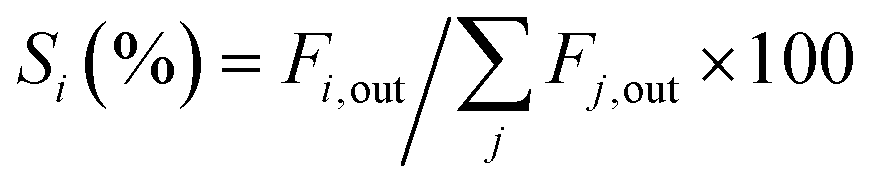InNi3C0.5@C-derived InNi3 alloy as a coke-resistant low-temperature catalyst for selective butadiene hydrogenation†
Received
11th September 2023
, Accepted 14th November 2023
First published on 15th November 2023
Abstract
The development of non-precious metal catalysts to replace Pd-based industrial catalysts for 1,3-butadiene hydrogenation is highly desired. However, the cheap transition metal-based candidates developed to date, including Ni, Cu, Co, and Fe, show lower activity than precious metal catalysts, and typically suffer from deactivation due to coke deposition. In this contribution, an InNi3 alloy catalyst is designed via depletion of the layers of a carbon-capped interstitial compound InNi3C0.5 by two different approaches, hydrogenation and air oxidation, under varying conditions. The carbon depletion processes are followed by combined techniques to determine the full structural and compositional evolution processes, and the resulting catalysts are thoroughly characterized and evaluated for the selective hydrogenation of 1,3-butadiene. The InNi3 alloy serves as a coke-resistant low-temperature catalyst for 1,3-butadiene hydrogenation, achieving >94% total butene selectivity at >96% conversion at a mild temperature of 318 K. Post characterization of the catalyst after long-term testing verifies the structural robustness and excludes the accumulation of carbonaceous deposits, thus confirming the InNi3 alloy as a novel and promising system for selective hydrogenations.
Introduction
The selective hydrogenation of 1,3-butadiene (BD) is vital to the purification of alkadienes in the upgrading of C4 species to polymers and other valuable chemicals, in which the content of BD residuals should be strictly controlled, as even a trace amount (>200 ppm) will cause catalyst deactivation in the polymerization units.1–4 In this context, selective hydrogenation is the most effective approach, and depends on the use of efficient catalysts to hydrogenate only one C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of the BD molecule while avoiding over-hydrogenating the other valuable olefins in the C4 stream. To date, precious metals based on Pd,5–8 Au,9–11 and Pt (ref. 12–14) have been identified as the most effective catalysts, among which promoter-modified Pd catalysts have been applied in industry. On the other hand, replacing precious metal catalysts with cheap candidates is attractive in heterogeneous catalysis. For BD hydrogenation, distinct catalytic systems based on cheap transition metals have been reported in the last decade, including Cu,15–18 Ni,19–21 Fe,22 Co,23,24 and Mo.25,26 Nonetheless, catalyst deactivation owing to the deposition of carbonaceous materials is frequently reported in the literature (Cu,17,18 Ni,20 Fe,22 Co,23,24 Mo (ref. 25 and 26)), causing a drop in activity and alteration in the product distributions. Furthermore, most of these catalysts are operated at relatively high temperatures (>373 K) in order to achieve a reasonable performance.
C bond of the BD molecule while avoiding over-hydrogenating the other valuable olefins in the C4 stream. To date, precious metals based on Pd,5–8 Au,9–11 and Pt (ref. 12–14) have been identified as the most effective catalysts, among which promoter-modified Pd catalysts have been applied in industry. On the other hand, replacing precious metal catalysts with cheap candidates is attractive in heterogeneous catalysis. For BD hydrogenation, distinct catalytic systems based on cheap transition metals have been reported in the last decade, including Cu,15–18 Ni,19–21 Fe,22 Co,23,24 and Mo.25,26 Nonetheless, catalyst deactivation owing to the deposition of carbonaceous materials is frequently reported in the literature (Cu,17,18 Ni,20 Fe,22 Co,23,24 Mo (ref. 25 and 26)), causing a drop in activity and alteration in the product distributions. Furthermore, most of these catalysts are operated at relatively high temperatures (>373 K) in order to achieve a reasonable performance.
Nickel is well-known for diverse hydrogenations, but its applications in BD hydrogenation are still rare. Previous studies reveal that catalyst deactivation and over-hydrogenation, particularly at high conversions, are the key concerns for Ni particles.19–21 Coating supported Ni catalysts with ionic liquids19,20 and designing ZnNi3C0.7 (ref. 21) interstitial compounds have been proven as effective methods to enhance the performance, which is attributed to the electronic effects induced by the ionic liquids or the segmentation atoms leading to altered adsorptions of the substrates.19,27 Meanwhile, the potentials of InNi3C0.5 in selective hydrogenations (CO2,28–30 dimethyl oxalate31) have only recently been recognized. Inspired by these promising literature results, herein, for the first time, we report InNi3 derived from carbon shell-capped InNi3C0.5 particles as a highly coke-resistant non-precious metal catalyst for low-temperature BD hydrogenation. The structure and composition evolution of the hybrids during depletion of the carbon shells via both air oxidation and H2 reduction are unveiled by combined characterization techniques. In contrast to the previous cheap metal catalysts, the InNi3 alloy featured high resistance to coking and good butene selectivity under mild reaction conditions (>90% butene yield at 318 K).
