
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStability of ZnMOx–SAPO-11 (OXZEO) composite catalysts for syngas conversion to gasoline†
Jingyao
Feng
ab,
Dengyun
Miao
*ab,
Yilun
Ding
ab,
Feng
Jiao
 ab,
Xiulian
Pan
*ab and
Xinhe
Bao
ab
ab,
Xiulian
Pan
*ab and
Xinhe
Bao
ab
aState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China. E-mail: dengyunmiao@dicp.ac.cn; panxl@dicp.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 26th December 2023
Abstract
The catalyst concept of oxide–zeolite (OXZEO) has been demonstrated in an increasing number of studies to be promising to tackle the selectivity challenge in syngas conversion and CO2 hydrogenation. While significant progress has been achieved in product selectivity control, little work has been contributed to the catalyst stability of the OXZEO catalysts, which is essential for practical applications. Herein we studied the stability of ZnMOx–SAPO-11 (M = Al, Cr, Mn) in syngas-to-gasoline, and observed slow deactivation within the initial 100 h reaction on stream. CO conversion decreased by 10–20% in 100 h depending on different oxide components. It was shown that the crystal sizes of ZnMOx oxides increased slightly with time on stream. At the same time, accumulated carbonaceous species were also observed in the micropores of SAPO-11. A number of model reactions by exchanging the used and fresh metal oxides and zeotypes indicate that the changes of the physiochemical properties of both components during the reaction induced the decrease of CO conversion. Zeotypes can be regenerated by calcination in air.
1. Introduction
The catalytic conversion of syngas (a mixture of CO and H2) is vitally important for the utilization of non-petroleum carbon resources such as coal, natural gas, biomass, and even carbon dioxide.1 Fischer–Tropsch synthesis (FTS) is the classic syngas conversion technology, which enables syngas conversion to liquid fuels and is further developed for the synthesis of other hydrocarbons such as light olefins, alkenes and higher alcohols.2–6 However, the product distribution is limited by the Anderson–Schulz–Flory (ASF) distribution due to the surface polymerization mechanism over the classical metal and metal carbide catalyst surface.7 The maximum selectivity to C5–C11 is 48% according to the ASF distribution with mainly straight chain hydrocarbons and hence further upgrading is necessary for gasoline applications.8,9 Recently, a metal oxide–zeolite (OXZEO) catalyst concept has been proposed and shown to be an effective strategy for direct syngas conversion to light olefins, aromatics, gasoline-range hydrocarbons and even oxygenates with the selectivity surpassing the limit by a large margin.10–17 In addition, the OXZEO catalyst concept was also successfully applied for CO2 hydrogenation to different hydrocarbons.18–23 More importantly, the resulting gasoline-range hydrocarbons can be tuned with a high fraction of isoparaffins, which are much more environmentally friendly as octane number contributors for gasoline by fine tuning the structure and properties of OXZEO catalysts.13,14,24 This was not achieved in one step by Fischer–Tropsch synthesis.Great efforts have been made to optimize OXZEO catalysts to increase the fraction of isoparaffins. For instance, a C5–C11 selectivity of 76.7% was achieved over a ZnMnOx–SAPO-11 composite catalyst in syngas conversion, and the isoparaffins to n-paraffins ratio reached up to 15.13 The iso/n-paraffin ratio can be further tuned to as high as 48 with a C5–C11 selectivity of around 80% by modulating the acid site distribution of SAPO-11 zeotype.14 Wang and coworkers also reported an iso/n-paraffin ratio of 13 over ZnAlOx combined with a SAPO-11 zeotype at a C5–C11 selectivity of 77% in syngas conversion.25 More recently, we demonstrated that upon phosphorous modification of MCM-22, it allowed the synthesis of gasoline enriched with isoparaffins, particularly the multi-branched isoparaffins.24 Following a simple hydrogenation in the same reactor, the content of isoparaffins reached 67% among the gasoline-range hydrocarbons and the fraction of multi-branched isoparaffins was as high as 27.5%.24 In addition, by using the combination of In2O3 oxide and hierarchical HZSM-5, CO2 can be directly converted to gasoline-range hydrocarbons with a selectivity of 78.6% and the iso/n-paraffin ratio was 16.8.20 Although the feasibility of this new route for one-step synthesis of gasoline-range isoparaffins is well demonstrated, the stability was rarely studied during longer-term reaction.
