DFT insights into crystal plane effects of molybdenum phosphide (MoP) on the catalytic performance in deoxygenation of palmitic acid†
Received
24th August 2023
, Accepted 23rd November 2023
First published on 25th November 2023
Abstract
Deoxygenation (DX) of bio-oil surrogates via hydrotreating has attracted considerable attention in bio-oil treatment research. This study investigates DX reaction mechanisms of palmitic acid on two distinct molybdenum phosphide (MoP) planes: (001) and (101). Through density functional theory (DFT) calculations, we aim to understand how different crystal planes influence these DX mechanisms. We classified the DX mechanisms on both surfaces into two potential pathways: (i) hydrodeoxygenation (HDO), where oxygen is removed as water, yielding an alkane product with an equal carbon count as the reactant and (ii) decarbonylation (DCO), which eliminates oxygen as CO, producing an alkane product with one fewer carbon atom. Our results reveal that the MoP (101) surface is more active than the MoP (001) surface, making it the optimal surface for DX reactions. The DX reaction mechanism on the MoP (101) surface also shows a preference for the HDO pathway over the DCO pathway (1.43 vs. 2.93 eV). The activation energy (Ea) for the HDO pathway on the MoP (101) surface is lower than that on the MoP (001) surface (1.43 vs. 1.64 eV), due to different hydrogen atom adsorption sites on both surfaces. Kinetic studies show that the reaction rate for both pathways increases with temperature on both surfaces. These findings align with experimental data indicating that the HDO reaction over MoP is more feasible than the DCO pathway. Our research significantly contributes to our understanding of DX reactions over MoP catalysts.
1. Introduction
In the past century, petroleum was a critical fossil fuel source in the transportation and chemical industries. It is expected that the worldwide energy demand will continue to increase because of two main reasons: (1) continuous increase in terms of world population and (2) rapid growth of world economic development.1 The century-long utilization of petroleum has caused serious impacts on the global environment and has contributed to worldwide climate change. This is largely due to the unprecedented quantity of pollutants released as fuel combustion by-products. However, the dwindling petroleum deposits and increasingly serious environmental problems in recent years have boosted the development of renewable clean energy sources from biomass. Through thermochemical methods such as pyrolysis and gasification, biomass can be converted into bio-oils as an alternative fuel due to its pollution-free properties and cost competitiveness.2–6 However, biomass-derived bio-oils cannot be directly used as fuel because of their high oxygen content (35–40 wt%) compared to fossil fuels.7 High oxygen content leads to numerous issues, such as high corrosivity, high viscosity, low volatility, low heating value, and thermal instability.2,8–10 Therefore, the bio-oil upgrading process and/or removal of oxygen components is an essential step required to convert bio-oil into high-performing fuels.
The bio-oil upgrading process can be achieved through a deoxygenation reaction (DX), which is a process of oxygen removal in the form of water (H2O), carbon monoxide (CO), and/or carbon dioxide (CO2) under a high pressure of hydrogen thereby increasing energy density and eliminating its unwanted properties.11,12 The DX mechanisms were classified into three reaction pathways: (I) hydrodeoxygenation (HDO) pathway, (II) decarbonylation (DCO) pathway, and (III) decarboxylation (DCO2) pathway. The DCO and DCO2 pathways are the removal of oxygen in the form of CO and CO2 molecules, respectively, resulting in a higher carbon loss, reducing the potential hydrocarbon product yield. Therefore, the HDO pathway is preferred in the DX process for the conversion of biomass into long-chain hydrocarbons as a clean fuel.4–6,12–16
Generally, heterogeneous catalysts such as transition-metal sulfides (TMSs), noble metals, and metal oxides (MOs) are usually used as catalysts in the DX reaction of bio-oils because of their excellent activities.5,6,17–22 However, they have a high cost, are poisonous and corrosive, and have negative effects on industrial processes, the environment, and human health. The high costs and limited availability are potential concerns for practical applications in the production of cost-effective biomass fuels. In addition, most of these catalysts tend to occur via the DCO pathway, which reduces the carbon chain length.19–22 This will cause a potential reduction in the hydrocarbon product yield via the DCO pathway. Therefore, it is necessary to find catalysts with low cost, high activity, high selectivity, and high resistance to contamination that are possible and suitable for industrial applications.
One of the interesting catalysts was molybdenum-based catalysts such as MoP and Mo2C, which have been widely studied for their low cost and superior catalytic activities in many catalysis processes, such as syngas conversion to hydrocarbons or oxygenated chemicals, biorefineries, the hydrogen evolution reaction (HER) and hydrotreating processes.23–28 Moreover, molybdenum-based catalysts have shown selectivity for alcohol production; Mo2O3, Mo2C and MoP have been explored for syngas conversion, which demonstrates the possibility of MoP and doped MoP compounds for the hydrotreating process and syngas conversion.29 Recently, M. Peroni and colleagues14 investigated the DX reaction of palmitic acid on an unsupported MoP catalyst. They discovered that hexadecane was the most abundant primary product, while pentadecane was the secondary product. Hexadecane and pentadecane are the final products of the HDO and DCO routes, respectively. Moreover, the dominant route for the conversion of palmitic acid on the MoP surface is the HDO pathway. In this experiment, it was found that the conversion of palmitic acid into various products occurs in the temperature range of 200–300 °C. Therefore, it can be concluded that the MoP catalyst has higher catalytic activity for the HDO pathway than for the DCO pathway. Additionally, CO2 was not found on the MoP catalyst, indicating that the DCO2 pathway was unfavorable on this surface. These findings suggested that the MoP surface was a promising catalyst because of its high selectivity and high-performance HDO catalytic activity. However, the underlying DX reaction mechanism of palmitic acid under hydrotreating conditions on the MoP surface has not yet been reported.
Here, we have used DFT calculations to thoroughly study the DX reaction mechanism of palmitic acid under hydrotreating conditions on the MoP surfaces. Nonetheless, because the MoP (101) surface has the maximum intensity on XRD profiles of the MoP surface30–33 and the MoP (001) surface has drawn the most interest in prior theoretical research,23–25,34–38 only these surfaces were considered for the DX reaction mechanism. In the DX reaction mechanism, we use butyric acid as a representative of palmitic acid to obtain results that could be extrapolated to long-chain hydrocarbons, which are suitable representatives of palmitic acid on several surfaces in previous theoretical studies.2,4–6,13,39–46 Based on previous experimental work,14 we focused on the DX reaction mechanism of butyric acid via the HDO and DCO pathways. The potential optimal reaction path and the rate-determining step on the MoP (001) and (101) surfaces were identified using calculated thermodynamic and kinetic data to comprehend the effects of the different planes on the DX reaction mechanism. Furthermore, kinetic studies were investigated to predict product formation rates over various temperatures. Our findings might provide new insights into the DX reaction of butyric acid on the various planes of the MoP surface, which is substantially beneficial for developing catalysts in high-quality biofuel production.