Results and discussion
Synthesis and characterization of InNi3C0.5@C
The bimetallic interstitial compound preserved in carbon shells (InNi3C0.5@C) was prepared by a simple one-pot solid-state reaction between melamine, Ni(OH)2 and In(OH)3 in H2 (see the Experimental section). In contrast to the previous approach, which typically involved the preparation of a bimetallic alloy followed by the insertion of interstitial carbon atoms by carbonization with C2H2, the developed method is more straightforward and without the use of any solvents. Powder X-ray diffraction patterns (PXRD) reveal that all the diffraction patterns of the resulting product can be perfectly indexed to InNi3C0.5 (PDF#28-0468, Fig. S1†). N2 sorption analysis showed a specific surface area (SBET) of 60 m2 g−1 for the sample (Fig. S2†). The relatively high SBET is likely because of the deposition of porous carbon shells on the InNi3C0.5 crystals. To prove the core–shell structures, we performed high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM, Fig. 1). The high-resolution images in Fig. 1a and b clearly display carbon shells of several nanometres in size, surrounding the InNi3C0.5 particles. The elemental color mapping images in Fig. 1c further illustrate the geometrical relevance of Ni and In, and the broader distribution of C around these particles, thus unambiguously pointing to the formation of InNi3C0.5@C core–shell structures.
 |
| | Fig. 1 a and b, High-resolution STEM images of the as-prepared InNi3C0.5@C with, c, elemental color mapping results. | |
Depleting the shells of InNi3C0.5@C
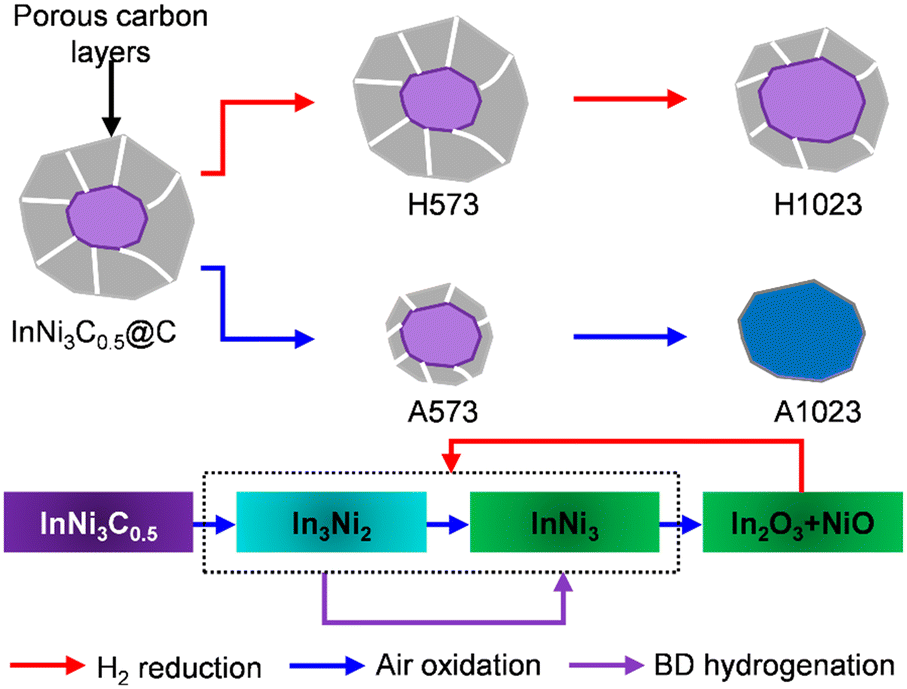
To remove the carbon shells, two different methods were adopted: air oxidation and methanation in H2. The derived solids were denoted as AX or HX, respectively, where X represents the applied temperature in K. PXRD revealed the typical facets of InNi3C0.5 for HX and A573, whereas mixed In2O3 and NiO were detected for A1023 (Fig. 2a). Nonetheless, the effectiveness of H2 reduction for carbon removal was confirmed by STEM observation of H1023, showing clearly reduced carbon layers surrounding the metal particles (Fig. S3†). In situ PXRD was further performed to track the phase evolution of InNi3C0.5@C during air oxidation (Fig. 2b). In this case, the two most intense diffraction peaks gradually weakened in intensity and shifted to lower 2θ with increasing temperature. Formation of In3Ni2 was clearly evidenced at 823 K, as well as the appearance of In2O3 and InNi3. When further increasing the temperature to 1073 K, the intensities of In3Ni2 decreased while In2O3 and InNi3 predominated, highlighting the different thermal stabilities. The above PXRD results confirmed the more aggressive destruction of InNi3C0.5@C by air oxidation than H2 reduction, which was corroborated by Raman spectroscopy (Fig. 2c). The typical D and G bands at 1350 and 1580 cm−1, respectively, were found for HX and A573, suggesting still the presence of carbonaceous residuals on these samples. Meanwhile, additional bands at 570 cm−1 likely relating to Ni–In alloys appeared for A573 and H1023, due to the partial depletion of the shells. In contrast, those carbon-defect related bands were not detected for A1023, and new bands at 518, 1069, and 1460 cm−1 associated with In2O3 (ref. 32) and NiO (ref. 33) appeared, suggesting the complete removal of the carbon shells. These results were further corroborated by X-ray photoelectron spectroscopy (XPS). Elemental analysis showed a significant increase of surface metal species for A1023 as compared to InNi3C0.5@C (4.44 vs. 1.21% for Ni, and 5.53 vs. 1.08% for In), plus the disappearance of N signals (Fig. S4†). Additionally, the Ni0 and In0 species, typically observed for InNi3C0.5 in previous studies,29,30 also disappeared after the oxidation treatment (Fig. S5†). Thermogravimetric analysis (TGA) was performed to quantify the carbon residuals (Fig. 2d). The TGA profiles differed greatly from each other, and showed sequential weight loss and weight gain due to the complicated carbon combustion and oxidation of the metallic components. By supposing that InNi3C0.5@C was completely oxidized into In2O3, NiO, and CO2, the carbon residuals were calculated to be 21%, 20%, 15%, 10%, and 0, for InNi3C0.5@C, H573, H1023, A573, and A1023, respectively (see the Experimental section for details).
