
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanistic insights into methanol carbonylation to methyl acetate over an efficient organic template-free Cu-exchanged mordenite
L. A.
Luque-Álvarez
 *,
J.
González-Arias
,
F.
Romero-Sarria
,
T. R.
Reina
,
L. F.
Bobadilla
and
J. A.
Odriozola
*,
J.
González-Arias
,
F.
Romero-Sarria
,
T. R.
Reina
,
L. F.
Bobadilla
and
J. A.
Odriozola
Department of Inorganic Chemistry and Institute of Materials Science of Seville, CSIC – University of Sevilla, Av. Américo Vespucio 49, 41092 Sevilla, Spain. E-mail: llalvarez@us.es
First published on 15th November 2023
Abstract
Currently, acetic acid is produced via the carbonylation reaction of methanol with the main route entailing the use of homogeneous noble metal-based catalysts, which has certain limitations, including the use of polluting alkyl halide promoters and difficulty in catalyst recovery. To overcome these challenges, the exploration of alternative methods utilizing heterogeneous catalysts, particularly zeolites with copper as a redox center, has gained attention. Nonetheless, the conversion and selectivity obtained are sought after to compete against the homogeneous route. Therefore, a deeper understanding of the reaction and mechanism is needed to determine the weak points and overcome them. In this study, we propose the use of time-resolved operando DRIFTS-MS to study the methanol carbonylation reaction over a Cu–H-MOR catalyst. The study aimed to propose a reaction mechanism through an investigation of the behavior of the catalyst, including potential identification of the location of the copper redox center in the zeolite. The catalytic performance of the Cu–H-MOR catalyst was also evaluated, demonstrating its activity and stability in the methanol carbonylation reaction. The operando DRIFTS-MS results provide insights into the reaction mechanism and the involvement of the acid and redox centers in the process. Based on the findings, we propose a reaction mechanism for methanol carbonylation on Cu–H-MOR zeolite: (i) methanol dehydration, (ii) CO insertion into methoxide groups, (iii) reaction between dimethyl ether and acetyl groups, and (iv) hydrolysis of methyl acetate. Overall, we believe that this work contributes to a deeper understanding of the heterogeneous route for acetic acid production and offers potential avenues for optimizing the process.
Introduction
The insertion of carbonyl groups into organic molecules to produce high-value added chemical compounds is a crucial process in the chemical industry. One prominent example of such processes is the carbonylation reaction of methanol to produce acetic acid or methyl acetate.1,2 Currently, the industrial production of acetic acid is around 10 million ton per year, and it is primarily utilized in the manufacturing of vinyl acetate, acetic anhydride, and acetic acid esters.2 Acetic acid is a building block used in many chemical industries: (i) in the textile industry for polyester production; (ii) in the plastic industry for terephthalate production; (iii) in the food and beverage industry; and (iv) in the construction and automobile sectors to produce polymers, adhesives, paints, and coatings.3Despite the initial adverse effects of the COVID-19 pandemic on the global acetic acid market, it is projected to experience significant growth in the upcoming years, primarily driven by a substantial surge in demand.4 In fact, the market volume of acetic acid has increased from 13.5 to approximately 18 million metric ton from 2015 to 2023.5 Nowadays, about 60% of the total world acetic acid manufactured is produced via the methanol carbonylation reaction.6 For more than 50 years, this reaction has been industrially carried out through the Monsanto and BP Cativa™ processes. These processes use homogeneous noble metal-based catalysts (Rh and Ir) and an alkyl halide (CH3I) as a promoter agent.7 The typical operating conditions of these processes are shown in Table 1.
| Process | Catalyst | T (°C) | P (atm) | CO/MeOH | Selectivity to acetic acid |
|---|---|---|---|---|---|
| Monsanto | cis-[Rh(CO)2I2]− | 150–200 | 30–60 | 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
>99% |
| BP Cativa™ | [Ir(CO)2I2]− | 190 | 28 | 2:1 |
>99% |
While rhodium- and iridium-catalyzed processes for methanol carbonylation demonstrate remarkable selectivity in producing acetic acid (as indicated in Table 1), it is important to note they have certain limitations. Among these, the most important are: (i) the co-catalyst or promoter agent that is used in the process (CH3I) is a highly polluting alkyl halide, and (ii) the catalysts themselves, which are homogeneous, make their recovery difficult and costly. Furthermore, those catalysts are based on precious metals that are expensive to use on an industrial scale. The limitations mentioned above have prompted the exploration of alternative methods utilizing heterogeneous catalysts that can be conveniently recovered and reused. The heterogeneous catalysts of major interest for this process are zeolites, such as mordenite (MOR), ferrierite (FER), and faujasite (FAU), or ZSM-5 doped with transition metals since these materials are low-cost aluminosilicates with an adequate porous structure for this reaction.2 The choice to utilize zeolites stems from their compatibility with the requirements of the process. These materials exhibit a bifunctional behavior, meaning they possess both acid sites for methanol dehydration and metal sites that activate carbon monoxide, enabling its insertion into the adsorbed methyl (–CH3) groups located at neighboring acid sites.9,10 Due to the presence of Brønsted acid sites in zeolites and the potential inclusion of charge compensation cations, these compounds are well-suited for utilization in carbonylation reactions.11–13
