the desire to maximize yield, modifications to the reaction conditions were implemented. Specifically, the addition of a suitable solvent, particularly toluene, was used to enhance the solubility of the reactants, leading to a substantial increase in yield, up to 78% yield. Additional optimization steps including increasing reaction temperature, reaction time, and base and catalyst loading ultimately afforded an outstanding 85% yield of the desired N-alkylated aniline. The optimized reaction conditions were found to be Cat. I (1.5 mol%), a reaction temperature of 140 °C, KOtBu (50 mol%), and 1 mL of toluene as solvent, refluxed for 24 hours.
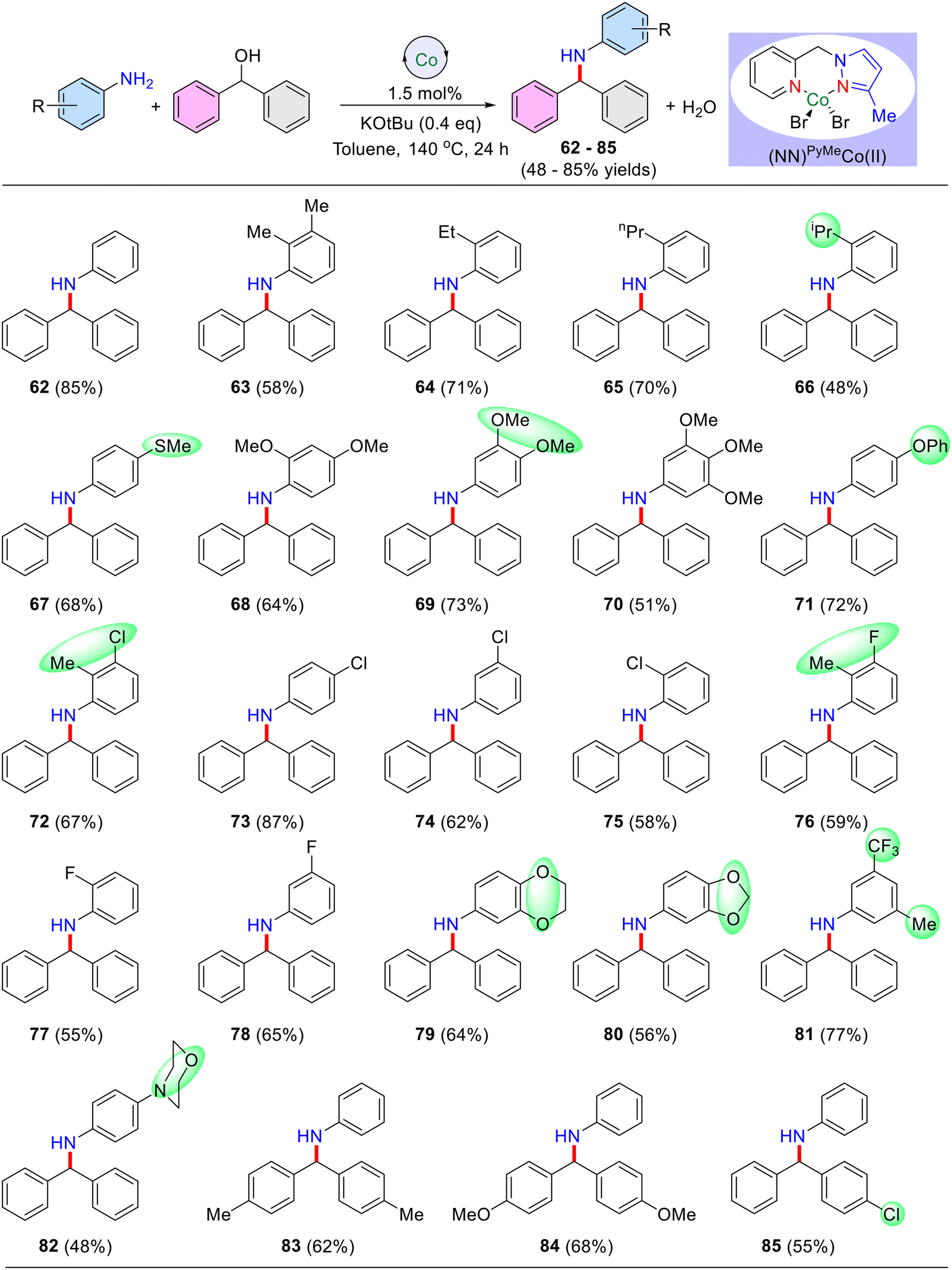
Having established the optimal reaction conditions, a broad range of anilines were selectively alkylated using benzhydrol (secondary alcohol) as the alkylating agent (Table 5). Notably, unsubstituted aniline reacted effectively, affording the desired N-alkylated product 62 in an excellent yield of 85%. Anilines bearing electron-donating groups such as –Me, –OMe, –iPr, –Pr, −Et, –OPh, and –SMe were well-tolerated, and the corresponding N-alkylated products 63–71 (Table 5) were obtained in yields ranging from 48% to 84%. The ortho-substituted aniline displayed lower yields due to its steric nature, however, long-chain ethyl and propyl groups at the ortho position yielded better results (up to 71% yield) than the simple methyl group (Table 5, product 63).