2. Computational details
2.1. DFT calculations
All calculations were performed using the plane-wave based periodic DFT method implemented in the Vienna ab initio simulation package (VASP),47,48 where the ionic cores are described by the projector augmented wave (PAW) method.49,50 The exchange and correlation energies are computed using the Perdew–Burke–Ernzerhof (PBE) functional51 along with the dispersion correction (PBE-D3).52,53 The plane-wave cutoff energy of 400 eV and the Gaussian electron smearing method with σ = 0.10 eV were used. Geometry optimization was performed until the force acting on each atom was <0.05 eV Å−1 with an electronic self-consistent field (SCF) iteration threshold of 10−5 eV. The lattice parameters for the MoP bulk were determined by minimizing the total energy of the unit cell using a conjugated-gradient algorithm to relax the ions and unit cell. The 5 × 5 supercells with two-layer slabs of the MoP (001) and MoP (101) surfaces were repeated periodically on the x–y plane, and a vacuum region of 21 Å was applied along the z-direction to avoid the spurious interactions between repeating slabs. The bottom-most atomic layer was kept fixed during geometry optimization to simulate bulk constraints. The Brillouin zone integrations were performed using the Monkhorst–Pack grids of 9 × 9 × 9 and 3 × 3 × 1 for the bulk and surface calculations, respectively. All transition state structures were located using the climbing image nudged elastic band (CI-NEB) method.54 For each optimized stationary point, we carried out vibrational analysis at the same level of theory to ensure that each transition state has only one imaginary frequency along the reaction coordinate.
2.2. Energy analysis
The adsorption energies (Eads) of butanal or butanol on the MoP (001) and (101) surfaces were estimated using the following equation:| | | Eads = Emolecule/surface − Esurface − Emolecule | (1) |
where Emolecule/surface is the total energy of the surface with the adsorbed species in relaxed geometries, Esurface is the total energy of the surface, and Emolecule is the total energy of the free adsorbate molecule in the gas phase. Therefore, the more negative the Eads value, the stronger the adsorption.
The activation energy barrier (Ea) and the reaction energy (Er) were also calculated using the following equations:
where
EIS,
ETS, and
EFS are the energies of the corresponding initial state (IS), transition state (TS) and final state (FS), respectively.
2.3. Kinetic studies
The reaction rate constants (k) of the HDO and DCO pathways on the MoP (001) and (101) surfaces were estimated using the standard Arrhenius equation:55–60where k is the reaction rate constant; A is the pre-exponential factor; Ea is the activation energy required for the reaction in eV; R is the gas constant (8.314 J K−1 mol−1); T is the absolute temperature in K.
3. Results and discussion
3.1. Structural and electronic properties
According to the previous experimental reports,30,31 several peaks such as (101), (100), (001), (110), (111), and (102) are observable in the XRD profiles of the MoP catalyst. For the purposes of this study, we focused on the MoP (001) and MoP (101) surfaces in our investigation of the DX reaction mechanism. This decision was based on two factors: the MoP (001) surface has been the focus of many previous theoretical studies,23–25,34–38 and the MoP (101) surface exhibits the highest intensity on the XRD profiles. Fig. 1 shows the optimized structures of the MoP (001) and MoP (101) surfaces. As illustrated in Fig. 1a and b, four potential adsorption sites exist on the Mo-terminations of the MoP (001) surface and the Mo/P mixed terminations of the MoP (101) surface. These are labeled as top, bridge, hollow1, and hollow2 sites. Furthermore, we calculated the average Bader charge of Mo atoms (QMo) on the MoP (001) and (101) surfaces to analyze the electronic properties of these surfaces. The results revealed a QMo of +0.46e on the MoP (001) surface, which is more positive than the QMo of +0.35e on the MoP (101) surface. This suggests stronger adsorption oxygenated molecules on the MoP (001) surface. It's crucial to note that numerous factors can influence the overall catalytic performance, including the reaction pathway and surface adsorption characteristics, and kinetics, which will be discussed in greater detail in the subsequent sections.
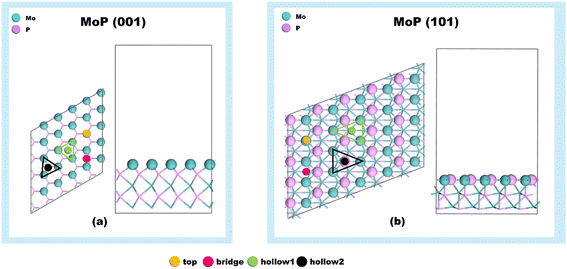 |
| | Fig. 1 The optimized structures of the (a) MoP (001) and (b) MoP (101) surfaces. | |
3.2. Adsorption of reactants, intermediates, and products
To explore the adsorption capabilities of the MoP (001) and (101) surfaces, we examined the adsorption of reactants, intermediates, and products along the DX reaction mechanisms on these surfaces. These included butyric acid, butanal, butanol, butane, and propane molecules. All potential adsorption structures are illustrated in Fig. S1–S4.† For each adsorbate, we primarily showcased the adsorption sites and structures that exhibited the greatest adsorption energy, despite assessing several more adsorption sites. Fig. 2 and 3 depict the most stable structures and corresponding Eads values for butyric acid, butanal, butanol, butane, and propane adsorption on the MoP (001) and (101) surfaces. Regarding butyric acid adsorption on both surfaces, there are two stable adsorption modes: (i) the carbonyl mode and (ii) the hydroxyl mode. In the carbonyl mode of butyric acid adsorption, as displayed in Fig. 2a and 3a, the O atom of the carbonyl group in the butyric molecule interacts with one Mo atom on the MoP (001) surface (Eads = −1.49 eV) and the Mo–O bond distance of 2.14 Å, while it binds to two Mo atoms on the MoP (101) surface (Eads = −1.03 eV) and the Mo1–O and the Mo2–O bond distances are 2.23 and 2.37 Å, respectively. This suggests that butyric acid adsorption in the carbonyl mode is more stable on the MoP (001) surface than on the MoP (101) surface. In contrast, butyric acid adsorption in the hydroxyl mode on the MoP (001) and (101) surfaces, as shown in Fig. 2b and 3b, involves an interaction between the O atom of the hydroxyl group in the butyric acid molecule and one Mo atom on the surfaces. The Eads values are −1.23 and −0.80 eV, respectively. These values are higher than those for the carbonyl mode on both surfaces (−1.49 and −1.03 eV), indicating that butyric acid adsorption in the carbonyl mode is more favorable on both surfaces. Consequently, we only considered butyric adsorption in the carbonyl mode on both surfaces for the DX reaction mechanisms on the MoP (001) and (101) surfaces. For butanal adsorption (Fig. 2c and 3c), the O atom of butanal interacts with the hollow1 (h1) site of the MoP (001) surface (Eads = −2.56 eV) and the bridge (b) site of the MoP (101) surface (Eads = −1.43 eV). This suggests that butanal adsorption is more favorable on the MoP (001) surface than on the MoP (101) surface.