Among the transition metals that act as redox centers in carbonylation reactions catalyzed by zeolites (e.g., Cu, Co, Zn, Ni, Ag, and Fe), Cu has been the most studied due to its low cost and interesting redox properties.14 In 1984, Fujimoto et al. were pioneers describing the use of zeolites partially exchanged with Cu (Cu–H-ZSM-5 and Cu–H-MOR) for the methanol carbonylation reaction.15 Ten years later, researchers from BP Chemicals re-examined the catalytic performance of H-MOR and Cu–H-MOR in the methanol carbonylation reaction and achieved remarkable yields with the Cu–H-MOR catalyst.16 Since then, MOR-type zeolites have been extensively studied to correlate the characteristic topology of these materials with their catalytic behavior.17,18 The elimination of the separation issue associated with homogeneous precious metal complexes as well as the avoidance of costly and corrosive liquid halides are some of the advantages obtained by using these heterogeneous catalysts. Furthermore, the conventional synthesis method of zeolites requires the use of a template to form the crystalline structure. The use of templates can introduce residual impurities into the synthesized zeolites, negatively affecting their purity and quality. The use of a template can also limit the diversity of the compositions and morphologies that can be obtained in the zeolites' synthesis. By removing this constraint, a template-free method would allow for better flexibility in synthesis, facilitating the production of zeolites with specific characteristics tailored to customized applications. Likewise, eliminating the required template removal step would reduce the overall synthesis time and simplify the process. This would result in increased efficiency and productivity in zeolite production, particularly for large-scale applications. In this sense, a template-free synthesis method for zeolites offers clear advantages over the conventional approach. The elimination of the template removal step, obtaining purer zeolites, and the flexibility in composition and morphology are positive aspects that can promote greater efficiency and quality in the synthesis process. These benefits support the adoption of this strategy in zeolite production for the carbonylation reaction proposed in this work. These benefits alone present promising opportunities for process optimization and cost reduction in carbonylation reactions. Nonetheless, further research and practical experiments are needed to validate the theoretical projections and to explore the wider application of these catalysts in carbonylation reactions. A more comprehensive understanding of the Cu–H-MOR catalyst capabilities may allow for the development of novel and efficient processes.
As above-mentioned, many works have previously studied the heterogeneous route for acetic acid production. However, the conversion and selectivity obtained in the heterogeneous alternative are far from those offered via the homogeneous catalytic process. In line with this, a deeper understanding of the reaction mechanism and the catalyst behavior is needed to advance the potential commercial use of heterogeneous acetic acid production. To this end, in this work we propose the use of time-resolved operando DRIFTS-MS for the methanol carbonylation reaction over a Cu–H-MOR catalyst. This study allows us to understand the behavior of the catalyst during the reaction, and hence to propose potential modifications on the catalyst to improve the reaction performance. Furthermore, the location of the Cu redox center in the MOR-type zeolite is still not clear. As shown in previous works, the active sites for methanol carbonylation are Brønsted acid sites located within the zeolitic cavity. Cu ions positioned near these sites enhance the catalyst's activity, making it less susceptible to deactivation by water.19 In the MOR structure, the Brønsted sites are found in 8-membered rings (8-MR) connected to 12-membered ring channels (12-MR). Interestingly, the carbonylation of methanol to acetic acid predominantly occurs selectively within the 8-MR pockets, while hydrocarbon formation, leading to deactivation, is more likely to happen in the 12-MR pockets.20 Nonetheless, the effect of the Cu location has not been studied in depth before. In this work, we reveal where this redox center is located via an in situ-FTIR study of CO adsorption. To achieve these two objectives (i.e., (i) the investigation of the reaction mechanism and (ii) the study of the Cu location), this work is organized as follow: first, the synthesis of the catalyst and the experimental setup used in this work are described, followed by the physicochemical characterization, including the in situ-FTIR CO adsorption. Afterwards, the time-resolved operando DRIFTS-MS experiment is analyzed, along with the experimental results of the carbonylation reaction using the selected Cu–H-MOR catalyst. We finish this work by proposing a potential reaction mechanism for the reaction studied.
Experimental
Catalyst synthesis
In the synthesis of mordenite-type zeolites, sodium hydroxide pellets (NaOH, Panreac), sodium aluminate (NaAlO2, Sigma-Aldrich, 99%), and high-purity silica (SiO2, Sigma-Aldrich, 99.5%) were utilized without further purification. Ammonium chloride (NH4Cl, Panreac) and hydrated copper acetate(II) (Cu(CH3COO)2·xH2O, Panreac) were used in the subsequent stages of cationic exchange.A mordenite zeolite in its sodium form was synthetized with the molar composition of 6Na2O:Al2O3; 30SiO2;780H2O and the ratio X = Si/Al of 15 via hydrothermal synthesis in the absence of a template, as previously described in the literature.21 Mordenite in its protonic form (H-MOR–X) was prepared by cation exchange with ammonium chloride and a subsequent calcination step. Finally, the zeolite in its proton–copper form was prepared by a cation-exchange process with hydrated copper(II) acetate according to a procedure previously reported in the literature.22 The solid obtained was calcined at 350 °C for 3 h with a heating ramp of 2 °C min−1.