| a Reaction conditions: aniline derivatives (0.5 mmol), diphenylmethanol (0.6 mmol), catalyst I (1.5 mol%), and KOtBu (0.25 mmol) heated at 140 °C (silicon oil-bath temperature) in 1 mL toluene for 24 h. Yield of isolated products are in parentheses. |
|---|
|
Notably, aniline with halide functionalities such as –Cl and –F reacted favourably, yielding the products up to 69%. Difunctional anilines containing 2-Me,3-Cl (Table 5, 72) and 2-Me,3-F (76) displayed respectable isolated yields of 67% and 59%, respectively. Moreover, 1,4-dioxanone, 1,3-dioxanone, trifluoro, and para-morpholine substituted anilines were effectively alkylated, affording the desired products (Table 5), 79 (64% yield), 80 (56% yield), 81 (77% yield), and 82 (48% yield), respectively. The other symmetrical diphenylmethanol bearing –Me, and –OMe groups gave the N-alkylated products 83 in 62%, and 84 in 68% yields, respectively. In addition, unsymmetrical diphenylmethanol having a p-chloro substituent yielded 55% of the product 85 under optimized reaction conditions. Notably, other secondary alcohols such as cyclopentanol, cyclohexanol and 1-phenylethanol did not afford the desired secondary amine under present reaction conditions and their corresponding dehydrogenated carbonyl compounds were observed (∼35%; GC and GCMS analyses).
In addition, we also investigated the alkylation of benzyl alcohol with diamine using diaminobenzene (1,2-, and 1,3-phenylenediamine) and 1,8-naphthylenediamine. The optimized reaction conditions led to the formation of both mono and bis-alkylated products (Scheme 3, products 86–91). Notably, upon increasing the equivalent of benzyl alcohol, the yield of the bis-alkylated product increased while the yield of the mono-alkylated product decreased, as summarized in Scheme 3.
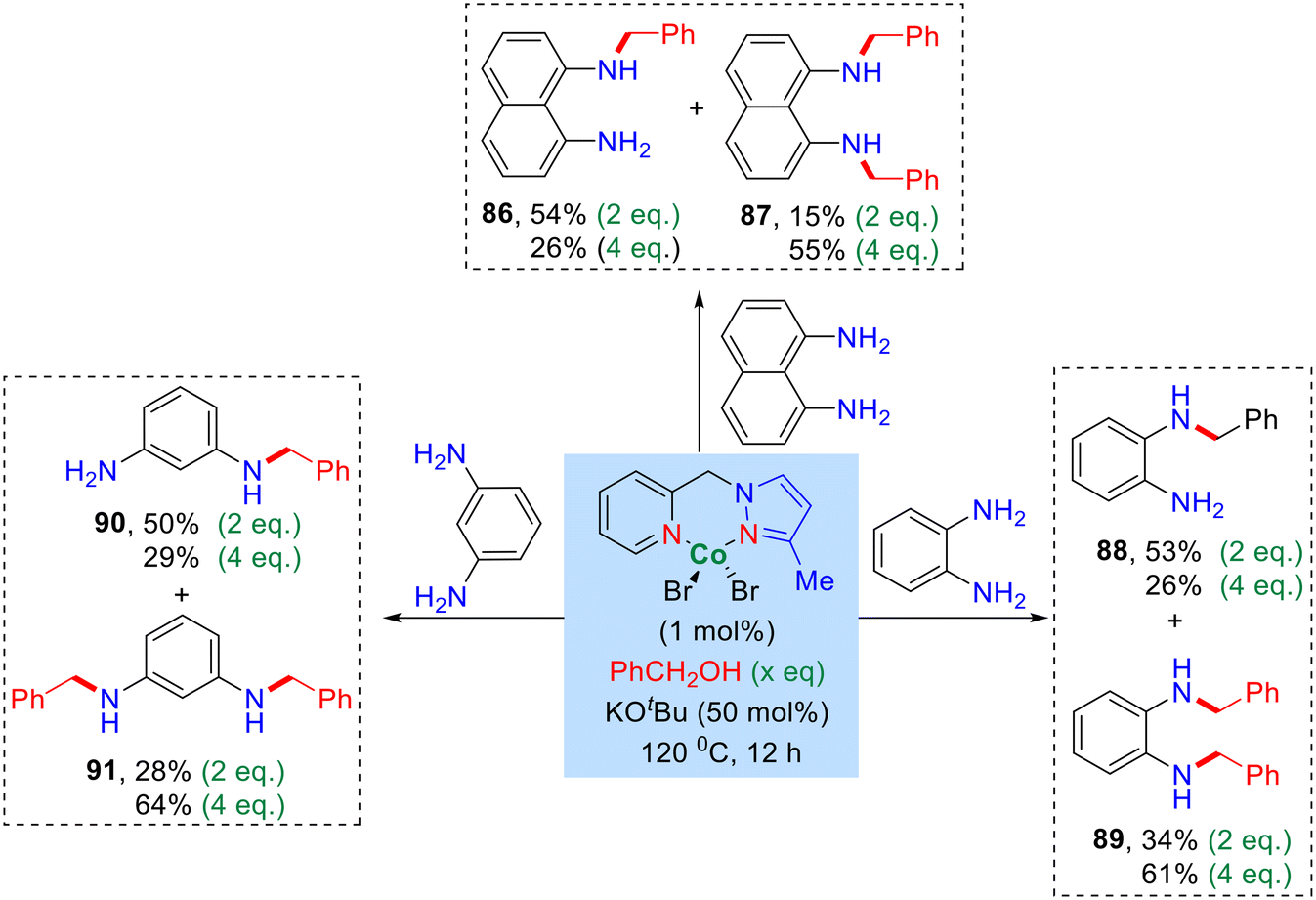 | ||
| Scheme 3 (NN)PyMeCo(II)-catalysed N-akylation of diamines with benzyl alcohols.a aReaction conditions: diamines (0.5 mmol), benzyl alcohol (x eq.), catalyst I (1 mol%), and KOtBu (0.25 mmol) heated at 120 °C (silicon oil-bath temperature) for 12 h. All are isolated yields of the products. | ||
We have also shown the scalability of the molecularly defined Co(II)-complex catalysed N-alkylation reactions. In this regard, the present amination of alcohols catalysed by a (NN)Co(II)-bidentate complex under solventless conditions was examined for the large-scale (5.0 mmol scale) synthesis of diverse N-alkylated amines and it worked smoothly with excellent isolated yield of the expected products (Scheme 4).