 |
| | Fig. 2 Optimized structures and related Eads values (eV) of butyric acid (a) and (b), butanal (c), butanol (d), butane (e), and propane (f) adsorption on the MoP (001) surface. | |
 |
| | Fig. 3 Optimized structures and related Eads values (eV) of butyric acid (a) and (b), butanal (c), butanol (d), butane (e), and propane (f) adsorption on the MoP (101) surface. | |
As shown in Fig. 2d and 3d, for butanol adsorption, the O atom of the hydroxyl group in the butanol molecule binds to the top site of the MoP (001) and (101) surfaces (Eads = −1.42 and −1.16 eV, respectively). This implies that butanol adsorption on the MoP (001) surface is more stable than that on the MoP (101) surface. For butane and propane adsorption on both surfaces (Fig. 2e and f and 3e and f), the molecules are situated above the surfaces, with the shortest distance of 2.67 Å between the H atom of the alkane and the Mo atom on the surface. This suggests that the Mo–H interaction of alkane products is likely to be physisorption on the MoP (001) and (101) surfaces. The Eads values of butane and propane on the MoP (001) and (101) surfaces are −0.75, −0.41, −0.47, and −0.23 eV, respectively, indicating that the adsorption of butane and propane on the MoP (001) surface is more stable than on the MoP (101) surface. In summary, our results suggest that the adsorption of butyric acid, butanal, butanol, butane, and propane is more favorable on the MoP (001) surface than on the MoP (101) surface, which corresponds with the QMo values on both surfaces. Note that the negative Qiso value represents the electron transfer from the surface to gas molecules.
3.3. DX reaction mechanism
Based on previous experimental work,14 the DX reaction mechanisms of palmitic acid on the MoP catalyst can be classified into 2 pathways: (I) HDO and (II) DCO pathways. To understand these reaction mechanisms on the MoP (001) and (101) surfaces, we studied the HDO and DCO reaction pathways of butyric acid, a representative molecule of palmitic acid for calculations.5,6,13Fig. 4 illustrates the HDO and DCO reaction mechanisms on the MoP (001) and (101) surfaces. In the HDO pathway, butyric acid is converted into butanal via hydrogenolysis (Fig. 4a). Butanal then undergoes hydrogenation to become butanol (Fig. 4b). Subsequently, butanol is transformed into butane through a hydrogenolysis reaction (Fig. 4c). Detailed descriptions of the HDO reaction are provided in section 3.3.2. In the DCO pathway, butyric acid undergoes a hydrogenolysis reaction into butanal (Fig. 4a), which then transforms into propane by decarbonylation in the second step (Fig. 4d), Detailed descriptions of the DCO reaction are provided in section 3.3.1. Additionally, the H2 dissociation on both surfaces was investigated as shown in Fig. S5.† The results reveal that the H2 dissociation readily proceeds on the MoP (001) and (101) surfaces with Ea values of 0.55 and 0.19 eV, respectively, corresponding to previous studies.5,6,13,44
 |
| | Fig. 4 Proposed reaction mechanisms for (a) butyric acid conversion to butanal, (b) butanal conversion to butanol, (c) butanol conversion to butane, and (d) butanal conversion to propane on the MoP (001) and (101) surfaces. | |
3.3.1. Decarbonylation (DCO) pathway.
The DCO pathway involves two critical steps, the conversion of butyric acid to butanal (Fig. 5), which is detailed in section a) conversion of butyric acid to butanal, followed by the transformation of butanal into propane through decarbonylation (Fig. 6), as outlined in section b) conversion of butanal acid to propane. This sequence starts with the hydrogenation of butyric acid, resulting in the formation of butanal, subsequently undergoing a decarbonylation process to propane, releasing CO in the second step as shown in Fig. 5 and 6.
 |
| | Fig. 5 The energy profile of the butyric acid conversion to butanal pathway on the MoP (001) and (101) surfaces. | |
 |
| | Fig. 6 The energy profile of the butanal conversion to propane pathway on the MoP (001) and (101) surfaces. | |
Similarly, the conversion process on the MoP (001) surface was explored, with associated structures illustrated in Fig. S6.† Our findings indicate that the reaction mechanism on the MoP (001) surface mirrors that on the MoP (101) surface. However, the reaction on the MoP (001) surface is more exothermic with a ΔEr of −0.06 eV, as compared to −2.25 eV on the MoP (101) surface. Moreover, on the MoP (001) surface, the rate-determining step is the H2O formation with an Ea of 1.08 eV, lower than the 1.61 eV required on the MoP (101) surface. These results revealed that the butyric acid conversion to butanal on the MoP (001) surface is more thermodynamically and kinetically favorable than that on the MoP (101) surface. Nevertheless, the formation of butanal through hydrogenolysis is feasible on both surfaces.
b) Conversion of butanal acid to propane.