Physicochemical characterization
The prepared catalyst was characterized by X-ray fluorescence (XRF), N2 adsorption, X-ray diffraction (XRD), scanning electron microscopy (SEM), and the in situ adsorption of CO at 77 K followed by infrared spectroscopy (FTIR). The chemical composition of the samples was characterized by XRF in a Zetium Minerals device. Nitrogen adsorption/desorption isotherms were measured at liquid nitrogen temperature (77 K) using a Micromeritics TRIFLEX instrument. The textural properties were studied using the Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH) methods. Initially, the sample was degassed for 4 h at 250 °C under vacuum. X-ray diffraction was carried out using an X'Pert Pro PANalytical diffractometer with Cu Kα radiation (40 mA, 45 kV) in the 2θ range 5–80°. To obtain the micrographs, a HITACHI S-4800 SEM-FEG (Scanning Electron Microscopy-Field Emission Gun) microscope was used, equipped with secondary and backscattered electron detectors for elemental analysis on the surface of the material.In situ CO adsorption at 77 K followed by IR spectroscopy in transmission mode was carried out using a vacuum line. In this system, the sample was placed in a cell in the form of a 13 mm-diameter wafer, and, after a pretreatment of 12 h at 200 °C under vacuum of 10−5 mbar, CO was introduced in small doses at 77 K until complete saturation was reached. For each dose of CO, the spectrum was recorded on a THERMO NICOLET Avatar 380 spectrophotometer, equipped with a DTGS/KBr detector. In each spectrum, 128 scans were accumulated at a resolution of 4 cm−1.
Catalytic performance
The catalytic performance was evaluated in a Hastelloy 9 mm-diameter tubular reactor coupled to a microactivity PID Eng&Tech device. Thus, ca. 80 mg (100–200 μm) were loaded in the reactor and pre-activated at 400 °C under 5 vol% O2/He during 1 h. Then, the catalytic activity was recorded at 8 bar with a CO/MeOH molar ratio of 5 in the temperature range 220–300 °C. Evaluation of the catalytic performance was carried out using the space–time yield (STY) metric, quantified as the production of μmol of methyl acetate per gram of catalyst per hour of the reaction. Furthermore, catalytic stability tests were performed at 300 °C. The inlet and outlet gas mixtures composition were analyzed online with an Agilent 6890N gas chromatograph equipped with an Agilent J&W DB-FATWAX Ultra Inert column.Operando DRIFTS-MS experiments
Time-resolved operando DRIFTS-MS studies were conducted in a Praying Mantis (Harrick) optical system with ZnSe windows that supported a high-temperature reaction cell where the sample was located (40 mg). The spectra were taken with a resolution of 4 cm−1 and an average scan of 64 scans using a Thermo Nicolet iS50 FTIR spectrometer equipped with an MCT detector cooled with liquid nitrogen. The spectra were corrected with the background initially taken on the spectrometer using an aluminum mirror; and all the mathematical treatments of the spectra were performed in OMNIC software version 9.0. The gas phase was analyzed on-line by mass spectrometry coupled to the IR equipment. The reactants were fed using a system of mass flow controllers and mixers to prepare the streams both in the activation of the catalyst (Ar) and in the reaction (CO + methanol + Ar). According to the experimental protocol followed, an initial pretreatment of the sample consisting of in situ activation at 350 °C with an Ar flow of 25 mL min−1 was carried out in order to remove the absorbed water and completely dehydrate the zeolite. The system was then cooled to 200 °C and the carbonylation reaction was carried out at atmospheric pressure by passing through the cell a flow of 30 mL min−1 of 17% v/v of CO in Ar saturated in methanol. The reaction time was 60 min and throughout the period the IR spectra were recorded continuously.Results and discussion
Structure, morphology, and textural properties
The structure and morphology of the synthesized catalyst were analyzed by XRD and SEM, respectively. Fig. 1A shows the XRD patterns of the prepared catalyst in its sodium form, in its proton form, and after the cation exchange. It can be observed that the catalyst in its sodium form presented a mordenite-like crystal structure (JCPDS: 043-0171) and this framework remained for both the H-MOR and Cu–H-MOR exchanged zeolites. Therefore, after cation exchange and calcination, the crystal structure of mordenite remained unchanged and no copper aggregates were observed. This allows us to assume that the metal was highly dispersed in the cavities of the material. | ||
| Fig. 1 (A) The XRD patterns of the prepared sodium-mordenite, H-mordenite, and Cu–H-mordenite; (B) SEM micrographs of H-MOR and Cu–H-MOR. | ||
Concerning the morphology, the SEM micrographs of the H-MOR and Cu–H-MOR samples are presented in Fig. 1B. The analysis of the morphology revealed that the crystals presented the characteristic shape of mordenites according to the conditions of synthesis; that is, needle-shaped crystals elongated on the z-axis.23 Furthermore, after exchange with copper, no significant changes in the particles were induced.