For example, CO conversion of ZnAlOx–SAPO-11 composite catalyst decreased from 34.3 to 30.6% during a 150 h time on stream test.14 Zhou et al. observed a decrease of CO conversion from 22 to 12% in a 100 h reaction in syngas conversion to aromatics over a Mo–ZrO2–ZSM-5 composite catalyst.26 The loss of activity was ascribed to the leaching of Mo.26 Wang and co-workers showed that the protonic sites of H-ZSM-5 can be neutralized by mobile indium and zinc species via a solid state ion-exchange mechanism, resulting in a drastic decreased C2+ hydrocarbon formation rate over In2O3/H-ZSM-5 and ZnO/H-ZSM-5 in CO2 hydrogenation.27 Further studies are needed to elucidate the deactivation mechanism in order to optimize the OXZEO catalysts. To do so, three bimetallic oxides ZnMOx (M = Al, Cr, Mn), which have been widely reported in syngas conversion,13,28–32 were coupled with SAPO-11 by physical mixing as catalysts for direct syngas conversion to gasoline. The stability was studied, and the deactivation mechanism and methods for regeneration were explored.
2. Experimental section
2.1 Catalyst preparation
All chemicals were of analytical grade and used without further purification. The oxides ZnAlOx, ZnCrOx, and ZnMnOx were prepared by a co-precipitation method adapted from previously reported methods.10,13,14 Typically, Zn(NO3)2·6H2O and Al(NO3)3·9H2O with a Zn/Al molar ratio of 0.5 were used to synthesize ZnAlOx. Zn(NO3)2·6H2O and Cr(NO3)3·9H2O with a Zn/Cr molar ratio of 2.0 were used as the precursors of ZnCrOx. Zn(NO3)2·6H2O and 50 wt% Mn(NO3)2 aqueous solution with a Zn/Mn molar ratio of 2.0 were used for the synthesis of ZnMnOx. The resulting precipitates were washed with deionized water, dried at 110 °C, and then calcined at 500 °C for 2 h in a muffle furnace in air. Commercial SAPO-11 was purchased from Nankai University Catalyst Co. Ltd. The oxide-SAPO-11 composite catalyst was obtained by physical mixing of oxide particles and SAPO-11 zeotype particles of 0.90–0.45 mm (20–40 mesh) with a mass ratio of 1/1 unless otherwise stated.After reaction for 100 h in syngas conversion, we separated the oxide and zeotype particles, which were named as ZnAlOx-100 h (representing oxide particles separated from used ZnAlOx–SAPO-11 composites), ZEO(Al)-AR (representing zeotype particles separated from used ZnAlOx–SAPO-11 composites), ZnCrOx-100 h, ZEO(Cr)-AR, ZnMnOx-100 h, and ZEO(Mn)-AR, respectively. Regeneration of metal oxides by air calcination was explored at 500 °C for 3 h in a muffle furnace. The resulting samples were denoted as R-ZnAlOx, R-ZnCrOx, and R-ZnMnOx, respectively. The regeneration of zeotypes by air calcination was studied at 600 °C for 4 h in a muffle furnace. The resulting samples were named as R-ZEO(Al), R-ZEO(Cr), and R-ZEO(Mn), respectively. Regeneration was also studied by treating the composite catalysts at 500 °C for 3 h in flowing air (30 mL min−1), and the regenerated catalysts were named as Cat(M)-R. The details of these catalysts are listed in Fig. 1 and Table 1.
 | ||
| Fig. 1 Combination of different composite catalysts for the study of the deactivation and regeneration of ZnMOx-SAPO-11 in syngas conversion to gasoline. Note that the compositions of the catalysts are listed in Table 1. | ||
| OXZEO catalysts | Oxide component | Zeotype component |
|---|---|---|
| a “M” in the brackets stands for Al, Cr or Mn. | ||
| Cat(M)-fresh | Fresh ZnMOx | Fresh SAPO-11 |
| Cat(M)-AR-100 h | ZnMOx-100 h | ZEO(M)-AR |
| Cat(M)-OX-R | R-ZnMOx | Fresh SAPO-11 |
| Cat(M)-OX-AR | ZnMOx-100 h | Fresh SAPO-11 |
| Cat(M)-ZEO-R | Fresh ZnMOx | R-ZEO(M) |
| Cat(M)-ZEO-AR | Fresh ZnMOx | ZEO(M)-AR |
| Cat(M)-R | Regeneration of Cat(M)-AR-100 h | |
2.2 Characterization
X-ray diffraction (XRD) was performed on a PANalytical Empyrean-100 equipped with a Cu Kα radiation source (λ = 1.5418 Å), operated at 40 mA and 40 kV. Temperature-programmed-desorption of NH3 (NH3-TPD) was performed on a Micromeritics AutoChem 2910 instrument equipped with a thermal conductivity detector (TCD) following a reported procedure.14 Before analysis, the sample was pretreated at 500 °C for 0.5 h under argon (Ar). After cooling down to 100 °C under Ar, the sample was exposed to 5% NH3–Ar at 100 °C. The temperature was then increased from 100 to 800 °C at a ramping rate of 10 °C min−1 under helium, during which the signal was recorded. For the used SAPO-11 samples (ZEO(M)-AR), the pretreatment temperature was 300 °C.The textural properties of the samples were characterized by nitrogen adsorption–desorption on a Quantachrome NOVA 4200e. Thermal gravimetric (TG) analysis was carried out on an STA 449 F3 instrument. The accumulated carbonaceous species were characterized by gas chromatography-mass spectrometry (GC-MS) on an Agilent 8890-7250 equipped with an HP-5 capillary column and a flame ionization detector (FID).