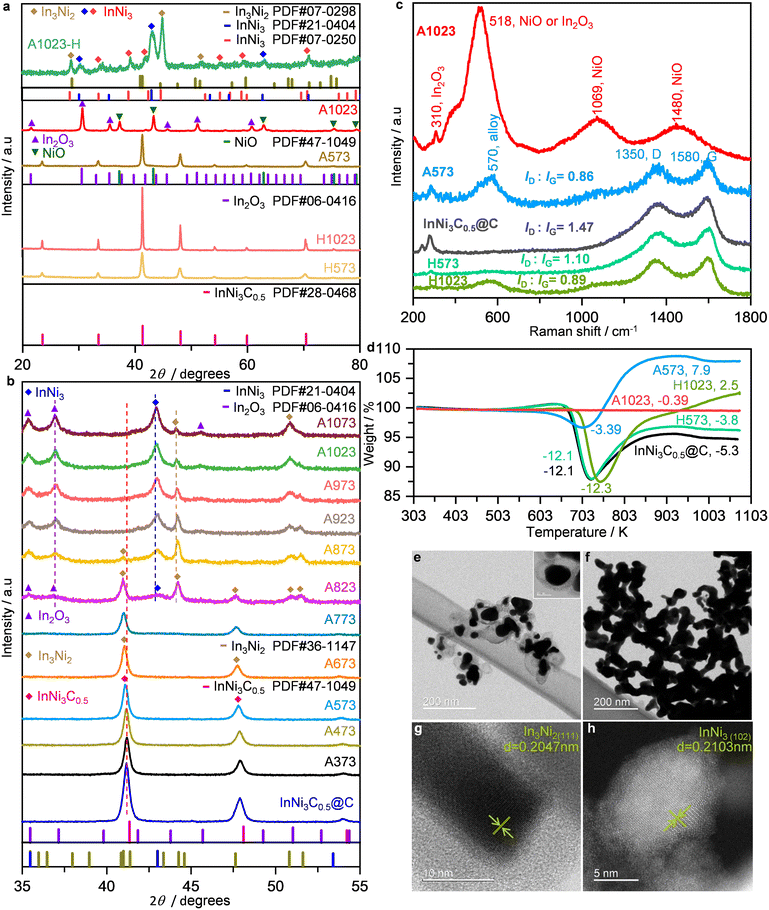 |
| | Fig. 2 a, PXRD; b, in situ PXRD of InNi3C0.5@C in air; c, Raman spectra; d, TGA profiles. STEM images of e, InNi3C0.5@C and f–h, A1023 after H2 reduction. The inset in e shows the magnified image. | |
Since air oxidation at 1023 K can completely remove the carbon shells, the compositions of A1023 after H2 reduction at 673 K were studied. STEM was used to visualize the aggregated particles without any carbon residuals, and the lattice fringes of two alloy phases (In3Ni2 and InNi3) were verified (Fig. 2e–h), agreeing well with the PXRD result (Fig. 2a). Combining all the above characterization results, the full structure and composition evolution of InNi3C0.5@C can be depicted as in Fig. 3. Air oxidation removes the carbon shells. The interstitial composition can be preserved in H2 reduction but gradual phase transformation from InNi3C0.5 to a mixed Ni–In alloy and finally to mixed oxides occurred during oxidation. The alloys can be easily restored by subsequent reduction of the mixed oxides.