The N2 adsorption–desorption isotherms at 77 K for the mordenite samples (i.e., H-MOR and Cu–H-MOR) are shown in Fig. 2. Following the IUPAC classification,24 typical type I isotherms were found in both samples, indicative of microporous solids with a narrow porous distribution. Table 2 contains details of the specific surface area, micropore surface area, and micropore volume parameters as well as data from the XRF chemical analysis. It is noteworthy that the H-MOR catalyst presented a higher surface area, whereas Cu–H-MOR presented a larger micropore surface area. This fact was primarily attributed to the unique breathing phenomenon observed in zeolites when metal species are present inside the cavities.25–27
 | ||
| Fig. 2 (A) N2 adsorption–desorption isotherms at 77 K for mordenite samples and (B) the pore-size distribution. | ||
| Surface area (SBET)/m2 g−1 | t-plot micropore surface area/m2 g−1 | t-plot micropore volume/cm3 g−1 | Pore size (Å) | Cu/molar % | SiO2/Al2O3 | |
|---|---|---|---|---|---|---|
| H-MOR | 441 | 330 | 0.17 | 24.3 | — | 17 |
| Cu–H-MOR | 436 | 357 | 0.18 | 22.8 | 0.54 | 17 |
Location of Cu sites: in situ-FTIR CO adsorption study at a temperature of 77 K
The adsorption of CO at 77 K followed by FTIR spectroscopy is a widely employed technique for characterizing solid surfaces. This method allows investigation of the interaction of CO with both Brønsted and Lewis acid sites present on the catalyst's surface.28Fig. 3 shows the FTIR results for the adsorption of CO at 77 K for the H-MOR and the Cu–H-MOR samples. As can be seen, three bands appeared in both samples: 2137, 2160, and 2178 cm−1, whereas in Cu–H-MOR, an additional band appeared at 2156 cm−1. The 2137 cm−1 band could be attributed to CO physiosorbed inside the pores of zeolite.29,30 The bands observed at 2160 and 2178 cm−1 could be assigned to the interaction of CO with hydroxyls located inside both cavities, i.e., in the main channels (2178 cm−1) and in the side pockets (2160 cm−1).30–32 Meanwhile, the additional band observed at 2156 cm−1 in the Cu–H-MOR catalyst was associated with the interaction of CO with copper species forming monocarbonyls Cu+–(CO). According to previous works, the presence of Cu+ was due to the fact that during calcination or in the ion-exchange process itself, copper is reduced from Cu(II) to Cu(I), and Cu(I) is stabilized inside the cavities of the mordenites.33 The deconvolution of the saturation spectra of both samples suggested that copper species were mainly located in the small cavities, whereas they only partially filled the main cavities. As will be further discussed, the location of copper species plays a key role in the catalytic performance. | ||
| Fig. 3 The FTIR study of adsorption of CO at 77 K over (A) H-MOR and (B) Cu–H-MOR samples. | ||
Catalytic performance
Fig. 4a shows the catalytic activity in terms of methyl acetate production (STY/μmol g−1 h−1) exhibited by the Cu–H-MOR sample in the methanol carbonylation reaction as a function of temperature, ranging from 220–300 °C. As can be seen, the production of methyl acetate increased with the temperature. Significantly, only DME and methyl acetate were produced during the reaction. This observation is in agreement with the results reported by Blasco et al.,19 in which the production of acetic acid was inhibited in Cu-exchanged mordenite. The active sites for methanol carbonylation are Brønsted sites inside the zeolitic cavity, so when placing Cu+ close to these sites, the catalyst activity increased and the catalyst was less prone to be deactivated by water.34 Brønsted sites are located in eight membered rings (8-MRs), connected to 12-ring channel in mordenite. However, the carbonylation of methanol occurs selectively in the 8-MR pockets, while hydrocarbon formation resulting in deactivation is easier in 12-MR pockets.35,36 | ||
| Fig. 4 (a) Catalytic performance as a function of the temperature and (b) catalytic stability test during 20 h at 300 °C for the methanol carbonylation reaction in terms of methyl acetate productivity over the Cu–H-MOR catalyst. | ||
In order to evaluate the long-term stability of the catalyst, additional tests were conducted over a period of 20 h to investigate any potential deactivation. Understanding the deactivation behavior of zeolitic heterogeneous catalysts is critical for optimizing their performance and extending their lifetime in practical applications. In this context, Fig. 4b illustrates the results of the stability tests performed on the Cu–H-MOR catalyst at 300 °C. The results obtained demonstrated that the catalyst rarely exhibited minimal deactivation over time, indicating a high level of stability. The stability of the catalysts can be compromised by the formation of coke in the zeolitic cavities, since zeolites are also active for the transformation of DME into hydrocarbons. By controlling the acidity, the dehydration rate may be enhanced with respect to coke formation.34 Moreover, the temperature control of the reaction also plays a key role since the hydrocarbon formation occurs at higher temperatures than the dehydration process and therefore the control of hot spots should improve the selectivity.
Time-resolved operando DRIFTS-MS during the methanol carbonylation reaction
In order to identify the possible reaction intermediates in methanol carbonylation, the reaction was studied by DRIFT spectroscopy coupled to mass spectrometry by passing the reactants (CO/methanol/Ar) at 200 °C for 1 h over the sample Cu–H-MOR, which was initially thermally pretreated at 350 °C for 2 h. These reaction conditions were selected to ensure a high concentration of surface intermediates.Fig. 5 shows the results obtained during the activation thermal pretreatment in the 4000–800 cm−1 fundamental region (a) and in the NIR region, including the deconvolution of the combination bands associated with hydroxyl groups in the final spectrum (b).