2.3 Catalytic performance
Syngas conversion was performed in a fixed-bed stainless steel reactor equipped with a quartz lining. A 420 mg composite catalyst (20–40 mesh) with a mass ratio of oxide/zeolite = 1/1 was loaded in the reactor and heated to the desired temperature under H2. Syngas with CO/H2 = 1 (v/v) (containing 5% Ar as the internal standard for online GC analysis) was then introduced into the reactor. The typical reaction conditions were 360 °C, 4 MPa, and GHSV = 1000 mL g−1 h−1. The methods for product analysis and the calculation of conversion and selectivity were the same as in our previous study.14 The carbon balance in all reactions was 100 ± 5%.3. Results and discussion
3.1 Syngas conversion over ZnMOx–SAPO-11
The selectivity of CO2 in carbon-containing products over all catalysts was very similar (around 50% in Fig. S1†) and therefore the selectivity of different hydrocarbons was reported excluding CO2 to simplify the discussion in this study. Furthermore, hydrocarbons with carbon numbers higher than 12, i.e. C12+ hydrocarbons, were hardly detected in the reaction.As shown in Fig. 2a, the main products are the gasoline-range hydrocarbons (C5–C11) over three OXZEO catalysts. This is consistent with previously reported OXZEO catalysts using SAPO-11 as the zeotype component.13,14,25 It is worth noting that the gasoline selectivity varies with the oxide component. ZnAlOx–SAPO-11 and ZnCrOx–SAPO-11 give a gasoline selectivity of 76% and 78%, respectively, whereas ZnMnOx–SAPO-11 is less selective towards gasoline-range hydrocarbons (69%). The distribution of C5–C11 hydrocarbons in Fig. S2† shows that isoparaffins are the main products in the gasoline-range hydrocarbons over all three ZnMOx–SAPO-11 catalysts. The fraction of isoparaffins is 56.3%, 53.8% and 44.4% over ZnAlOx–SAPO-11, ZnCrOx–SAPO-11 and ZnMnOx–SAPO-11, respectively. In comparison, the fractions of normal paraffins (n-paraffins) are all below 4%. As a result, the ratios of isoparaffins to n-paraffins (iso/n-paraffins) in C5–C11 hydrocarbons are around 14 among three OXZEO catalysts. Except for the selectivity to gasoline-range hydrocarbons, ZnMOx–SAPO-11 exhibits different CO conversion. ZnAlOx–SAPO-11 gives the highest CO conversion of 36.3% among the three catalysts whereas ZnMnOx–SAPO-11 shows a half CO conversion under the same conditions. Fig. 2b shows that after the induction period, CO conversion decreases gradually over the three catalysts with reaction time on stream (TOS). For example, it decreases from 36.3 to 33.9% over ZnAlOx–SAPO-11, from 31.8 to 29.6% over ZnCrOx–SAPO-11, and from 16.7 to 14.0% over ZnMnOx–SAPO-11, respectively. In comparison, the product selectivity remains stable with reaction time on stream, particularly the selectivity of gasoline among hydrocarbons remains almost unchanged after the induction period (Fig. S1†). The above results indicate that the ZnMOx–SAPO-11 composite catalysts deactivate slowly during the reaction.