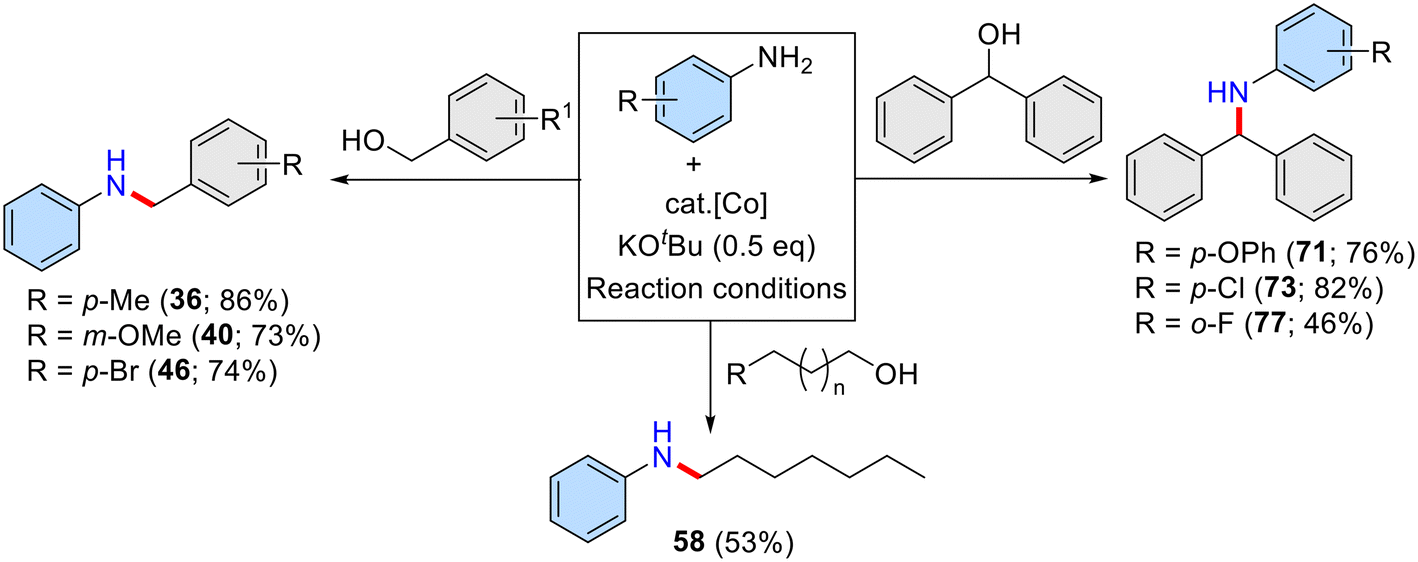 | ||
| Scheme 4 Large-scale synthesis & reusability of the homogeneous Co-catalytic system. Reaction conditions: aniline derivatives (5 mmol), alcohols (6 mmol), Cat. I (2 mol%), and KOtBu (2.5 mmol) heated for 36 h (120 °C and in the absence of solvent for compounds 36, 40, 46 and 58; 140 °C in 6 mL toluene for compounds 71, 73 and 77) and the yield are isolated yields. | ||
Gratifyingly, the reusability of the present homogeneous Co-catalyzed N-alkylation of amines using alcohols was successfully demonstrated (Fig. 2). Thus, the reaction of p-toluidine with benzyl alcohol under standard reaction conditions was carried out by externally adding starting materials into the reaction mixture (without additional (NN)PyMecobalt complex I) after every 12 h and monitored the catalytic efficiency of the cobalt catalyst. Interestingly, the desired N-alkylated product (N-benzyl-4-methylaniline, 3) was obtained in moderate yield after the 3rd cycle. The decrease in the catalytic activity after the 3rd cycle can be attributed to the gradual decomposition of the metal complex. Indeed, the reusability of soluble homogeneous catalytic systems for dehydrogenation and related reactions is rarely reported in the literature.60–63
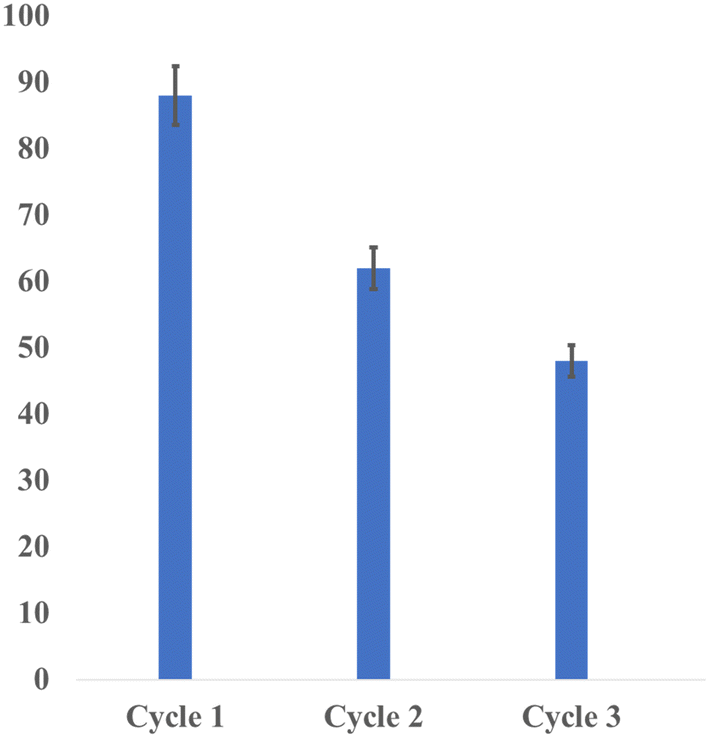 | ||
| Fig. 2 Recyclability test of the cobalt-catalysed N-alkylation reaction (of p-toluidine with benzyl alcohol). | ||
For a detailed understanding of the bidentate Co-catalyzed N-alkylation reaction of aniline with primary and secondary alcohols, several control experiments were carried out under standard reaction conditions (Scheme 5). To investigate the dehydrogenation step, different alcohols (benzyl alcohol, 1-pentanol, and benzhydrol) were reacted in the absence of aniline, leading to the isolation of the