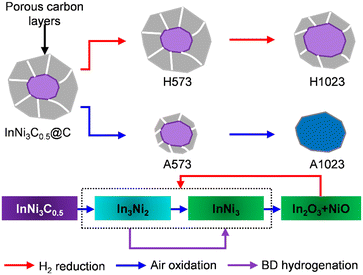 |
| | Fig. 3 Schematic illustrations of the structure and composition evolution of InNi3C0.5@C subject to different treatments. | |
Catalytic performance in BD hydrogenation
Pristine InNi3C0.5@C and the H2- or air-treated sample were applied for the first time to BD hydrogenation in a fixed-bed reactor. The impact of the H2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :BD ratio was first evaluated in a temperature-ramping study on InNi3C0.5@C, showing a shift of the activity curves to lower temperatures and slightly reduced selectivity to total butene when the ratio was increased from 20 to 50, and no significant change at 100 (Fig. 4a and b). Further comparisons of the performances of the developed catalysts were conducted at H2:BD = 50 (Fig. 4c and d). Divergent catalytic behaviors were observed for the catalysts subject to different treatments. Namely, the air-oxidized samples showed much higher activity and slightly improved selectivity to total butene as compared with the pristine InNi3C0.5@C. For example, the temperature corresponding to 96% BD conversion was markedly lower, by 75 K (from 423 to 348 K). In contrast, the H2-treated samples showed lower activity in comparison to InNi3C0.5@C, although the total butene selectivity was enhanced. These different catalytic responses can be understood by considering the different structures and compositions of the catalysts as revealed in the previous sections. The activities of the four catalysts with InNi3C0.5 particles preserved by carbon shells followed the order A573 > InNi3C0.5@C > H573 > H1023, hinting at likely interplays between the carbon residuals and the particle sizes (e.g., the greatly sharpened PXRD diffractions of H1023 suggested enlarged particle sizes of InNi3C0.5). A1023 displayed a higher activity, even though N2 sorption isotherms revealed much lower specific surface areas of A1023 than InNi3C0.5@C (11 vs. 60 m2 g−1, Fig. S2†), thus indicating reasonable hydrogenation potentials of the Ni–In alloys. Temperature-programmed desorption of H2 (H2-TPD) was further performed to understand the catalytic differences (Fig. S6†). A1023 without the carbon shells showed three desorption peaks at 433, 801, and 1107 K, whereas the pristine InNi3C0.5@C only displayed a low-temperature desorption peak at 380 K. Therefore, the stronger H2 adsorption propensity of A1073 agrees well with the better hydrogenation activity. Given the superior catalytic performance of A1023, the long-term stability was evaluated. Negligible deactivation was observed under the high conversion conditions (XBD > 98%) and the total butene selectivity was maintained at >87% (Fig. 4e). The catalyst stability was also verified at lower conversions (ca. 80%) by increasing the gas hourly space velocity (GHSV) from 90000 to 300000 cm3 gcat−1 h−1. The catalyst remained essentially stable in the 40 h test (Fig. S7†), although a slow activity drop in the first 16 h was observed, which might be related to the phase transition from In3Ni2 to InNi3 (vide infra). The GHSV was further varied and near full conversion was reached at 318 K and 30000 cm3 gcat−1 h−1 (Fig. S8†). To assess the catalyst low-temperature potential, the stability at 318 K was evaluated (Fig. 4f). After a very small activity drop in the first 12 h accompanied by an increase in butene selectivity, a stable performance was achieved in the next 24 h with >94% total selectivity to butene at >96% BD conversion.
:BD ratio was first evaluated in a temperature-ramping study on InNi3C0.5@C, showing a shift of the activity curves to lower temperatures and slightly reduced selectivity to total butene when the ratio was increased from 20 to 50, and no significant change at 100 (Fig. 4a and b). Further comparisons of the performances of the developed catalysts were conducted at H2:BD = 50 (Fig. 4c and d). Divergent catalytic behaviors were observed for the catalysts subject to different treatments. Namely, the air-oxidized samples showed much higher activity and slightly improved selectivity to total butene as compared with the pristine InNi3C0.5@C. For example, the temperature corresponding to 96% BD conversion was markedly lower, by 75 K (from 423 to 348 K). In contrast, the H2-treated samples showed lower activity in comparison to InNi3C0.5@C, although the total butene selectivity was enhanced. These different catalytic responses can be understood by considering the different structures and compositions of the catalysts as revealed in the previous sections. The activities of the four catalysts with InNi3C0.5 particles preserved by carbon shells followed the order A573 > InNi3C0.5@C > H573 > H1023, hinting at likely interplays between the carbon residuals and the particle sizes (e.g., the greatly sharpened PXRD diffractions of H1023 suggested enlarged particle sizes of InNi3C0.5). A1023 displayed a higher activity, even though N2 sorption isotherms revealed much lower specific surface areas of A1023 than InNi3C0.5@C (11 vs. 60 m2 g−1, Fig. S2†), thus indicating reasonable hydrogenation potentials of the Ni–In alloys. Temperature-programmed desorption of H2 (H2-TPD) was further performed to understand the catalytic differences (Fig. S6†). A1023 without the carbon shells showed three desorption peaks at 433, 801, and 1107 K, whereas the pristine InNi3C0.5@C only displayed a low-temperature desorption peak at 380 K. Therefore, the stronger H2 adsorption propensity of A1073 agrees well with the better hydrogenation activity. Given the superior catalytic performance of A1023, the long-term stability was evaluated. Negligible deactivation was observed under the high conversion conditions (XBD > 98%) and the total butene selectivity was maintained at >87% (Fig. 4e). The catalyst stability was also verified at lower conversions (ca. 80%) by increasing the gas hourly space velocity (GHSV) from 90000 to 300000 cm3 gcat−1 h−1. The catalyst remained essentially stable in the 40 h test (Fig. S7†), although a slow activity drop in the first 16 h was observed, which might be related to the phase transition from In3Ni2 to InNi3 (vide infra). The GHSV was further varied and near full conversion was reached at 318 K and 30000 cm3 gcat−1 h−1 (Fig. S8†). To assess the catalyst low-temperature potential, the stability at 318 K was evaluated (Fig. 4f). After a very small activity drop in the first 12 h accompanied by an increase in butene selectivity, a stable performance was achieved in the next 24 h with >94% total selectivity to butene at >96% BD conversion.