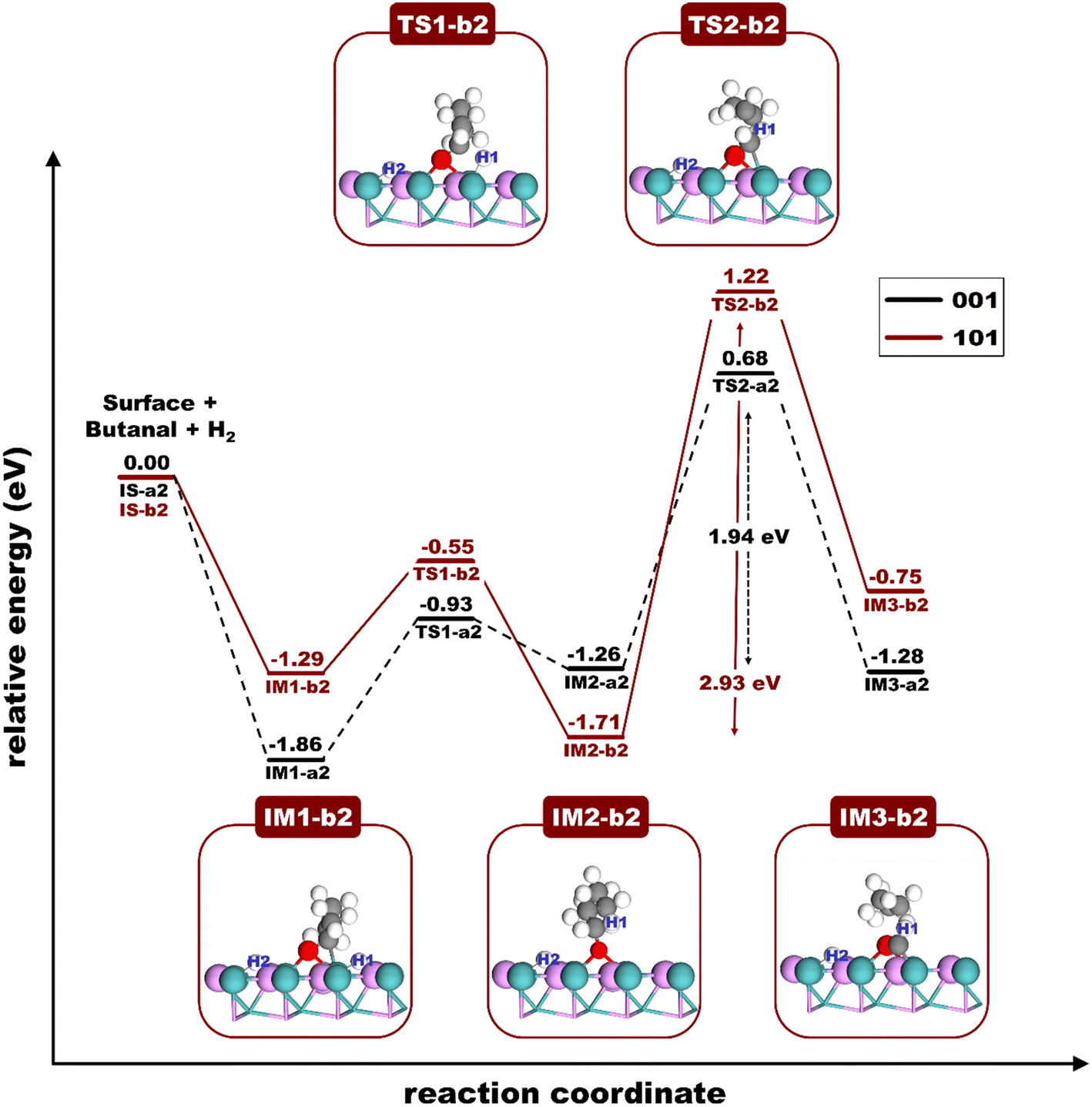
This section focused on the butanal conversion to propane via the hydrogenation reaction, in which butanal reacts with an H atom to form a propane molecule. Fig. 6 presents the energy profiles and accompanying structural shifts along this transformation pathway across two different surfaces. On the MoP (101) surface, the conversion process starts with butanal adsorption at the Mo-bridge site with the Eads of −1.43 eV. This is followed by the adsorption of two H atoms settled onto the two Mo-bridge sites nearby butanal, forming the IM1-b2 state with an Eads of −1.29 eV; it's evident that their adhesion to the surface is firmly established. As the reaction progresses, the H1 atom forms a bond with the α-C atom of the butanal's carbonyl group, transitioning into the IM2-b2 state, requiring an Ea of 0.74 eV. Finally, the H1 atom is transferred from α-C to β-C followed by the C–C bond breaking to produce the propane molecule and HCO intermediate (IM3-b2), requiring a very high Ea of 2.93 eV. In summary, the butanal conversion to propane on the MoP (101) surface is an exothermic reaction with ΔEr = −0.75 eV. The rate-determining step of this reaction on the MoP (101) surface is the IM2-b2 → TS2-b2 step with an Ea of 2.93 eV, which is the transfer hydrogenolysis reaction.
Meanwhile, the conversion of butanal to propane on the MoP (001) surface was studied. Fig. S7† depicts the structures along this reaction pathway. Our observations indicate that the conversion mechanisms for butanal to propane on the MoP (001) surface mirror those on the MoP (101) surface. Notably, the conversion on the MoP (001) surface is exothermic with a ΔEr of −1.28 eV, a value slightly more favorable than its counterpart on the MoP (101) surface (−0.75 eV). Moreover, the rate-determining step of the conversion of butanal to propane on the MoP (001) surface is the transfer hydrogenolysis reaction with an Ea of 1.94 eV, which is lower than that on the MoP (101) surface (2.93 eV). A comparative analysis of the TS2 structures shows that the bond distances between the α-C atom of the butanal molecule and the Mo atom on the MoP (001) and (101) surfaces significantly influence the reaction's energetics. Notably, the bond length between the α-C atom of butanal and the Mo atom on the MoP (001) surface is more compressed at 2.5 Å compared to 3.2 Å on the MoP (101) surface. This suggests a more stable interaction between the butanal's α-C atom and the Mo atom on the MoP (001) surface, as depicted in TS2-b2 in Fig. 6. The enhanced stability of this interaction on the MoP (001) surface facilitates a lower energy barrier for breaking the α-C to β-C bond within the butanal molecule, thus making the formation of propane more energetically favorable. These insights collectively hint that the MoP (001) surface is more thermodynamically and kinetically favorable for butanal conversion to propane than the MoP (101) surface. Overall, the rate-determining steps in the DCO pathway on the MoP (101) and MoP (001) surfaces are the transfer hydrogenolysis process in the production of propane with Ea values of 2.93 and 1.94 eV, respectively. These are notably higher than those seen in the HDO pathway (1.43 and 1.64 eV), which will be elaborated upon in the next section.