corresponding aldehyde and ketone as products (92–94). The formation of hydrogen gas was detected using gas chromatography (Scheme 5a). Performing the reaction for 4 hours, both the N-alkylated product (major) and the corresponding imine intermediate (minor) (95) were observed in GC and GC–MS analyses (Scheme 5b). Furthermore, treatment of the imine intermediate with the corresponding alcohol under the optimized reaction conditions resulted in the desired N-alkylated product in excellent yield (Scheme 5c).
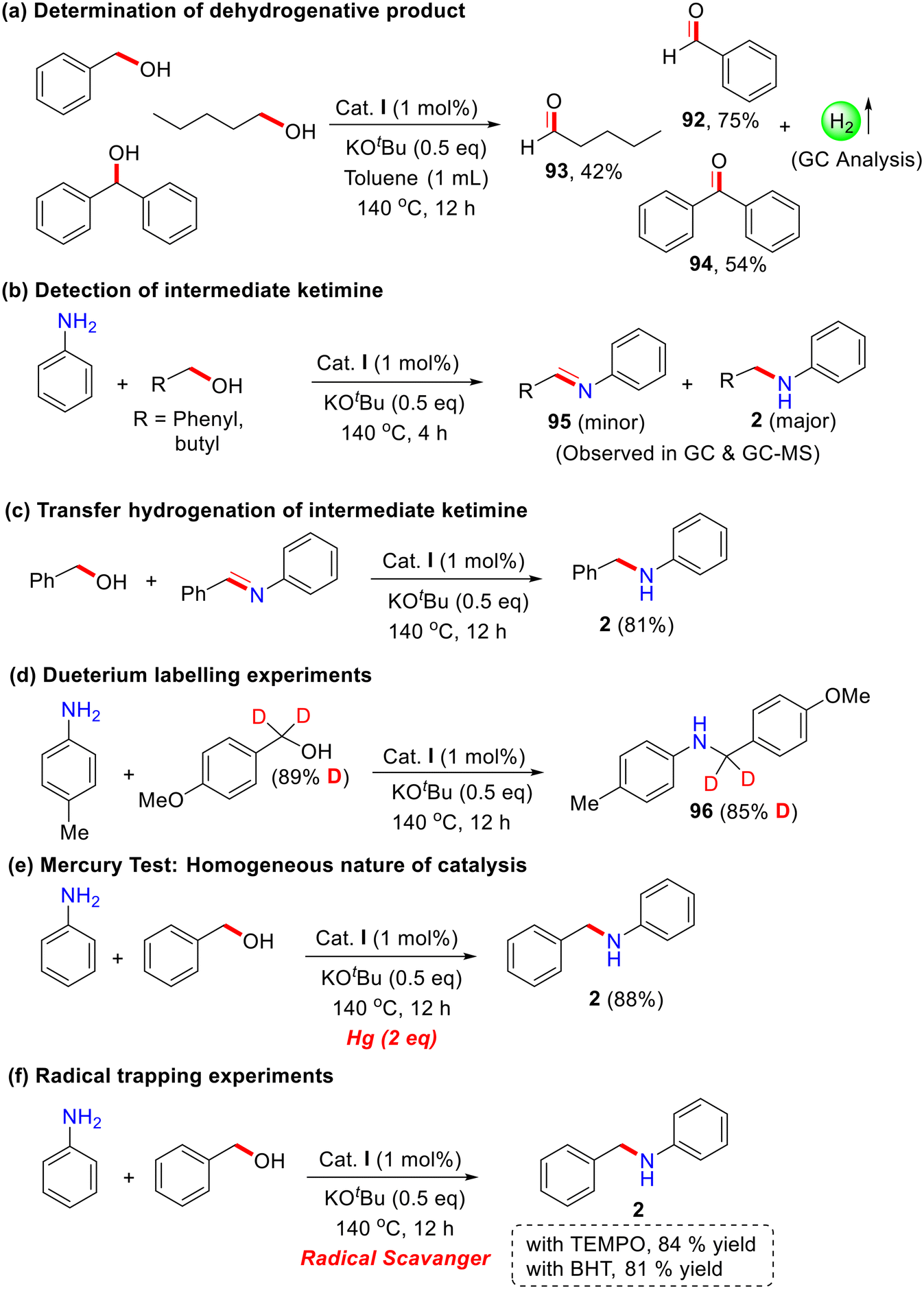 | ||
| Scheme 5 Control experiments (a–f). | ||
The independent deuterium labeling experiments were carried out; almost 85% deuterium incorporated was observed at the α-methylene carbon of N-alkylation products (96) under the present catalytic condition These results demonstrated that the reaction proceeds via the BH/HT pathway. Additionally, the presence of mercury under standard reaction conditions led to an excellent yield of 88% of the N-alkylated product, indicating that the present Co-catalyst is homogeneous in nature (Scheme 5e). Indeed, we didn't observe any nanoparticle formation after the catalytic reaction (SEM analysis). Moreover, in the presence of radical quenchers, the reaction proceeded smoothly and didn't affect the product yields (Scheme 5f). These results suggest that this Co-catalysis does not follow the single electron transfer (SET) or free radical pathway. Various reports describe that the Co–hydride complex is an important intermediate in dehydrogenation and related reactions (for example, C–C and C–N bond formation) using amine and alcohol as alkylating agents via the borrowing hydrogenation strategy.55,64,65 Recently, our research group also contributed to cobalt catalysis.51,53,66,67 Plausibly, we predict the in situ formation of a Co–H intermediate