 |
| | Fig. 4 The catalytic performance of a and b, pristine, and c and d, partially carbon shell-removed InNi3C0.5@C in BD hydrogenation. e and f Time-on-stream performance of A1023. Reaction conditions: GHSV = 90000 cm3 gcat−1 h−1, 0.69 vol% BD balanced in N2, a and b, H2:BD = 20–100, c–e, H2:BD = 50, e, T = 353 K, f, 318 K, H2:BD = 100, GHSV = 30000 cm3 gcat−1 h−1. | |
Characterization of the used catalysts and kinetics
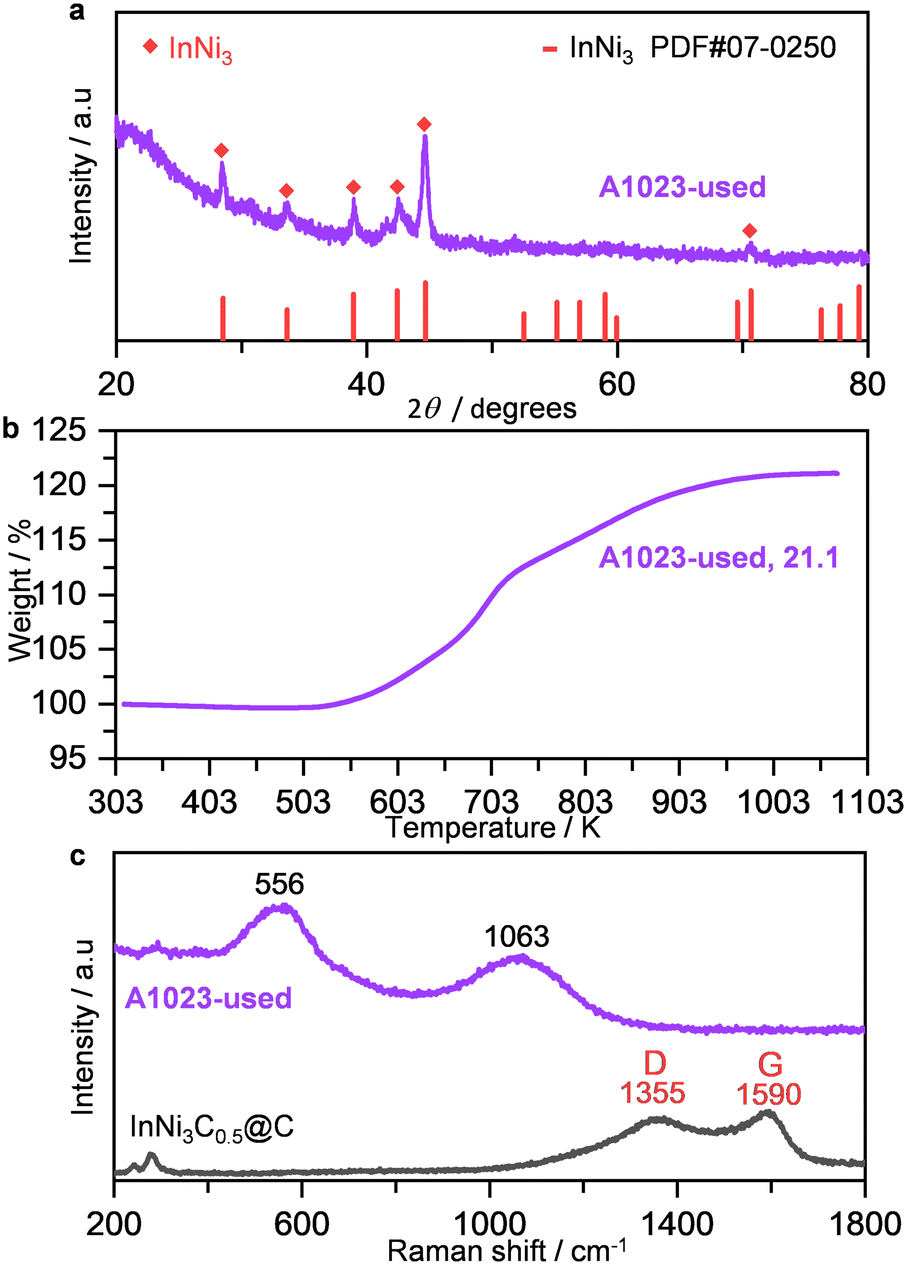
To probe the properties of the used catalyst, A1023 after the 24 h stability test was characterized using different techniques. PXRD revealed that all the diffraction lines can be indexed to InNi3, and the other alloy, In3Ni2, in the reduced sample was not observed (Fig. 5a), thus supporting our earlier speculation on the activity drop. TEM observations evidenced the similar structures of the interconnected nanoparticles, as well as the distinct lattice fringes of the (101) and (201) facets of the InNi3 alloys (Fig. S9†). Additionally, coke deposition was excluded by TGA and Raman spectra (Fig. 5b and c), showing weight gain of 21.1%, close to the theoretical values (24.7% for InNi3 and 22.5% for In3Ni2), and the absence of Raman features for carbonaceous materials. Similar characterization results were acquired for the catalyst evaluated at 318 K for 36 h (Fig. S10†), except that both InNi3 and In3Ni2 were preserved, hinting that the phase transition was much favored at high temperatures.