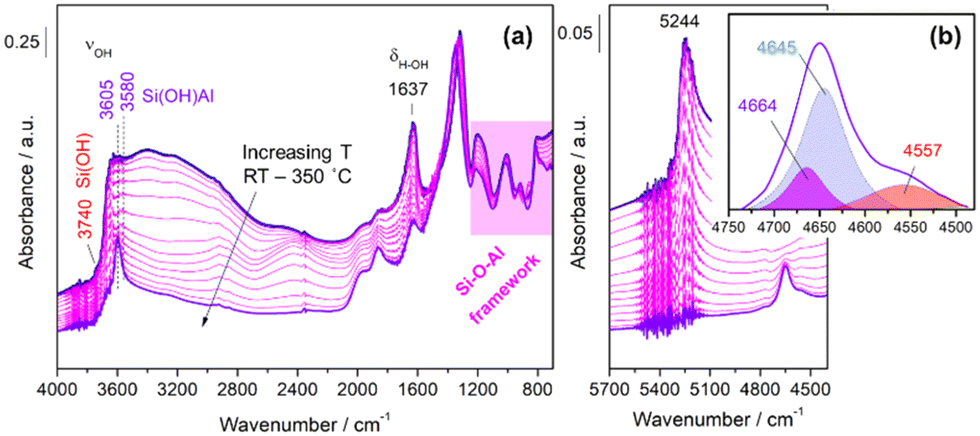 | ||
| Fig. 5 (a) The evolution of the IR spectra during the activation pretreatment in the fundamental region 4000–800 cm−1 and (b) in the NIR region, including the deconvolution of the combination bands associated with hydroxyl groups in the final spectrum. | ||
In the fundamental region (Fig. 5a), at RT, a strong band at 1637 cm−1 and a broad complex band centered at 3200 cm−1 could be observed, and both were associated with the presence of physisorbed water interacting with hydroxyl groups in the cavities.31 Increasing the temperature led to the disappearance of these bands, giving rise to another band around 3600 cm−1 with two components at 3605 and 3580 cm−1, assigned to the vibration of Si(OH)Al bridge-type hydroxyl groups inside the main cavities (12-MR) and in the side pockets (8-MR), respectively.31 In addition, a less intense band appeared at 3740 cm−1, corresponding to the vibration of the terminal Si(OH) silanol hydroxyl groups located, probably, on the exterior of the cavities.30,37 On the other hand, the region between 1400 and 750 cm−1 was characteristic of Si–O and Al–O vibrations in the structural framework of the zeolite.31 A summary of the band assignments is provided in Table 3.
| Temperature | Vibration frequency | Band assignment |
|---|---|---|
| RT | 1637 cm−1 | H2O (bending) |
| 3200 cm−1 | HO⋯OH2 | |
| 350 °C | 3580 cm−1 | Si(OH)Al (8-MR) |
| 3605 cm−1 | Si(OH)Al (12-MR) | |
| 3740 cm−1 | SiOH (external framework) | |
| RT-350 °C | 1400–750 cm−1 | Si–O; Al–O (framework) |
In the NIR region (Fig. 5b), at RT, the characteristic combination band (ν + δ)HOH of physisorbed water molecules inside the zeolite cavities38 at 5244 cm−1 was present. With temperature treatment, three bands appeared at 4557, 4645, and 4664 cm−1, associated with the combination of the vibrational stretching and bending modes (ν + δ)OH of different hydroxyl groups.39,40 The 4557 cm−1 band was related to the hydroxyls of the terminal silanol groups, while the bands at 4645 and 4664 cm−1 corresponded to hydroxyls in the main cavities and side pockets, respectively.39 A summary of the band assignments is provided in Table 4.
| Vibration frequency | Band assignment |
|---|---|
| 4557 cm−1 | SiOH |
| 4645 cm−1 | Si(OH)Al (12-MR) |
| 4664 cm−1 | Si(OH)Al (8-MR) |
Considering the areas obtained from the deconvolution, it could be deduced that the number of hydroxyl groups in the main cavities was greater than the number of hydroxyl groups in the side pockets. This observation was in good agreement with the results obtained through CO adsorption, where it was concluded that the proportion of Cu atoms incorporated in the side pockets was higher than in the main cavities. Consequently, there will be a higher concentration of Brønsted acid sites in the main cavities compared to the side pockets.