from alcohol; however, it is very challenging to isolate due to its highly unstable nature. The Chirik group reported similar cobalt–hydride species using bis(silylene)pyridine cobalt(III) precatalyst using strong reducing agents like NaHBEt3 and LiHBEt3.68 Using the same reaction procedure, several reactions were performed for the isolation and characterization of Co–H species with our catalytic system, using the NNPyMe–Co(II) bi-dentate complex and LiHBEt3 as a hydride donor. However, our attempts were unsuccessful. We propose two possible reasons for this outcome: (i) the research group of Chirik used a pincer complex, which is much more stable than the bidentate system we employed. The use of LiHBEt3 as a hydride donor might have decomposed our catalyst system. (ii) The Co(II) species is paramagnetic, which makes the NMR characterization of the cobalt complex challenging.
Based on control experiments and literature precedents,51,55,69 we have proposed a plausible catalytic cycle for the N,N-bidentate Co-catalysed amination of primary and secondary alcohols to aniline, as shown in Scheme 6. Initially, the alkoxide complex (I) was observed to form under the applied catalytic conditions. Subsequently, β-hydride elimination yielded the Co–hydride complex (II) with the generation of the corresponding carbonyl compound (III). The condensation of aniline with a carbonyl compound (III) resulted in imine formation, which further coordinated to give complex (IV). Reduction of the imine bond led to the amino–cobalt complex (V). Furthermore, abstraction of the proton of the alcohol resulted in the desired N-alkylated product, regenerating complex (I) and completing the catalytic cycle. Indeed, the isolation of the proposed intermediates and detailed mechanistic investigation are in progress in our laboratory.