 |
| | Fig. 5 Characterization of A1023 after the 24 h evaluation in BD hydrogenation: a, PXRD; b, TGA; c, Raman spectra. | |
To shed light on the reaction mechanism, the reaction kinetics were further studied. By varying the partial pressures of the respective reactants while keeping the same total flow rate, the partial reaction orders of H2 and BD on A1023 were determined to be 1.1 and −1.2, respectively (Fig. 6). The negative BD order suggests the significantly stronger adsorption of the alkadiene on the alloy surface than H2. This trend agrees well with those observed for Pd- and Pt-based catalysts.8,34 Therefore, it is likely that the hydrogenation on the InNi3 alloy also follows the classic Horiuti–Polanyi mechanism for alkyne hydrogenation on Pd-based catalysts.35 Since BD hydrogenation is a chain reaction, the timely desorption of butene is crucial in order to achieve high selectivity, as well as to avoid alkene oligomerization, which is suspected to induce coke accumulation. In this study, the main efforts were placed on understanding the impacts of the carbon shell depletion of the interstitial compounds. A joint study coupling the synthesis of structurally well-defined catalysts and density functional theory calculations is expected to produce more fundamental insights into the nature of the active sites and reaction mechanisms.
 |
| | Fig. 6 The partial reaction orders of the respective reactants in BD hydrogenation on A1023. | |
Conclusions
In summary, we have reported an InNi3 alloy as a new and stable non-precious metal catalyst for the selective hydrogenation of 1,3-butadiene. This alloy was derived from a bimetallic interstitial compound, InNi3C0.5, preserved in a carbon shell. With the aim of removing the carbon shell, the evolution of the structure and composition during air oxidation and H2 reduction was studied in detail. Complete depletion of the shell was achieved by oxidation at 1023 K, accompanied by the formation of mixed metal oxides, which can be converted to In–Ni alloys by H2 reduction. When evaluated in BD hydrogenation, a remarkable performance was achieved at a low temperature of 318 K, with >94% selectivity to total butene at >96% BD conversion. In contrast to the other known non-precious metal catalysts that frequently suffer from quick deactivation, InNi3 showed high coke resistance in the hydrogenation reaction, thus offering relatively stable operation.
Experimental
Materials
Melamine (C3H6N6, 99%) was purchased from Aladdin. Ni(OH)2 (Ni 61%) and In(OH)3 (99.99%) were purchased from Macklin. All reagents were obtained from commercial suppliers without further purification prior to use.
Catalyst synthesis
InNi3C0.5@C: the bimetallic interstitial compound InNi3C0.5 preserved in carbon shells (InNi3C0.5@C) was prepared by a one-pot solid-state synthetic approach. Typically, a mixture of commercial nickel hydroxide and indium hydroxide was used as the metal precursors, with melamine as the carbon precursor, where the mass ratio of the metal precursors to melamine was fixed at 1:5. By mechanical mixing, 0.3735 g of In(OH)3, 0.6265 g of Ni(OH)2, and 5 g of melamine were ground in a mortar for 15 min until uniformly mixed. The obtained greenish powders were placed in a tubular furnace under a flow of H2 (60 mL min−1) and annealed at 973 K (duration 4 h, ramp rate 5 K min−1). After cooling to room temperature, the solids were stored in air.
Decarbonized InNi3C0.5@C: to remove the carbon shells, the as-prepared InNi3C0.5@C solids were placed in a tubular furnace and treated by two different methods. The first method was oxidation in static air for at 573 and 1023 K (duration 3 h, ramp rate 5 K min−1). The derived samples were denoted by A573 and A1023. The other method was methanation via H2 reduction. The solids were treated in flowing H2 (30 mL min−1) at 573 and 1023 K (duration 3 h, ramp rate 5 K min−1). The obtained materials were denoted by H573 and H1023.
Characterization methods
N2 sorption was performed at 77 K on a Quanta chrome NT3LX-2 instrument. Prior to the analysis, the powder samples were degassed at 573 K for 3 h. The specific surface areas (SBET) were calculated by using the Brunauer–Emmett–Teller (BET) method. Powder X-ray diffraction (PXRD) patterns were conducted on an X'Pert 3, PANalytical X-ray diffractometer using Cu Kα radiation in a scanning angle (2θ) range of 20–80° at a speed of 2° min−1. In situ PXRD patterns were recorded with a Rigaku SmartLab-9 kW D/teX Ultra250 diffractometer using a Cu Kα radiation source operated at 40 kV and 200 mA. The powder samples were spread into a high-temperature chamber and the diffraction patterns were recorded at 2θ of 30–90° with a scanning rate of 10° min−1 at room temperature. Then, the samples were heated to 1073 K successively at a rate of 10 K min−1 under an air flow of 30 mL min−1. Thermogravimetric analyses (TGA) of the catalysts were carried out using the NETZSCH STA 449 F5 system with a heating rate of 10 K min−1 (from room temperature to 1073 K) under flowing air. The samples were placed in an α-Al2O3 crucible. Raman spectra were acquired on a confocal laser micro-Raman spectrometer (HORIBA, Lab RAM HR Evolution) with a wavelength of 532 nm, 60 s transits per sample, and a spectral resolution of 1 cm−1. Transmission electron microscopy (TEM) was conducted on a FEI Talos F200S. The high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) measurements were carried out on a JEOL-ARM200F electron microscope. The sample powders were dispersed in ethanol by ultrasonication and then the species were obtained by dropping a droplet suspension on a carbon film supported on a copper grid for the analysis. Temperature-programmed desorption of H2 (H2-TPD) was conducted on a Micromeritics Autochem II 2920 chemisorber equipped with a thermal conductivity detector and mass spectrometer. Typically, the sample (100 mg) was pretreated in a quartz U-tube reactor with a He gas stream (50 cm3 min−1) at 473 K for 1 h. After cooling to room temperature, a 10% H2/Ar flow (cm3 min−1) was introduced and the sample was heated to 1273 K at a rate of 10 K min−1. The X-ray photoelectron spectroscopy (XPS) measurements were carried out on a Thermo ESCALAB 250Xi spectrometer (15 kV Al Kα X-ray source). The C 1s peaks were calibrated at the binding energy of 284.8 eV.