 | ||
| Fig. 2 Catalytic performance of composite catalysts in syngas conversion to gasoline. (a) CO conversion and hydrocarbon distribution at a time on stream (TOS) of 16 h; (b) CO conversion as a function of reaction time on stream (TOS). Reaction conditions: OX/SAPO-11 = 1/1 (mass ratio), H2/CO = 1/1, 360 °C, 4 MPa, GHSV = 1000 mL g−1 h−1. | ||
3.2 Regeneration of ZnMOx–SAPO-11
To understand the deactivation mechanism of these composite catalysts, we carefully separated the oxide and zeotype particles after reaction. Then the used oxide was mixed with fresh zeotype particles and the used zeotype was mixed with fresh oxide, and their performance was tested under the same conditions again. The details of these catalysts are listed in Fig. 1 and Table 1.As shown in Fig. 3a, CO conversion is 34.4% over Cat(Al)-OX-AR, i.e. used ZnAlOx-100 h and fresh SAPO-11 (Fig. 3a), which is 94.8% of that obtained over the fresh ZnAlOx–SAPO-11. The composite of fresh ZnAlOx and used SAPO-11 (Cat(Al)-ZEO-AR in Fig. 3a) gives 32.2% CO conversion, which is 88.4% of the fresh one. Similar results are observed over ZnCrOx–SAPO-11 and ZnMnOx–SAPO-11 catalysts. This indicates that the decreased CO conversion of the composite catalysts in syngas conversion can be ascribed to both ZnMOx and SAPO-11.
 | ||
| Fig. 3 Catalytic performance of different composite catalysts in syngas conversion. (a) ZnAlOx–SAPO-11; (b) ZnCrOx–SAPO-11; (c) ZnMnOx–SAPO-11. The red numbers in the figure denote the relative CO conversion with regard to the initial value of the fresh catalyst. | ||
Fig. 3 also shows that the regeneration of ZnAlOx–SAPO-11 and ZnMnOx–SAPO-11 composite catalysts (Cat(M)-R) can recover the CO conversion to 94.6% and 95.2% of their initial values, whereas the regeneration of ZnCrOx–SAPO-11 is less effective as the CO conversion is only 88.4% of its initial value. We then regenerated the used oxide and the used zeolite by calcination separately. Fig. 3 shows that the composite by coupling the regenerated oxide and fresh SAPO-11 (Cat(M)-OX-R) gives a lower CO conversion than that of the fresh OXZEO catalyst. In comparison, the composite by mixing the regenerated SAPO-11 and fresh oxides (Cat(M)-ZEO-R) gives practically the same CO conversion and product selectivity as the fresh OXZEO catalyst. The above results indicate that neither the oxides alone nor the composite catalysts can be regenerated simply by air calcination. In contrast, SAPO-11 alone can be regenerated by calcination.
3.3 The deactivation mechanism
The above results indicated that carbon deposition may not be the main reason for the deactivation of metal oxides. To prove this, different oxides were characterized by thermogravimetric analysis (TG) as shown in Fig. S3.† The TG profiles after the reaction for 30 min (ZnMOx-30 min) and for 100 h (ZnMOx-100 h) for the same ZnMOx are quite similar. The differences between the TG profiles of ZnMOx-30 min and ZnMOx-100 h are only around 0.6% for the three oxides, which further indicates that there are almost no carbon deposits on the oxides after reaction for 100 h. Carbon deposition is not the main reason for the deactivation of metal oxides. Su et al. also indicated that deposited carbon species were more likely to be located on the zeotype (SAPO-34) than the oxide (ZnCrOx).28In order to investigate the deactivation mechanism of oxides, we further characterized the fresh oxides and the used ones by XRD and N2 physisorption. Fig. 4 shows the XRD patterns of different oxides. The fresh ZnAlOx oxide exhibits a typical crystalline phase of the ZnAl2O4 spinel and it remains the same after reaction for 30 min (ZnAlOx-30 min) or even for 100 h (ZnAlOx-100 h in Fig. 4a).33 However, analysis of the crystal size, estimated with Scherrer's equation, indicates that it increases from 3.1 to 4.9 nm after reaction for 100 h, as listed in Table 2. For the ZnCrOx oxide, we did the same analysis. XRD shows that the fresh catalyst exhibits ZnO and ZnCr2O4 spinel phases (Fig. 4b).34 After syngas conversion for 30 min or 100 h reaction, the crystal phases do not change (Fig. 4b). Similarly, aggregation is also observed. The average sizes of ZnO and the ZnCr2O4 spinel are 13.7 and 8.8 nm, respectively at the beginning of the reaction. They increase to 14.7 and 10.0 nm after the reaction. By contrast, the ZnMnOx oxide behaves differently from the other two oxides. The fresh ZnMnOx oxide exhibits a typical ZnMnO3 phase. It transforms to a mixture of ZnO and MnO upon reaction in syngas conversion for 30 min, which is maintained during the initial 100 h syngas conversion (Fig. 4c). This is consistent with the previous observation.13 The average crystal sizes of the ZnO and MnO phases at the beginning of the reaction are 17.5 and 8.8 nm, which increased to 22.8 and 10.8 nm, respectively, after the reaction for 100 h, as listed in Table 2.