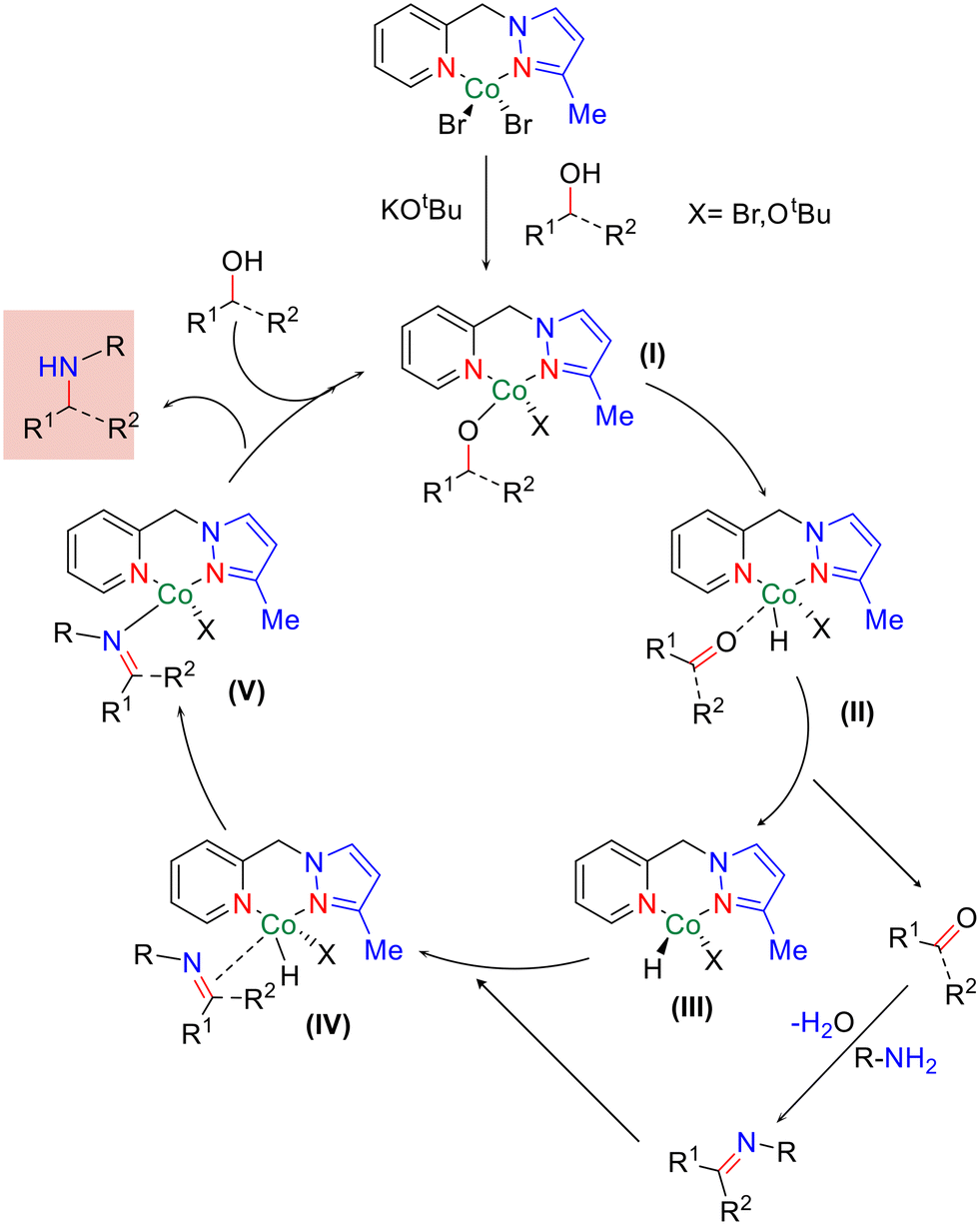 | ||
| Scheme 6 A plausible mechanism. | ||
In summary, we present an environmentally benign and highly selective direct synthesis of N-alkylation of anilines using primary (including unactivated aliphatic) and sterically demanding secondary alcohols (benzhydrol) via the borrowing hydrogen strategy using an air-stable, phosphine-free, non-precious cobalt(II)–NN–bidentate complex. The N-alkylation reaction demonstrated significant efficiency, as evidenced by the high degree of substrate and functional group tolerance observed. The reaction operates under solventless conditions with a low catalyst loading. Large-scale synthesis of N-alkylated amines and reusability of the present homogeneous Co-catalysis are additional advantages of the present strategy. Furthermore, the replacement of tridentate pincer ligands with bidentate ligands in the metal complex system represents a promising avenue for future research.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work is supported by the CSIR (Project No.: 01/(3030)/21/EMR-II). E. B. acknowledges funding from the Swarnajayanti Fellowship (SERB/F/5892/2020-2021). E. B. is an Alexander-von-Humboldt (AvH) fellow. R. K. thanks the IISER-Tirupati for fellowship.References
- S. A. Lawrence, Org. Process Res. Dev., 2005, 9, 1016 Search PubMed.
- A. Ricci, Amino group chemistry: from synthesis to the life sciences, John Wiley & Sons, 2008 Search PubMed.
- J. L. McGuire, Eur. J. Med. Chem., 2001, 36, 967–968 Search PubMed.
- V. Froidevaux, C. Negrell, S. Caillol, J.-P. Pascault and B. Boutevin, Chem. Rev., 2016, 116, 14181–14224 CrossRef CAS PubMed.
- R. Vardanyan and V. Hruby, Synthesis of Best-Seller Drugs, Elsevier Science, 2016 Search PubMed.
- J. Magano and J. R. Dunetz, Chem. Rev., 2011, 111, 2177–2250 CrossRef CAS PubMed.
- R. Dorel, C. P. Grugel and A. M. Haydl, Angew. Chem., Int. Ed., 2019, 58, 17118–17129 CrossRef CAS PubMed.