Catalytic testing
Butadiene hydrogenation was evaluated in a home-made fixed-bed quartz reactor (4 mm internal diameter, 400 mm length) at ambient pressure. Pellet catalysts of 20–40 mesh in size were used for the catalytic evaluation. Typically, the catalysts were packed between glass wool in the middle of the reactor, which was vertically placed in an electronic oven equipped with a temperature controller. The catalysts were first reduced by H2 (50 cm3 min−1) for 1 h. After cooling to room temperature, the reactive gases composed of 4.03 vol% BD/N2, 10 vol% H2/N2, and balancing N2 were introduced into the reactor. The tail gas from the outlet of the reactor was periodically analyzed by using an Agilent 7890B chromatograph equipped with a CEC-Alumina column (30 m × 0.53 mm × 10 μm) and a flame ionization detector. For the kinetic measurements, the BD conversions were restricted to <25% in order to minimize the gradient of the reactants in the feed. The reaction rates were measured at 323 K by varying the relative partial pressures of the respective reactants (PH2 = 0.17–0.52, PBD = 0.0086–0.014).
The BD conversion (XBD) and the product selectivity (Si) were determined by eqn (1) and (2), respectively:
| | | XBD (%) = (FBD,in − FBD,out)/FBD,in × 100 | (1) |
| |  | (2) |
where
FBD,in and
FBD,out are the BD flow rates (mol min
−1) from the inlet and outlet, respectively, and
Fi,out and
Fj,out are the outlet flow rates (mol min
−1) of the reaction products.
i (
j) = 1-butene,
trans-2-butene,
cis-2-butene, and butane.
The carbon balances (ε, %), estimated by eqn (3), were typically within 100 ± 2 for all the tests:
| |  | (3) |
Estimation of the carbon residual content of InNi3C0.5@C
i) Supposing that the content of the carbon shell (not the total carbon content) is x, then the share of InNi3C0.5 is 1 − x.
ii) According to the PXRD of the completely oxidized samples (see Fig. 2a), the pristine sample is finally converted into NiO and In2O3. Therefore, the chemical reaction during the combustion of InNi3C0.5 can be expressed by eqn (4):
| | | InNi3C0.5 + 2.75O2 → 0.5In2O3 + 3NiO + 0.5CO2 | (4) |
Based on
eqn (4), the weight gain
y is 20.2%, determined by
eqn (5):
| | | y = (4.5 × Mw(O) − 1.2 × Mw(C))/Mw(InNi3C0.5) | (5) |
The final weight change (
z) in the TGA is shown in
Fig. 2d.
Then z can be determined by eqn (6):
Therefore:
x
= 0.202 − z/1.202.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank the National Natural Science Foundation of China (22372150), Jinhua Science and Technology Plan Project (2022-1-078), National Key Research and Development Project of China (2022YFB3805600), and Zhejiang Normal University for providing financial support (ZZ323205020521005039, KYJ51020910, and YS304320036).
References
- C. Louis and L. Delannoy, Adv. Catal., 2019, 1–88, DOI:10.1016/bs.acat.2019.08.002.
-
P. R. Selvakannan, L. Hoang, V. V. Kumar, D. Dumbre, D. Jampaiah, J. Das and S. K. Bhargava, in Catalysis for clean energy and environmental sustainability: Petrochemicals and refining processes, ed. K. K. Pant, S. K. Gupta and E. Ahmad, Springer International Publishing, Cham, 2021, vol. 2, pp. 205–228 Search PubMed.
- M. Wang, Y. Wang, X. Mou, R. Lin and Y. Ding, Chin. J. Catal., 2022, 43, 1017–1041 CrossRef CAS.
- Q. C. Wei, Y. Chen, Z. Wang, D. Z. Yu, W. H. Wang, J. Q. Li, L. H. Chen, Y. Li and B. L. Su, Angew. Chem., 2022, 61, e202210573 CrossRef CAS.
- H. Yi, H. Du, Y. Hu, H. Yan, H.-L. Jiang and J. Lu, ACS Catal., 2015, 5, 2735–2739 CrossRef CAS.
- E. Castillejos-Lopez, G. Agostini, M. Di Michel, A. Iglesias-Juez and B. Bachiller-Baeza, ACS Catal., 2017, 7, 796–811 CrossRef CAS.
- H. Yan, H. Cheng, H. Yi, Y. Lin, T. Yao, C. Wang, J. Li, S. Wei and J. Lu, J. Am. Chem. Soc., 2015, 137, 10484–10487 CrossRef CAS PubMed.
- Y. Wang, Q. Yuan, L. Shi, Z. Chen, X. Mou, M. Li, J. Dai, X. Song, R. Lin and Y. Ding, Mol. Catal., 2023, 546, 113278 CrossRef CAS.