 | ||
| Fig. 4 XRD patterns of different oxides. (a) ZnAlOx; (b) ZnCrOx; (c) ZnMnOx. The “ZnMOx-30 min” means the oxide subjected to syngas conversion for 30 min; the “ZnMOx-100 h” stands for the oxide after 100 h syngas conversion. | ||
With the increased crystal size after the reaction, the specific surface areas of the oxides were reduced, as evidenced by N2 physisorption (Fig. S4 and Table S1†). This could result in the loss of the active sites for the activation of CO and H2. Therefore, the growing crystal size of the oxides could be one of the reason for an inferior activity of Cat(M)-OX-AR than Cat(M)-fresh (Fig. 3).
The reaction results in Fig. 3 further show that the change of SAPO-11 during the reaction is also one of the important reasons that induce decreased CO conversion. Our previous work shows that there may be carbonaceous species accumulated in the micropores of the zeolite component in the OXZEO catalyst.35 To verify this, thermogravimetric analysis (TG) was used to characterize the used SAPO-11 (ZEO(M)-AR). TG analysis in Fig. 5a indicates that there exist carbonaceous species in SAPO-11 after reaction. The total mass loss was around 4% for ZEO(Al)-AR and ZEO(Cr)-AR, respectively. And it is a little higher over ZEO(Mn)-AR (5.2%). The carbonaceous species were further analyzed by GC-MS. The results are listed in Fig. S5a.† Polyalkyl aromatics, long-chain hydrocarbons, and polycyclic aromatic hydrocarbons are observed on ZEO(Al)-AR, ZEO(Cr)-AR, and ZEO(Mn)-AR. In addition, the composition of the accumulated carbonaceous species is a bit different (Fig. S5b†). The fraction of aromatics and aromatic-ring containing compounds is higher over ZEO(Mn)-AR (44.3%) than that over ZEO(Al)-AR (37.8%) and ZEO(Cr)-AR (38.5%). Compared with chain-like hydrocarbons, aromatics and aromatic-ring containing compounds show higher diffusion resistance in the micropore of SAPO-11. Hence, after the reaction, more carbonaceous species are accumulated in ZEO(Mn)-AR (Fig. 5a).
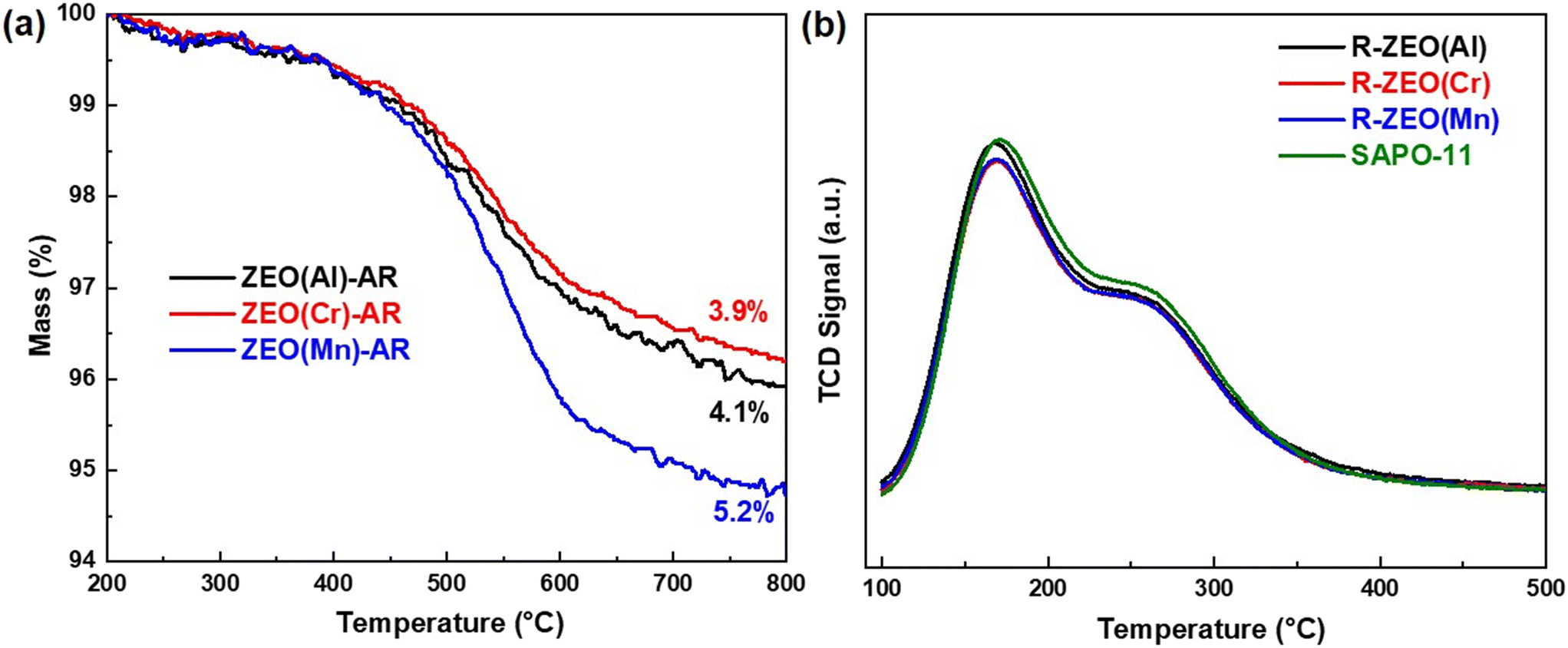 | ||
| Fig. 5 Characterization of SAPO-11 samples. (a) TG profiles of used SAPO-11; (b) NH3-TPD profiles of fresh and regenerated SAPO-11. | ||
To further understand the effect of these carbonaceous species on SAPO-11, we further characterized the acidic properties of the used SAPO-11. As shown in Fig. S6,† the acid site density is reduced apparently after the reaction of syngas conversion to gasoline. The acid sites of SAPO-11 catalyze the conversion of the intermediates generated on ZnMOx oxides to gasoline. Hence, the decreased acid site density may induce the decreased CO conversion. More interestingly, after regeneration by calcination in air at 600 °C for 4 h in a muffle furnace (R-ZEO(M)), the acid sites recover (Fig. 5b). This demonstrates that air calcination can remove the carbonaceous species and restore the acidity properties of SAPO-11. This is corroborated by the comparable catalytic performance of Cat(M)-ZEO-R with Cat(M)-fresh in Fig. 3a.