3.3.2. Hydrodeoxygenation (HDO) pathway.
In the HDO pathway, the reaction starts with butyric acid undergoing hydrogenolysis to form butanal. Subsequently, a hydrogenation process converts it into butanol in section a) conversion of butanal to butanol, which is then transformed into butane in section b) conversion of butanol to butane, as illustrated in Fig. 7 and 8.
 |
| | Fig. 7 The energy profile of the butanal conversion to butanol pathway on the MoP (001) and (101) surfaces. | |
 |
| | Fig. 8 The energy profile of the butanol conversion to butane pathway on the MoP (001) and (101) surfaces. | |
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond, activating the O atom in butanal for a stronger interaction with the Mo atoms on the surface. Following this, the H2 atom engages with the O atom, resulting in the formation of a butanol molecule (IM3-b3 state) with an Ea of 0.84 eV. In summary, the butanal conversion to butanol on the MoP (101) surface is an exothermic reaction with ΔEr = −1.16 eV. The rate-determining step of this reaction on the MoP (101) surface is the IM2-b3 → TS2-b3 step with an Ea of 0.84 eV, the second hydrogenation reaction. Similarly, the butanal conversion to butanol on the MoP (001) surface was investigated. The structures along this reaction pathway are shown in Fig. S8.† The results demonstrate that the reaction mechanism of the butanal conversion to butanol on the MoP (001) surface is similar to that on the MoP (101) surface. The butanal conversion to butanol on the MoP (001) surface is an exothermic reaction with ΔEr = −1.28 eV, which is slightly lower than that on the MoP (101) surface (−1.16 eV). This suggested that the butanal conversion to butanol on the MoP (001) surface is more thermodynamically favorable than that on the MoP (101) surface. However, the rate-determining step on the MoP (001) surface exhibits higher Ea (0.93 eV) compared to the MoP (101) surface (0.84 eV), revealing that the butanal conversion to butanol on the MoP (101) surface is more kinetically favorable than that on the MoP (001) surface. Nonetheless, butanol production via a hydrogenation reaction is possible on both surfaces.
b) Butanol to butane conversion.
We study the conversion of butanol to butane through a hydrogenation reaction, where butanol interacts with two H atoms, resulting in the formation of a butane molecule accompanied by water removal. Fig. 8 presents the energy profiles and structures characterizing this conversion on both surfaces. The reaction on the MoP (101) surface begins with the adsorption of butanol on the Mo top site with an Eads of −1.16 eV. The IM1-b4 state is then generated by adsorbing two H atoms on the Mo bridge site of the MoP (101) surface. The Eads value of butanol and two hydrogen atoms on the MoP (101) surface in the IM1-b4 state is −3.08 eV, suggesting that their adsorption on this surface is stable. Following this, the H atom from the butanol molecule transfers from the O atom of the carbonyl group to the α-C, causing a C–O bond cleavage, while the H1 atom from the surface engages with the O atom from the carbonyl group, giving rise to butane and an OH group on the surface, requiring an Ea of 1.43 eV. This step is the rate-determining step of the butanol conversion to butane on the MoP (101) surface. Then, the H2 atom moves to a bridge site near the OH group on the surface with an Ea of 0.71 eV. Finally, the H2 atom bonds with the O atom of the OH group on the surface to produce the H2O molecule, requiring an Ea of 1.41 eV. In summary, the butanol conversion to butane on the MoP (101) surface is an exothermic reaction with ΔEr = −2.93 eV. The rate-determining step of this reaction step on the MoP (101) surface is the IM1-b4 → IM2-b4 step with an Ea of 1.43 eV, which is the transfer hydrogenolysis reaction. These findings demonstrate that the rate-determining step of the HDO pathway on the MoP (101) surface is the transfer hydrogenolysis process in the production of butane.
O bond, activating the O atom in butanal for a stronger interaction with the Mo atoms on the surface. Following this, the H2 atom engages with the O atom, resulting in the formation of a butanol molecule (IM3-b3 state) with an Ea of 0.84 eV. In summary, the butanal conversion to butanol on the MoP (101) surface is an exothermic reaction with ΔEr = −1.16 eV. The rate-determining step of this reaction on the MoP (101) surface is the IM2-b3 → TS2-b3 step with an Ea of 0.84 eV, the second hydrogenation reaction. Similarly, the butanal conversion to butanol on the MoP (001) surface was investigated. The structures along this reaction pathway are shown in Fig. S8.† The results demonstrate that the reaction mechanism of the butanal conversion to butanol on the MoP (001) surface is similar to that on the MoP (101) surface. The butanal conversion to butanol on the MoP (001) surface is an exothermic reaction with ΔEr = −1.28 eV, which is slightly lower than that on the MoP (101) surface (−1.16 eV). This suggested that the butanal conversion to butanol on the MoP (001) surface is more thermodynamically favorable than that on the MoP (101) surface. However, the rate-determining step on the MoP (001) surface exhibits higher Ea (0.93 eV) compared to the MoP (101) surface (0.84 eV), revealing that the butanal conversion to butanol on the MoP (101) surface is more kinetically favorable than that on the MoP (001) surface. Nonetheless, butanol production via a hydrogenation reaction is possible on both surfaces.
b) Butanol to butane conversion.
We study the conversion of butanol to butane through a hydrogenation reaction, where butanol interacts with two H atoms, resulting in the formation of a butane molecule accompanied by water removal. Fig. 8 presents the energy profiles and structures characterizing this conversion on both surfaces. The reaction on the MoP (101) surface begins with the adsorption of butanol on the Mo top site with an Eads of −1.16 eV. The IM1-b4 state is then generated by adsorbing two H atoms on the Mo bridge site of the MoP (101) surface. The Eads value of butanol and two hydrogen atoms on the MoP (101) surface in the IM1-b4 state is −3.08 eV, suggesting that their adsorption on this surface is stable. Following this, the H atom from the butanol molecule transfers from the O atom of the carbonyl group to the α-C, causing a C–O bond cleavage, while the H1 atom from the surface engages with the O atom from the carbonyl group, giving rise to butane and an OH group on the surface, requiring an Ea of 1.43 eV. This step is the rate-determining step of the butanol conversion to butane on the MoP (101) surface. Then, the H2 atom moves to a bridge site near the OH group on the surface with an Ea of 0.71 eV. Finally, the H2 atom bonds with the O atom of the OH group on the surface to produce the H2O molecule, requiring an Ea of 1.41 eV. In summary, the butanol conversion to butane on the MoP (101) surface is an exothermic reaction with ΔEr = −2.93 eV. The rate-determining step of this reaction step on the MoP (101) surface is the IM1-b4 → IM2-b4 step with an Ea of 1.43 eV, which is the transfer hydrogenolysis reaction. These findings demonstrate that the rate-determining step of the HDO pathway on the MoP (101) surface is the transfer hydrogenolysis process in the production of butane.