- Z. P. Liu, C. M. Wang and K. N. Fan, Angew. Chem., Int. Ed., 2006, 45, 6865–6868 CrossRef CAS.
- G. C. Bond, Gold Bull., 2016, 49, 53–61 CrossRef CAS.
- Y. Wang, F. Zhang, M. Wang, X. Mou, S. Liu, Z. Jiang, W. Liu, R. Lin and Y. Ding, Angew. Chem., Int. Ed., 2022, 61, e202214166 CrossRef CAS.
- Y. Wang, M. Wang, X. Mou, S. Wang, X. Jiang, Z. Chen, Z. Jiang, R. Lin and Y. Ding, Nanoscale, 2022, 14, 10506–10513 RSC.
- Z. Zhang, Y. Zhu, H. Asakura, B. Zhang, J. Zhang, M. Zhou, Y. Han, T. Tanaka, A. Wang, T. Zhang and N. Yan, Nat. Commun., 2017, 8, 16100 CrossRef CAS PubMed.
- K. Yang and B. Yang, J. Phys. Chem. C, 2018, 122, 10883–10891 CrossRef CAS.
- N. Hu, X.-Y. Li, S.-M. Liu, Z. Wang, X.-K. He, Y.-X. Hou, Y.-X. Wang, Z. Deng, L.-H. Chen and B.-L. Su, Chin. J. Catal., 2020, 41, 1081–1090 CrossRef CAS.
- G. Totarella, R. Beerthuis, N. Masoud, C. Louis, L. Delannoy and P. E. de Jongh, J. Phys. Chem. C, 2021, 125, 366–375 CrossRef CAS PubMed.
- A. Jalal, Y. Zhao and A. Uzun, Ind. Eng. Chem. Res., 2022, 61, 2068–2080 CrossRef CAS.
- G. Totarella, J. W. de Rijk, L. Delannoy and P. E. de Jongh, ChemCatChem, 2022, 14, e202200348 CrossRef CAS.
- A. Jalal and A. Uzun, J. Catal., 2017, 350, 86–96 CrossRef CAS.
- A. Jalal and A. Uzun, Appl. Catal., A, 2018, 562, 321–326 CrossRef CAS.
- S. Dong, Y. Pu, Y. Niu, L. Zhang, Y. Wang and B. Zhang, Acta Phys.-Chim. Sin., 2023, 39, 2301012 Search PubMed.
- Z. Wang, L. Santander de Soto, C. Méthivier, S. Casale, C. Louis and L. Delannoy, Chem. Commun., 2021, 57, 7031–7034 RSC.
- Y. Jiang, G. Liu, S. Wu, X. Zhang, X. Dai, Q. Sheng, Y. Wang and H. Wang, Micropor. Mesopor. Mat., 2019, 288, 109557 CrossRef CAS.
- Z. Chen, L. Guo, Y. Wang, X. Mou, Z. Chen, R. Lin and Y. Ding, Catal. Sci. Technol., 2023, 13, 968–974 RSC.
- R. Hou, K. Chang, J. G. Chen and T. Wang, Top. Catal., 2015, 58, 240–246 CrossRef CAS.
- Q. Yang, R. Qiu, X. Ma, R. Hou and K. Sun, Catal. Sci. Technol., 2020, 10, 3670–3680 RSC.
- C. S. Spanjers, J. T. Held, M. J. Jones, D. D. Stanley, R. S. Sim, M. J. Janik and R. M. Rioux, J. Catal., 2014, 316, 164–173 CrossRef CAS.
- C. Meng, G. Zhao, X.-R. Shi, Q. Nie, Y. Liu and Y. Lu, Appl. Catal., B, 2022, 316, 121699 CrossRef CAS.
- P. Chen, G. Zhao, X.-R. Shi, J. Zhu, J. Ding and Y. Lu, iScience, 2019, 17, 315–324 CrossRef CAS.
- C. Meng, G. Zhao, X. R. Shi, P. Chen, Y. Liu and Y. Lu, Sci. Adv., 2021, 7, 34348903 Search PubMed.
- J. Zhu, G. Zhao, C. Meng, P. Chen, X.-R. Shi and Y. Lu, Chem. Eng. J., 2021, 426, 130857 CrossRef CAS.
- R. Cuscó, T. Yamaguchi, E. Kluth, R. Goldhahn and M. Feneberg, Appl. Phys. Lett., 2022, 121, 062106 CrossRef.
- J. Qiu, T. H. Nguyen, Y. J. Lee, S. Kim, S. Kim, S.-J. Kim, M.-T. Song, W.-J. Huang, X.-B. Chen and I.-S. Yang, Spectrochim. Acta A Mol. Biomol., 2023, 297, 122700 CrossRef CAS.
- C. Yoon, M. X. Yang and G. A. Somorjai, Catal. Lett., 1997, 46, 37–41 CrossRef CAS.
- I. Horiuti and M. Polanyi, Trans. Faraday Soc., 1934, 30, 1164–1172 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Methods of catalyst synthesis, characterization, and testing, plus additional characterization results. See DOI: https://doi.org/10.1039/d3cy01260c |
| ‡ Equal contribution. |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *b,
Li
Yan
b,
Ronghe
Lin
*b,
Li
Yan
b,
Ronghe
Lin