4. Summary
In summary, ZnMOx–SAPO-11 (M = Al, Cr and Mn with Zn/Al molar ratio of 0.5, Zn/Cr and Zn/Mn of 2) composite catalysts were studied for syngas conversion to gasoline-range hydrocarbons with the focus on their stability in the initial 100 h on stream as well as regeneration. The results show that all three catalysts went through a slow degradation in CO conversion whereas the product selectivity remained almost constant. Characterization and model reactions indicate that it was attributed to the changed physiochemical properties of both components during the reaction. As for the oxides, a very slow aggregation of metal oxide crystals was observed. Meanwhile for SAPO-11, there exist accumulated carbonaceous species after reaction. These carbonaceous species can be removed by calcination in air, leading to a recovered activity. Hence, it is important to design oxides with high stability to improve the stability and regenerability of the catalyst. For example, the oxide can be stabilized by spatial confinement by covering the oxide with porous materials, such as silica,36,37 boron nitride,38,39 carbon materials,40etc. Surface confinement can also be utilized to stabilize the oxide, by increasing the interaction of the active component with the support through the rational design of the structure and optimization of the catalyst compositions. Our findings in this work provide an important guidance for the optimization of OXZEO catalysts, especially the stability, in the conversion of syngas and even in CO2 utilization.Author contributions
Jingyao Feng: conceptualization, methodology, investigation, data curation, writing – original draft; Dengyun Miao: supervision, formal analysis, writing – review & editing; Yilun Ding: formal analysis; Feng Jiao: formal analysis; Xiulian Pan: funding acquisition, supervision, formal analysis, writing – review & editing; Xinhe Bao: discussion.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the Ministry of Science and Technology of China (2018YFA0704503), the National Natural Science Foundation of China (22321002, 22008234, 22272167), the Natural Science Foundation of Liaoning Province (2022-MS-022), the Dalian High-level Talent Innovation Program (2021RQ111, 2022RY02) and the Innovation Research Fund of Dalian Institute of Chemical Physics (DICP I202240).References
- X. Pan, F. Jiao, D. Miao and X. Bao, Chem. Rev., 2021, 121, 6588–6609 CrossRef CAS PubMed.