Meanwhile, the conversion of butanol to butane on the MoP (001) surface was examined. Fig. S9† depicts the structures along this reaction pathway. The findings suggest that the reaction process for the conversion of butanol to butane on the MoP (001) surface is analogous to that on the MoP (101) surface. Nonetheless, only the H2O production process on both surfaces is different. The butanol conversion to butane on the MoP (001) surface is an exothermic reaction with ΔEr = −1.86 eV, which is higher than that on the MoP (101) surface (−2.93 eV). Furthermore, the rate-determining step of the conversion of butanol to butane on the MoP (001) surface is the transfer hydrogenolysis reaction with an Ea of 1.64 eV, which is greater than that on the MoP (101) surface (1.43 eV). Based on these results, the conversion of butanol to butane on the MoP (101) surface was shown to be more thermodynamically and kinetically favourable than on the MoP (001) surface. Overall, the transfer hydrogenolysis process in the production of butane is the rate-determining step in the HDO route on both surfaces. This is notably more favorable than the demands in the DCO pathway, which require 2.83 eV and 1.94 eV. Consequently, these computational findings indicate that the HDO pathway is more favored than the DCO pathway on both surfaces, aligning with experimental observations.14
3.4. Comparison of MoP (101) and MoP (001)
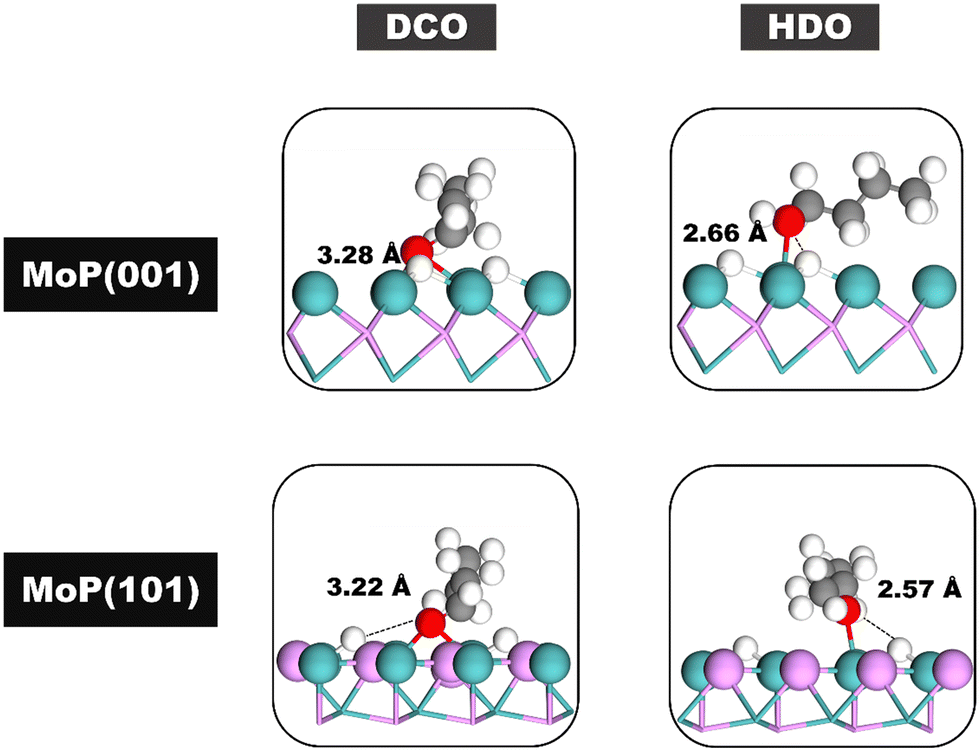
To understand the influence of different planes on the DX reaction, we analyzed the initial structures in the rate-determining steps of both the HDO and DCO pathways on the MoP (101) and MoP (001) surfaces. The most stable structures for the initial state in these rate-determining steps on both surfaces are illustrated in Fig. 9. Our energy profiles suggest that the DX mechanism on both the MoP (101) and MoP (001) surfaces is more favorable via the HDO pathway (with energy requirements of 1.43 eV and 1.64 eV, respectively) than the DCO pathway (2.93 eV and 1.94 eV). This is attributed to the shorter distance between the H atom and the O atom of butanol in the HDO pathway (measuring 2.57 Å and 2.66 Å for MoP (101) and MoP (001) respectively), compared to the corresponding distances in the DCO pathway for butanal, which are 3.22 Å and 3.28 Å. Furthermore, the DX reaction proceeds more efficiently via the HDO pathway on the MoP (101) surface than on the MoP (001) surface (1.43 eV versus 1.64 eV). This efficiency is due to the slightly shorter distance between the H atom and the O atom of butanol on the MoP (101) surface compared to the MoP (001) surface (2.57 Å versus 2.66 Å). These findings indicate that the diverse hydrogen adsorption sites on the two surfaces significantly influence the DX reaction mechanism.
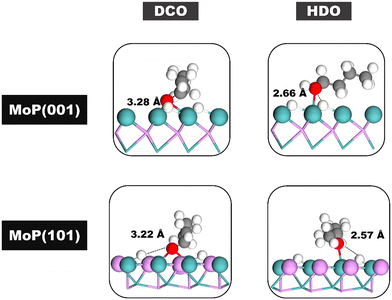 |
| | Fig. 9 The most stable structures of the initial state in the rate-determining steps of the HDO and DCO pathways on the MoP (001) and (101) surfaces. | |
3.5. Kinetic studies
To elucidate the impact of temperature on the DX reaction, we conducted kinetic studies of the HDO and DCO pathways on both the MoP (001) and MoP (101) surfaces using the standard Arrhenius equation. Fig. 10 depicts the relationship between the reaction rate constants (k) and temperatures (T) for the HDO and DCO pathways on the MoP (001) and (101) surfaces. The findings illustrate that the reaction rate constants for both pathways on each surface tend to increase with rising temperatures, implying that the DX reaction is more favorable at higher temperatures. Specifically, the reaction rate constants for the HDO pathway on both surfaces substantially exceed those of the DCO pathways within the temperature range of 273 to 700 K. Furthermore, the reaction rate for the HDO pathway on the MoP (101) surface consistently outpaces that on the surface across the entire temperature spectrum (273 to 700 K). These data underscore that the HDO pathway on the MoP (101) surface emerges as the most optimal reaction pathway within the studied temperature range. Moreover, to validate our findings that the MoP (101) surface serves as the optimal catalyst for the DX reaction, we performed microkinetic modeling to calculate the reaction rate of the DX reaction on the MoP (101) surface under experimental conditions. Fig. 11 displays the reaction rates for the DX reaction on the MoP (101) surface through both the HDO and DCO pathways, highlighting the temperature dependence. Our calculations indicate that the most favorable temperature for the DX reaction via both pathways is approximately 570 K, corresponding to previous experiments.14 Comparison of the two pathways reveals that the DX reaction on the MoP (101) surface is more favorable via the HDO pathway than the DCO pathway within the same temperature range.