- E. Sperotto, G. P. M. van Klink, G. van Koten and J. G. de Vries, Dalton Trans., 2010, 39, 10338–10351 RSC.
- G. Yashwantrao and S. Saha, Tetrahedron, 2021, 97, 132406 CrossRef CAS.
- M. J. West, J. W. B. Fyfe, J. C. Vantourout and A. J. B. Watson, Chem. Rev., 2019, 119, 12491–12523 CrossRef CAS.
- J.-Q. Chen, J.-H. Li and Z.-B. Dong, Adv. Synth. Catal., 2020, 362, 3311–3331 CrossRef CAS.
- L. Huang, M. Arndt, K. Gooßen, H. Heydt and L. J. Gooßen, Chem. Rev., 2015, 115, 2596–2697 CrossRef CAS PubMed.
- M. B. Smith and J. March, March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, Wiley, 2007 Search PubMed.
- T. E. Müller, K. C. Hultzsch, M. Yus, F. Foubelo and M. Tada, Chem. Rev., 2008, 108, 3795–3892 CrossRef.
- S. Zhu, N. Niljianskul and S. L. Buchwald, Nat. Chem., 2016, 8, 144–150 CrossRef CAS PubMed.
- K. Murugesan, T. Senthamarai, V. G. Chandrashekhar, K. Natte, P. C. J. Kamer, M. Beller and R. V. Jagadeesh, Chem. Soc. Rev., 2020, 49, 6273–6328 RSC.
- T. Irrgang and R. Kempe, Chem. Rev., 2020, 120, 9583–9674 CrossRef CAS PubMed.
- N. U. D. Reshi, V. B. Saptal, M. Beller and J. K. Bera, ACS Catal., 2021, 11, 13809–13837 CrossRef CAS.
- O. I. Afanasyev, E. Kuchuk, D. L. Usanov and D. Chusov, Chem. Rev., 2019, 119, 11857–11911 CrossRef CAS PubMed.
- G. Guillena, D. J. Ramón and M. Yus, Chem. Rev., 2010, 110, 1611–1641 CrossRef CAS PubMed.
- A. Corma, J. Navas and M. J. Sabater, Chem. Rev., 2018, 118, 1410–1459 CrossRef CAS PubMed.
- E. Podyacheva, O. I. Afanasyev, D. V. Vasilyev and D. Chusov, ACS Catal., 2022, 12, 7142–7198 CrossRef CAS.
- B. G. Reed-Berendt, K. Polidano and L. C. Morrill, Org. Biomol. Chem., 2019, 17, 1595–1607 RSC.
- B. G. Reed-Berendt, D. E. Latham, M. B. Dambatta and L. C. Morrill, ACS Cent. Sci., 2021, 7, 570–585 CrossRef CAS PubMed.
- S. Bähn, S. Imm, L. Neubert, M. Zhang, H. Neumann and M. Beller, ChemCatChem, 2011, 3, 1853–1864 CrossRef.
- M. Beller and C. Bolm, Transition Metals for Organic Synthesis: Building Blocks and Fine Chemicals, Wiley-VCH, 2004 Search PubMed.
- S. Pan and T. Shibata, ACS Catal., 2013, 3, 704–712 CrossRef CAS.
- G. Chelucci, Coord. Chem. Rev., 2017, 331, 1–36 CrossRef CAS.
- P. A. Slatford, M. K. Whittlesey and J. M. J. Williams, Tetrahedron Lett., 2006, 47, 6787–6789 CrossRef CAS.
- O. Saidi, A. J. Blacker, G. W. Lamb, S. P. Marsden, J. E. Taylor and J. M. J. Williams, Org. Process Res. Dev., 2010, 14, 1046–1049 CrossRef CAS.
- A. Nandakumar, S. P. Midya, V. G. Landge and E. Balaraman, Angew. Chem., Int. Ed., 2015, 54, 11022–11034 CrossRef CAS PubMed.
- A. Tillack, D. Hollmann, D. Michalik and M. Beller, Tetrahedron Lett., 2006, 47, 8881–8885 CrossRef CAS.
- D. Hollmann, A. Tillack, D. Michalik, R. Jackstell and M. Beller, Chem. – Asian J., 2007, 2, 403–410 CrossRef CAS PubMed.
- A. Tillack, D. Hollmann, K. Mevius, D. Michalik, S. Bähn and M. Beller, Eur. J. Org. Chem., 2008, 2008, 4745–4750 CrossRef.
- M. H. S. A. Hamid, C. L. Allen, G. W. Lamb, A. C. Maxwell, H. C. Maytum, A. J. A. Watson and J. M. J. Williams, J. Am. Chem. Soc., 2009, 131, 1766–1774 CrossRef CAS PubMed.
- K. O. Marichev and J. M. Takacs, ACS Catal., 2016, 6, 2205–2210 CrossRef CAS PubMed.
- T. T. Dang, B. Ramalingam, S. P. Shan and A. M. Seayad, ACS Catal., 2013, 3, 2536–2540 CrossRef CAS.
- T. T. Dang, S. P. Shan, B. Ramalingam and A. M. Seayad, RSC Adv., 2015, 5, 42399–42406 RSC.