- L. Zhong, F. Yu, Y. An, Y. Zhao, Y. Sun, Z. Li, T. Lin, Y. Lin, X. Qi, Y. Dai, L. Gu, J. Hu, S. Jin, Q. Shen and H. Wang, Nature, 2016, 538, 84–87 CrossRef CAS PubMed.
- P. Zhai, C. Xu, R. Gao, X. Liu, M. Li, W. Li, X. Fu, C. Jia, J. Xie, M. Zhao, X. Wang, Y. W. Li, Q. Zhang, X. D. Wen and D. Ma, Angew. Chem., Int. Ed., 2016, 55, 9902–9907 CrossRef CAS PubMed.
- J. Li, Y. He, L. Tan, P. Zhang, X. Peng, A. Oruganti, G. Yang, H. Abe, Y. Wang and N. Tsubaki, Nat. Catal., 2018, 1, 787–793 CrossRef CAS.
- Y. Chen, Y. Ni, Y. Liu, H. Liu, X. Ma, S. Liu, W. Zhu and Z. Liu, Catal. Sci. Technol., 2018, 8, 5943–5954 RSC.
- M. Ao, G. H. Pham, J. Sunarso, M. O. Tade and S. Liu, ACS Catal., 2018, 8, 7025–7050 CrossRef CAS.
- A. R. B. Friedel R A, J. Am. Chem. Soc., 1950, 72, 1212–1215 CrossRef.
- P. J. Flory, J. Am. Chem. Soc., 1936, 58, 1877–1885 CrossRef CAS.
- R. B. Anderson, J. Catal., 1978, 55, 114–115 CrossRef CAS.
- F. Jiao, J. Li, X. Pan, J. Xiao, H. Li, H. Ma, M. Wei, Y. Pan, Z. Zhou, M. Li, S. Miao, J. Li, Y. Zhu, D. Xiao, T. He, J. Yang, F. Qi, Q. Fu and X. Bao, Science, 2016, 351, 1065–1068 CrossRef CAS PubMed.
- K. Cheng, W. Zhou, J. Kang, S. He, S. Shi, Q. Zhang, Y. Pan, W. Wen and Y. Wang, Chem, 2017, 3, 334–347 CAS.
- J. Yang, X. Pan, F. Jiao, J. Li and X. Bao, Chem. Commun., 2017, 53, 11146–11149 RSC.
- N. Li, F. Jiao, X. Pan, Y. Chen, J. Feng, G. Li and X. Bao, Angew. Chem., Int. Ed., 2019, 58, 7400–7404 CrossRef CAS PubMed.
- J. Feng, D. Miao, Y. Ding, F. Jiao, X. Pan and X. Bao, ACS Energy Lett., 2022, 7, 1462–1468 CrossRef CAS.
- W. Zhou, J. Kang, K. Cheng, S. He, J. Shi, C. Zhou, Q. Zhang, J. Chen, L. Peng, M. Chen and Y. Wang, Angew. Chem., Int. Ed., 2018, 57, 12012–12016 CrossRef CAS PubMed.
- J. Kang, S. He, W. Zhou, Z. Shen, Y. Li, M. Chen, Q. Zhang and Y. Wang, Nat. Commun., 2020, 11, 827 CrossRef CAS PubMed.
- Y. Luo, S. Wang, S. Guo, K. Yuan, H. Wang, M. Dong, Z. Qin, W. Fan and J. Wang, Catal. Sci. Technol., 2021, 11, 338–348 RSC.
- S. Wang, L. Zhang, W. Zhang, P. Wang, Z. Qin, W. Yan, M. Dong, J. Li, J. Wang, L. He, U. Olsbye and W. Fan, Chem, 2020, 6, 3344–3363 CAS.
- J. Wei, R. Yao, Q. Ge, D. Xu, C. Fang, J. Zhang, H. Xu and J. Sun, Appl. Catal., B, 2021, 283, 119648 CrossRef CAS.
- P. Gao, S. Li, X. Bu, S. Dang, Z. Liu, H. Wang, L. Zhong, M. Qiu, C. Yang, J. Cai, W. Wei and Y. Sun, Nat. Chem., 2017, 9, 1019–1024 CrossRef CAS PubMed.
- Y. Ni, Z. Chen, Y. Fu, Y. Liu, W. Zhu and Z. Liu, Nat. Commun., 2018, 9, 3457 CrossRef PubMed.