Fig. 6a shows a three-dimensional representation of the evolution of the species adsorbed on the surface, and Fig. 6b shows the two-dimensional projection in which the contours reflect the evolution of the bands associated with the adsorbed species. The presence of vibrational bands related to νCH, δCH, and νCO vibrations could be observed, alongside the disappearance of negative bands attributed to hydroxyl groups resulting from their interaction with the reactants. Furthermore, a highly intense band could be observed, indicating the presence of a copper carbonyl complex.
 | ||
| Fig. 6 (a) The three-dimensional and (b) two-dimensional representations of the bands that appear and disappear as a function of time during the carbonylation reaction at 200 °C and 1 bar in the Cu–H-MOR sample. | ||
Fig. 7 provides a more detailed analysis of the observed changes in the IR spectra during the reaction, highlighting the most significant variations in the bands. In the OH region (3800–3500 cm−1), it was observed that the bands associated with the bridge hydroxyls Si(OH)Al in both cavities (3605 and 3580 cm−1) had disappeared. Concurrently, a distinct set of complex bands emerged between 2800 and 3010 cm−1, suggesting a potential correlation between these observations. It is well known that the reaction of methanol with the acid centers produces a dehydration reaction in which dimethyl ether (DME) and water are formed.41,42 The presence of DME could be indicated by the bands that appeared in the region νCH, and also the bands observed at 1730, 1560 and 1360 cm−1.43 Therefore, the dehydration of methanol at acid sites is one of the fundamental stages of the reaction.19 On the other hand, methanol interacted with the surface forming methoxide species with characteristic bands at 1194 and 1178 cm−1.44 It should also be noted that CO was activated forming carbonyl species in the centers of Cu. The band observed at 2156 cm−1 was characteristic of Cu+–(CO) species and it was detected at the beginning of the reaction. As the reaction time elapsed, the intensity of this band decreased and a new band appeared at 2132 cm−1. This band could be associated with the formation of intermediate acetyl species via CO insertion into methoxide species. Finally, the reaction between acetyl species and DME produced by dehydration produced methyl acetate, which was desorbed to the gas phase.
 | ||
| Fig. 7 The time evolution of the IR spectra during the carbonylation reaction at 200 °C and 1 bar with the Cu–H-MOR catalyst. | ||
Fig. 8 shows the evolution of the bands associated with the surface species involved in the reaction (Fig. 8A) and the evolution of the gas phase (Fig. 8B). There was a direct correlation between the production of DME (m/z = 45) and methanol consumption (m/z = 31) at the same time that the hydroxyls associated with the Brønsted acid centers disappeared (3580 and 3605 cm−1). This clearly indicates that the acid centers were responsible for the dehydration of methanol to produce DME. Conversely, methanol was adsorbed forming methoxide species (1194 cm−1) on the surface of the zeolitic catalyst and CO was activated by forming Cu+ monocarbonyls (2156 cm−1).45
 | ||
| Fig. 8 (A) The evolution of the bands associated with the surface species involved in the reaction and (B) evolution of the reactants and products in the gas phase analyzed online by MS during the reaction. | ||
As can be seen in Fig. 8A, during the first minutes of the reaction a new band appeared at 2132 cm−1. According to the evolution of the bands of Cu+(CO) at 2156 cm−1 and the one of the adsorbed methoxides at 1194 cm−1, it seems that the band at 2132 cm−1 appeared after a reaction between both surface species. In addition, it could be observed that the evolution of this band followed the trend of methyl acetate production (m/z = 43). According to these observations, an insertion of CO takes place on the methoxides catalyzed by the Cu+ redox centers to form an intermediate acetyl that leads to the production of methyl acetate by reaction with the DME and the subsequent regeneration of methoxides and Cu+ centers. These observations are supported by the results published by Corma's group19 and highlights the bifunctional behavior of the Cu–H-MOR catalyst, in which acid centers and redox centers are directly involved in the process, leading to the preferential absorption of carbon monoxide and dimethyl ether on the copper surface, ultimately resulting in the predominant production of methyl acetate, and proving the synergistic effect of the Brønsted acid sites and redox (Cu+) sites.
Proposed reaction mechanism
According to the results obtained, the reaction mechanism of methanol carbonylation on Cu–H-MOR zeolite can be described in a general way through the following stages:(i) Dehydration of methanol on the Brønsted acid centers to form methoxide (CH3O−) and dimethyl ether (DME) groups;
(ii) Insertion of CO into the methoxide groups forming acetyl groups (CH3–COO−);
(iii) Reaction between DME and acetyl groups to produce methyl acetate and regenerate methoxide groups;
(iv) Hydrolysis of methyl acetate to give acetic acid.
Fig. 9 includes a schematic representation of the proposed reaction mechanism. Corma et al.46 proposed that the acid centers located in the 8-MR cavities are responsible for the carbonylation reaction in H-MOR. Iglesia et al.34 also showed that methoxide groups formed inside 8-MR cavities react more quickly with CO than those formed in 12-MR cavities. Apparently, there are no differences in the reaction mechanism that takes place in the different cavities. The acetyl intermediate reacts with water or DME producing acetic acid or methyl acetate and restoring the Brønsted acid centers and methoxide groups.
 | ||
| Fig. 9 A schematic representation of the possible reaction mechanism of methanol carbonylation on Cu–H-MOR. | ||
Nevertheless, other reactions that compete with carbonylation and that do not occur in the 8-MR cavities due to steric impediment could also take place in the 12-MR cavities. For example, methoxide groups can react with DME to form a trimethyl oxonium cation (TMO+) leading to hydrocarbon production and coke formation.46 This reaction is much less favored in the 8-MR cavities. On the other hand, the carbonylation reaction can also be inhibited in the 8-MR cavities when there are many water molecules already present due to the formation of clusters blocking these cavities. This can be avoided by increasing the temperature of the reaction, but thermodynamically the carbonylation reaction is not favored at high temperatures.