- R. M. Bullock, Catalysis without Precious Metals, Wiley, 2011 Search PubMed.
- A. Mukherjee and D. Milstein, ACS Catal., 2018, 8, 11435–11469 CrossRef CAS.
- P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192–2452 CrossRef CAS PubMed.
- T. Irrgang and R. Kempe, Chem. Rev., 2019, 119, 2524–2549 CrossRef CAS PubMed.
- M. Hapke and G. Hilt, Cobalt Catalysis in Organic Synthesis: Methods and Reactions, Wiley, 2020 Search PubMed.
- Z. Yin, H. Zeng, J. Wu, S. Zheng and G. Zhang, ACS Catal., 2016, 6, 6546–6550 CrossRef CAS.
- A. Quintard and J. Rodriguez, ChemSusChem, 2016, 9, 28–30 CrossRef CAS PubMed.
- M. Mastalir, G. Tomsu, E. Pittenauer, G. Allmaier and K. Kirchner, Org. Lett., 2016, 18, 3462–3465 CrossRef CAS PubMed.
- G. Zhang, Z. Yin and S. Zheng, Org. Lett., 2016, 18, 300–303 CrossRef CAS PubMed.
- K. Paudel, S. Xu, O. Hietsoi, B. Pandey, C. Onuh and K. Ding, Organometallics, 2021, 40, 418–426 CrossRef CAS.
- A. Martínez-Asencio, D. J. Ramón and M. Yus, Tetrahedron, 2011, 67, 3140–3149 CrossRef.
- A. Singh, A. Maji, M. Joshi, A. R. Choudhury and K. Ghosh, Dalton Trans., 2021, 50, 8567–8587 RSC.
- S. P. Midya, J. Pitchaimani, V. G. Landge, V. Madhu and E. Balaraman, Catal. Sci. Technol., 2018, 8, 3469–3473 RSC.
- N. Şahin, İ. Yıldırım, N. Özdemir, N. Gürbüz and İ. Özdemir, J. Organomet. Chem., 2020, 918, 121285 CrossRef.
- S. P. Midya, A. Mondal, A. Begum and E. Balaraman, Synthesis, 2017, 49, 3957–3961 CrossRef CAS.
- S. Rösler, M. Ertl, T. Irrgang and R. Kempe, Angew. Chem., Int. Ed., 2015, 54, 15046–15050 CrossRef PubMed.
- I. Borthakur, A. Sau and S. Kundu, Coord. Chem. Rev., 2022, 451, 214257 CrossRef CAS.
- S. Yadav, D. Prabha, D. Ahluwalia, A. Bag and R. Gupta, Eur. J. Org. Chem., 2022, 2022, 22–33 Search PubMed.
- P. G. Nandi, P. Thombare, S. J. Prathapa and A. Kumar, Organometallics, 2022, 41, 3387–3398 CrossRef CAS.
- M. Jafarzadeh, S. H. Sobhani, K. Gajewski and E. Kianmehr, Org. Biomol. Chem., 2022, 20, 7713–7745 RSC.
- H. Tian, W. Xue, J. Wu, Z. Yang, H. Lu and C. Tang, Org. Chem. Front., 2022, 9, 4554–4560 RSC.
- P. Hu, Y. Diskin-Posner, Y. Ben-David and D. Milstein, ACS Catal., 2014, 4, 2649–2652 CrossRef CAS.
- B. Maji, A. Bhandari, D. Bhattacharya and J. Choudhury, Organometallics, 2022, 41, 1609–1620 CrossRef CAS.
- S. P. Midya, J. Rana, J. Pitchaimani, A. Nandakumar, V. Madhu and E. Balaraman, ChemSusChem, 2018, 11, 3911–3916 CrossRef CAS PubMed.
- Y. Han, Z. Wu, Z. Wei, Y. Zhai, S. Ru, Q. Zhao, J. Wang, S. Han and Y. Wei, Commun. Chem., 2019, 2, 1–7, DOI:10.1038/s42004-019-0109-4.
- L. Alig, M. Fritz and S. Schneider, Chem. Rev., 2019, 119(4), 2681–2751 CrossRef CAS PubMed.
- W. Ai, R. Zhong, X. Liu and Q. Liu, Chem. Rev., 2019, 119, 2876–2953 CrossRef CAS PubMed.
- S. P. Midya, V. G. Landge, M. K. Sahoo, J. Rana and E. Balaraman, Chem. Commun., 2018, 54, 90–93 RSC.
- V. G. Landge, J. Pitchaimani, S. P. Midya, M. Subaramanian, V. Madhu and E. Balaraman, Catal. Sci. Technol., 2018, 8, 428–433 RSC.
- R. Arevalo, T. P. Pabst and P. J. Chirik, Organometallics, 2020, 39, 2763–2773 CrossRef CAS PubMed.
- M. R. Elsby and R. T. Baker, Chem. Soc. Rev., 2020, 49, 8933–8987 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization of Co-complex, experimental and spectroscopic data, copies of 1H, and 13C NMR spectra. CCDC 2264033. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cy00809f |
| This journal is © The Royal Society of Chemistry 2024 |