 |
| | Fig. 10 The relation between reaction rate constants (k) and temperatures (T) of the HDO and DCO pathways on the MoP (001) and (101) surfaces. | |
 |
| | Fig. 11 Reaction rates for the DX reaction via the HDO and DCO pathways on MoP (101) as a function of temperature. | |
4. Conclusions
In our investigation of the DX reaction mechanism of butyric acid on the MoP (001) and (101) surfaces, DFT calculations elucidated the influence of differing planes on the DX reaction mechanism. We identified two principal pathways for the DX reaction mechanism on these surfaces: the HDO and DCO pathways. The rate-determining steps for these pathways on both surfaces hinge on the transfer hydrogenolysis process in alkane production, characterized by Ea values of 1.64, 1.94, 1.43, and 2.93 eV. The data reveal a preferential occurrence of the DX mechanism via the HDO pathway (with Ea values of 1.64 and 1.43 eV) over the DCO pathway (1.94 and 2.93 eV) on the MoP surfaces. This preference stems from a notably shorter distance between the H atom and the O atom of butanol in the HDO pathway (2.66 and 2.57 Å) compared to the distance in the DCO pathway with butanal (3.28 and 3.22 Å), a finding in alignment with previous experimental research.14 Interestingly, the DX reaction manifests greater efficiency via the HDO pathway on the MoP (101) surface than its counterpart, MoP (001), attributed to a marginally shorter H to O atom distance in butanol (2.57 vs. 2.66 Å). This suggests that hydrogen adsorption site variations on the surfaces critically influence the DX reaction's Ea. The respective Ea differences for the HDO and DCO pathways on the MoP (001) and (101) surfaces stand at 0.30 and 1.40 eV. This points to a strong selectivity of these surfaces toward the HDO pathway, with the MoP (101) surface emerging as particularly promising. From a kinetic perspective, DX mechanisms via both pathways on the surfaces are more favored at elevated temperatures. Nonetheless, the HDO pathway on the MoP (101) surface prevails as the optimal reaction pathway across a temperature range of 273 to 700 K. Given these observations, the MoP (101) surface presents itself as a promising candidate for the DX reaction catalyst. Such insights show significant potential in advancing the development of high-performance catalysts in biofuel production.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research project was supported by the NSRF via the Program Management Unit for Human Resources & Institutional Development, Research and Innovation (B05F640218). The authors are grateful to the department of chemistry at Ubon Ratchathani University and the NSTDA Supercomputer Center (ThaiSC) for providing computing resources. S. K. thanks the Thailand Graduate Institute of Science and Technology (TGIST) for financial support. A. W. would like to acknowledge partial financial support from Chiang Mai University.
References
- Y. Romero, F. Richard and S. Brunet, Appl. Catal., B, 2010, 98, 213–223 CrossRef CAS.
- S. Fu, Z.-M. Wang, L.-J. Liu, T.-H. Liu, D. Li, H.-Q. Yang and C.-W. Hu, Catal. Sci. Technol., 2021, 11, 6425–6437 RSC.
- X. Y. Ooi, W. Gao, H. C. Ong, H. V. Lee, J. C. Juan, W. H. Chen and K. T. Lee, Renewable Sustainable Energy Rev., 2019, 112, 834–852 CrossRef CAS.
- R. Li, H. Yan, Y. Dang, Y. Liu, X. Feng, X. Chen, X. Jin and C. Yang, Mol. Catal., 2019, 479, 110588 CrossRef.
- X.-J. Zhuang, Q.-N. Xia, Y.-Q. Wang, S. C. E. Tsang and X.-Q. Gong, Int. J. Hydrogen Energy, 2016, 41, 18502–18508 CrossRef CAS.
- Y. Shi, Y. Yang, Y.-W. Li and H. Jiao, Catal. Sci. Technol., 2016, 6, 4923–4936 RSC.
- W. Jin, L. Pastor-Pérez, D. Shen, A. Sepúlveda-Escribano, S. Gu and T. Ramirez Reina, ChemCatChem, 2019, 11, 924–960 CrossRef CAS.
- C.-C. Chiu, A. Genest, A. Borgna and N. Rösch, ACS Catal., 2014, 4, 4178–4188 CrossRef CAS.
- J. Lu, S. Behtash, O. Mamun and A. Heyden, ACS Catal., 2015, 5, 2423–2435 CrossRef CAS.
- J. Lu and A. Heyden, J. Catal., 2015, 321, 39–50 CrossRef CAS.
- K. Li, R. Wang and J. Chen, Energy Fuels, 2011, 25, 854–863 CrossRef CAS.
- H. Ren, W. Yu, M. Salciccioli, Y. Chen, Y. Huang, K. Xiong, D. G. Vlachos and J. G. Chen, ChemSusChem, 2013, 6, 798–801 CrossRef CAS PubMed.
- Y. Wongnongwa, S. Jungsuttiwong, M. Pimsuta, P. Khemthong and M. Kunaseth, Chem. Eng. J., 2020, 399, 125586 CrossRef CAS.
- M. Peroni, I. Lee, X. Huang, E. Baráth, O. Y. Gutiérrez and J. A. Lercher, ACS Catal., 2017, 7, 6331–6341 CrossRef CAS.
- A. Saraeian, M. W. Nolte and B. H. Shanks, Renewable Sustainable Energy Rev., 2019, 104, 262–280 CrossRef CAS.
- J. Chen, H. Shi, L. Li and K. Li, Appl. Catal., B, 2014, 144, 870–884 CrossRef CAS.
- M. R. De Brimont, C. Dupont, A. Daudin, C. Geantet and P. Raybaud, J. Catal., 2012, 286, 153–164 CrossRef.