- P. Gao, S. Dang, S. Li, X. Bu, Z. Liu, M. Qiu, C. Yang, H. Wang, L. Zhong, Y. Han, Q. Liu, W. Wei and Y. Sun, ACS Catal., 2018, 8, 571–578 CrossRef CAS.
- S. Wang, L. Zhang, P. Wang, X. Liu, Y. Chen, Z. Qin, M. Dong, J. Wang, L. He, U. Olsbye and W. Fan, Chem, 2022, 8, 1376–1394 CAS.
- Y. Ding, D. Miao, J. Feng, B. Bai, X. Pan and X. Bao, Appl. Catal., B, 2022, 316, 121628 CrossRef CAS.
- M. Wang, Y. Han, S. Liu, Z. Liu, D. An, Z. Zhang, K. Cheng, Q. Zhang and Y. Wang, Chin. J. Catal., 2021, 42, 2197–2205 CrossRef CAS.
- W. Zhou, S. Shi, Y. Wang, L. Zhang, Y. Wang, G. Zhang, X. Min, K. Cheng, Q. Zhang, J. Kang and Y. Wang, ChemCatChem, 2019, 11, 1681–1688 CrossRef CAS.
- Y. Wang, G. Wang, L. I. van der Wal, K. Cheng, Q. Zhang, K. P. de Jong and Y. Wang, Angew. Chem., Int. Ed., 2021, 60, 17735–17743 CrossRef CAS PubMed.
- J. Su, H. Zhou, S. Liu, C. Wang, W. Jiao, Y. Wang, C. Liu, Y. Ye, L. Zhang, Y. Zhao, H. Liu, D. Wang, W. Yang, Z. Xie and M. He, Nat. Commun., 2019, 10, 1297 CrossRef PubMed.
- M. T. Arslan, B. A. Qureshi, S. Z. A. Gilani, D. Cai, Y. Ma, M. Usman, X. Chen, Y. Wang and F. Wei, ACS Catal., 2019, 9, 2203–2212 CrossRef CAS.
- X. Liu, M. Wang, H. Yin, J. Hu, K. Cheng, J. Kang, Q. Zhang and Y. Wang, ACS Catal., 2020, 10, 8303–8314 CrossRef CAS.
- J. Su, L. Zhang, H. Zhou, Y. Ye, X. Zheng, C. Liu, S. Liu, W. Jiao, X. Liu, C. Wang, Y. Wang and Z. Xie, ACS Catal., 2023, 13, 2472–2481 CrossRef CAS.
- Y. Fu, Y. Ni, Z. Chen, W. Zhu and Z. Liu, J. Energy Chem., 2022, 66, 597–602 CrossRef CAS.
- W. Walerczyk, M. Zawadzki and J. Okal, Appl. Surf. Sci., 2011, 257, 2394–2400 CrossRef CAS.
- Y. Li, J. Hu, J. Xu, Y. Zheng, M. Chen, H. Wan, Q. Fu, F. Yang and X. Bao, Appl. Surf. Sci., 2019, 494, 353–360 CrossRef CAS.
- H. Wang, F. Jiao, Y. Ding, W. Liu, Z. Xu, X. Pan and X. Bao, Natl. Sci. Rev., 2022, 9, nwac146 CrossRef CAS PubMed.
- Q. Cheng, Y. Tian, S. Lyu, N. Zhao, K. Ma, T. Ding, Z. Jiang, L. Wang, J. Zhang, L. Zheng, F. Gao, L. Dong, N. Tsubaki and X. Li, Nat. Commun., 2018, 9, 3250 CrossRef PubMed.
- Y. Xu, H. Liang, R. Li, Z. Zhang, C. Qin, D. Xu, H. Fan, B. Hou, J. Wang, X.-K. Gu and M. Ding, Angew. Chem., Int. Ed., 2023, 62, e202306786 CrossRef CAS PubMed.
- J. Deng, K. Bu, Y. Shen, X. Zhang, J. Zhang, K. Faungnawakij and D. Zhang, Appl. Catal., B, 2022, 302, 120859 CrossRef CAS.
- J. Wu, L. Wang, B. Lv and J. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 14319–14327 CrossRef CAS PubMed.
- J. Hong, B. Wang, G. Xiao, N. Wang, Y. Zhang, A. Y. Khodakov and J. Li, ACS Catal., 2020, 10, 5554–5566 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cy01166f |
| This journal is © The Royal Society of Chemistry 2024 |