Conclusions
In this work, time-resolved operando DRIFTS-MS was used to investigate the methanol carbonylation reaction over a Cu–H-MOR catalyst hydrothermally synthesized in the absence of a template. The primary goal was to elucidate the reaction mechanism by studying the catalyst's behavior, including potential determination of the copper redox center's location within the zeolite. The catalytic performance of the Cu–H-MOR catalyst was also assessed, demonstrating its effectiveness and stability in the methanol carbonylation process. The operando DRIFTS-MS results provide valuable insights into the reaction mechanism and the involvement of the acid and redox centers during the reaction. The results obtained suggest that Cu–H-MOR follows a bifunctional mechanism, in which Brønsted acid sites favor the dehydration of methanol to form methoxides and DME, and redox sites (Cu+) are responsible for the activation of CO for insertion into methoxides, giving rise to an acetyl-like intermediate complex with which DME reacts to form methyl acetate, which can be further hydrolyzed to acetic acid. This approach provides new insights into the rational design of more efficient catalysts to replace conventional processes (BP Cativa™ and Monsanto) and for achieving sustainable acetic acid production. Overall, this work significantly contributes to enhancing our understanding of the heterogeneous approach for acetic acid production and opens potential avenues for optimizing the process. By addressing the weaknesses and uncovering the underlying mechanisms, this study paves the way for advancements in heterogeneous catalysis to more efficiently produce acetic acid and other valuable chemicals.Author contributions
L. F. Bobadilla, L. A. Luque-Álvarez: conceptualization, methodology. L. A. Luque-Álvarez, J. González-Arias: data curation, writing – original draft preparation. L. A. Luque-Álvarez: visualization, investigation. J. A. Odriozola and T. R. Reina: funding acquisition. L. F. Bobadilla and F. Romero-Sarria: supervision, writing – reviewing and validation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for this work has been obtained from the Spanish Ministerio de Ciencia e Innovación through the project SMART-FTS (PID2021-126876OB-I00). Ligia A. Luque-Álvarez thanks VI-PPITUS (University of Sevilla) for her current predoctoral contract. This work has been supported by the FJC2021-047672-I grant co-financed by MCIN/AEI/10.13039/501100011033 and the European Union Next Generation EU/PRTR funds.References
- F. Li, B. Chen, Z. Huang, T. Lu, Y. Yuan and G. Yuan, Green Chem., 2013, 15, 1600–1607 RSC.
- Y. Ni, L. Shi, H. Liu, W. Zhang, Y. Liu, W. Zhu and Z. Liu, Catal. Sci. Technol., 2017, 7, 4818–4822 RSC.
- G. Deshmukh and H. Manyar, Biotechnol. Appl. Biomass, 2020, 5 DOI:10.5772/INTECHOPEN.92289.
- Acetic Acid – Chemical Economics Handbook (CEH) | S&P Global, https://www.spglobal.com/commodityinsights/en/ci/products/acetic-acid-chemical-economics-handbook.html.
- Statista, Acetic acid global market volume 2015–2026, https://www.statista.com/statistics/1245203/acetic-acid-market-volume-worldwide/.
- J. H. Jones, Platinum Met. Rev., 2000, 44, 94–105 CAS.
- G. J. Sunley and D. J. Watson, Catal. Today, 2000, 58, 293–307 CrossRef CAS.
- J. H. Jones, Platinum Met. Rev., 2000, 44, 94–105 CAS.
- E. Zhan, Z. Xiong and W. Shen, J. Energy Chem., 2019, 36, 51–63 CrossRef.
- L. A. Luque-Álvarez, M. Serrano-Cruz, M. González-Castaño, L. F. Bobadilla and J. A. Odriozola, Microporous Mesoporous Mater., 2023, 360, 112725 CrossRef.
- R. Ahmad, D. Schrempp, S. Behrens, J. Sauer, M. Döring and U. Arnold, Fuel Process. Technol., 2014, 121, 38–46 CrossRef CAS.
- K. Sun, W. Lu, F. Qiu, S. Liu and X. Xu, Appl. Catal., A, 2003, 252, 243–249 CrossRef CAS.
- J. Ereña, R. Garoña, J. M. Arandes, A. T. Aguayo and J. Bilbao, Catal. Today, 2005, 107–108, 467–473 CrossRef.
- S. Huang, Y. Wang, Z. Wang, B. Yan, S. Wang, J. Gong and X. Ma, Appl. Catal., A, 2012, 417–418, 236–242 CrossRef CAS.
- K. Fujimoto, T. Shikada, K. Omata and H. Tominaga, Bull. Chem. Soc. Jpn., 1984, 2047–2050 CAS.
- B. Ellis, M. J. Howard, R. W. Joyner, K. N. Reddy, M. B. Padley and W. J. Smith, Stud. Surf. Sci. Catal., 1996, 101, 771–779 CrossRef CAS.
- Y. Chu, A.-Y. Lo, C. Wang and F. Deng, J. Phys. Chem. C, 2019, 123, 15503–15512 CrossRef CAS.