- M. F. Wagenhofer, E. Baráth, O. Y. Gutiérrez and J. A. Lercher, ACS Catal., 2017, 7, 1068–1076 CrossRef CAS.
- J. Fu, X. Lu and P. E. Savage, Energy Environ. Sci., 2010, 3, 311–317 RSC.
- K. Murata, Y. Liu, M. Inaba and I. Takahara, Energy Fuels, 2010, 24, 2404–2409 CrossRef CAS.
- E. Santillan-Jimenez, T. Morgan, J. Lacny, S. Mohapatra and M. Crocker, Fuel, 2013, 103, 1010–1017 CrossRef CAS.
- S. Behtash, J. Lu, M. Faheem and A. Heyden, Green Chem., 2014, 16, 605–616 RSC.
- Y. Zhang, H. Lei, D. Duan, E. Villota, C. Liu and R. Ruan, ACS Appl. Mater. Interfaces, 2018, 10, 20429–20439 CrossRef CAS PubMed.
- J. Ren, C.-F. Huo, X.-D. Wen, Z. Cao, J. Wang, Y.-W. Li and H. Jiao, J. Phys. Chem. B, 2006, 110, 22563–22569 CrossRef CAS PubMed.
- G. Li, H. Zhu, L. Zhao, W. Guo, H. Ma, Y. Yu, X. Lu and Y. Liu, J. Phys. Chem. C, 2016, 120, 23009–23023 CrossRef CAS.
- X. Tian, T. Wang, L. Fan, Y. Wang, H. Lu and Y. Mu, Appl. Surf. Sci., 2018, 427, 357–362 CrossRef CAS.
- X. Tian, J. Min and T. Wang, Appl. Surf. Sci., 2019, 480, 172–176 CrossRef CAS.
- X. Tian, P. Li and T. Wang, Mol. Catal., 2019, 464, 57–62 CrossRef CAS.
- M. Fields, C. Tsai, L. D. Chen, F. Abild-Pedersen, J. K. Nørskov and K. Chan, ACS Catal., 2017, 7, 2528–2534 CrossRef CAS.
- J. Zhu, S. Huang, J. Key, S. Nie, S. Ma and P. K. Shen, Catal. Sci. Technol., 2017, 7, 5974–5981 RSC.
- R. Cheng, Y. Shu, L. Li, M. Zheng, X. Wang, A. Wang and T. Zhang, Appl. Catal., A, 2007, 316, 160–168 CrossRef CAS.
- T. Wang, K. Du, W. Liu, Z. Zhu, Y. Shao and M. Li, J. Mater. Chem. A, 2015, 3, 4368–4373 RSC.
- S. S. Nkabinde, Z. B. Ndala, N. P. Shumbula, T. Kolokoto, O. Nchoe, G. N. Ngubeni, K. P. Mubiayi and N. Moloto, New J. Chem., 2020, 44, 14041–14049 RSC.
- H. Yang, X. Liu, A. She, Z. Zhao, F. Zhou, L. Niu, H. Li, M. Feng and D. Wang, Mater. Chem. Phys., 2022, 281, 125800 CrossRef CAS.
- Y. Li, W. Guo, H. Zhu, L. Zhao, M. Li, S. Li, D. Fu, X. Lu and H. Shan, Langmuir, 2012, 28, 3129–3137 CrossRef CAS PubMed.
- S. F. Zaman, M. Daous and L. Petrov, C. R. Acad. Bulg. Sci., 2014, 67, 125–132 Search PubMed.
- S. F. Zaman, M. Daous and L. Petrov, C. R. Acad. Bulg. Sci., 2013, 66, 1535–1540 CAS.
- S.-R. Li, X.-Q. Lu, H.-Y. Zhu and W.-Y. Guo, Acta Phys.-Chim. Sin., 2016, 32, 465–473 CAS.
- Y. K. Lugo-José, S. Behtash, M. Nicholson, J. R. Monnier, A. Heyden and C. T. Williams, J. Mol. Catal. A: Chem., 2015, 406, 85–93 CrossRef.
- H. Du, Q. Yu, G. Liu, J. Li, J. Zhang, W. Wang, G. Duan, Y. Meng and H. Xie, Fuel, 2022, 317, 123367 CrossRef CAS.
- H. H. Lamb, L. Sremaniak and J. L. Whitten, Surf. Sci., 2013, 607, 130–137 CrossRef CAS.
- X. Li, Y. Zhao, S. Wang, Y. Zhu and G. Yang, Catal. Lett., 2016, 146, 2015–2024 CrossRef CAS.
- S. K. Kim, J. Kim and S.-C. Lee, Catal. Commun., 2017, 99, 61–65 CrossRef CAS.
- R. R. Oliveira and A. B. Rocha, J. Mol. Model., 2019, 25, 1–10 CrossRef CAS PubMed.
- M. McEntee, W. Tang, M. Neurock and J. T. Yates Jr, J. Am. Chem. Soc., 2014, 136, 5116–5120 CrossRef CAS.
- J. Du, Y. Chen, L. Wang and M. Zhang, Catal. Sci. Technol., 2023, 13, 3158–3173 RSC.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed.
- G. Kresse, J. Furthmüller and J. Hafner, Europhys. Lett., 1995, 32, 729 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- L. Chen, Z. Wang, Z. Qin, G. Zhang, H. Yue, B. Liang and D. Luo, J. Mater. Res. Technol., 2021, 12, 1391–1402 CrossRef CAS.
- D. Kilinc, Int. J. Hydrogen Energy, 2022, 11894–11903 CrossRef CAS.
- J. Lv, J. Hong, B. Liang, E. Zhao, K. Zeng, M. Chen, J. Hu and G. Yang, Thermochim. Acta, 2020, 683, 178442 CrossRef CAS.
- Y. Zhang, J. Wang, S. Xue, Y. Wu, Z. Li and L. Chang, Korean J. Chem. Eng., 2016, 33, 862–872 CrossRef CAS.
- X. Zhang, F. Bi, Z. Zhu, Y. Yang, S. Zhao, J. Chen, X. Lv, Y. Wang, J. Xu and N. Liu, Appl. Catal., A, 2021, 297, 120393 CrossRef CAS.
- Q. Jiang, M. Huang, Y. Qian, Y. Miao and Z. Ao, Appl. Surf. Sci., 2021, 566, 150624 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 cd,
Manaschai
Kunaseth
cd,
Manaschai
Kunaseth