- K. Cai, S. Huang, Y. Li, Z. Cheng, J. Lv and X. Ma, ACS Sustainable Chem. Eng., 2019, 7, 2027–2034 CrossRef CAS.
- T. Blasco, M. Boronat, P. Concepción, A. Corma, D. Law and J. A. Vidal-Moya, Angew. Chem., Int. Ed., 2007, 46, 3938–3941 CrossRef CAS.
- L. F. Bobadilla, L. Azancot, L. A. Luque-Álvarez, G. Torres-Sempere, M. González-Castaño, L. Pastor-Pérez, J. Yu, T. Ramírez-Reina, S. Ivanova, M. A. Centeno and J. A. Odriozola, Chem, 2022, 4, 1250–1280 CAS.
- M. Bryan, Z. Gili and M. T. Conato, Mater. Res. Express, 2018, 6, 015515 CrossRef.
- S. H. Lee, J. K. Kang and E. D. Park, Korean J. Chem. Eng., 2018, 35, 2145–2149 CrossRef CAS.
- M. Hassnain, J. Mater. Sci. Nanotechnol., 2017, 3–19 Search PubMed.
- G. Leofanti, M. Padovan, G. Tozzola and B. Venturelli, Catal. Today, 1998, 41, 207–219 CrossRef CAS.
- M. Alhamami, H. Doan and C.-H. Cheng, Materials, 2014, 7, 3198–3250 CrossRef PubMed.
- A. J. Knorpp, A. B. Pinar, M. A. Newton, T. Li, A. Calbry-Muzyka and J. A. Van Bokhoven, RSC Adv., 2021, 11, 31058–31061 RSC.
- V. M. Georgieva, E. L. Bruce, M. C. Verbraeken, A. R. Scott, W. J. Casteel, S. Brandani and P. A. Wright, J. Am. Chem. Soc., 2019, 141, 12744–12759 CrossRef CAS.
- N. Sergent, P. Gélin, L. Périer-Camby, H. Praliaud and G. Thomas, Phys. Chem. Chem. Phys., 2002, 4, 4802–4808 RSC.
- S. Bordiga, E. E. Platero, C. O. Arean and C. Lamberti, J. Catal., 1992, 185, 179–185 CrossRef.
- S. Bordiga, C. Lamberti, F. Geobaldo, A. Zecchina, G. T. Palomino and C. O. Areán, Langmuir, 1995, 11, 527–533 CrossRef CAS.
- S. Bordiga, C. Lamberti, F. Bonino, A. Travert and F. Thibault-Starzyk, Chem. Soc. Rev., 2015, 44, 7262–7341 RSC.
- A. Zecchina, G. Spoto and S. Bordiga, Vibrational Spectroscopy of Zeolites, John Wiley & Sons, Ltd., 2006, pp. 3043–3071 Search PubMed.
- K. I. Hadjiivanov and G. N. Vayssilov, Adv. Catal., 2002, 47, 307–511 CAS.
- A. Bhan, A. D. Allian, G. J. Sunley, D. J. Law and E. Iglesia, J. Am. Chem. Soc., 2007, 129, 4919–4924 CrossRef CAS PubMed.
- M. Boronat, C. Martínez-Sánchez, D. Law and A. Corma, J. Am. Chem. Soc., 2008, 130, 16316–16323 CrossRef CAS.
- P. Feng, G. Zhang, X. Chen, K. Zang, X. Li and L. Xu, Appl. Catal., A, 2018, 557, 119–124 CrossRef CAS.
- R. Anquetil, J. Saussey and J. C. Lavalley, Phys. Chem. Chem. Phys., 1999, 1, 555–560 RSC.
- W. Hanke and K. Möller, Zeolites, 1984, 4, 244–250 CrossRef CAS.
- V. B. Kazansky, L. M. Kustov and V. Y. Borovkov, Zeolites, 1983, 3, 77–81 CrossRef CAS.
- A. Vimont, J. M. Goupil, J. C. Lavalley, M. Daturi, S. Surblé, C. Serre, F. Millange, G. Férey and N. Audebrand, J. Am. Chem. Soc., 2006, 128, 3218–3227 CrossRef CAS.
- Z. Azizi, M. Rezaeimanesh, T. Tohidian and M. R. Rahimpour, Chem. Eng. Process., 2014, 82, 150–172 CrossRef CAS.
- A. Brunetti, M. Migliori, D. Cozza, E. Catizzone, G. Giordano and G. Barbieri, ACS Sustainable Chem. Eng., 2020, 8, 10471–10479 CrossRef CAS.
- F. E. Celik, T. Kim, A. N. Mlinar and A. T. Bell, J. Catal., 2010, 274, 150–162 CrossRef CAS.
- S. G. Izmailova, I. V. Karetina, S. S. Khvoshchev and M. A. Shebaeva, J. Colloid Interface Sci., 1993, 165, 318–324 CrossRef.
- K. Hadjiivanov and H. Knözinger, J. Catal., 2000, 191, 480–485 CrossRef CAS.
- M. Boronat, C. Martínez and A. Corma, Phys. Chem. Chem. Phys., 2011, 13, 2603 RSC.
| This journal is © The Royal Society of Chemistry 2024 |