
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Promoted propane dehydrogenation over Co confined within core–shell silicalite-1 zeolite crystals†
Shohei
Kubota
a,
Tomoka
Sumi
a,
Haruna
Kitamura
a,
Koji
Miyake
 *ab,
Yoshiaki
Uchida
a and
Norikazu
Nishiyama
ab
*ab,
Yoshiaki
Uchida
a and
Norikazu
Nishiyama
ab
aDivision of Chemical Engineering, Graduate School of Engineering Science, Osaka University, 1-3 Machikaneyama, Toyonaka, Osaka 560-8531, Japan. E-mail: kojimiyake@cheng.es.osaka-u.ac.jp
bInnovative Catalysis Science Division, Institute for Open and Transdisciplinary Research Initiatives (ICS-OTRI), Osaka University, Suita, Osaka 565-0871, Japan
First published on 9th January 2024
Abstract
Catalytic propane dehydrogenation (PDH) is an attractive process that can meet the growing demand for propylene. Among the extensively studied PDH catalysts, Co-based catalysts are considered especially promising because of their high activity and low cost. However, Co-based catalysts are often degraded by the aggregation of Co species and the formation of carbon nanotubes. To overcome these drawbacks, we prepared Co-confined core–shell silicalite-1 zeolite crystals by coating Co-loaded silicalite-1 with silicalite-1 layers. We confirmed that the silicalite-1 shell layers suppressed the formation of aggregated Co nanoparticles and carbon nanotubes during the PDH reaction, leading to improved catalytic performance. The material design described in this study contributes to progress in the field of materials chemistry related to energy and sustainability.
1. Introduction
Propylene, a lower olefin, is essential for producing polypropylene, acetone, acrolein, propylene oxide, and other industrial products.1–3 Conventional propylene manufacturing methods include naphtha and light diesel steam cracking and fluid catalytic cracking. Given the rapid consumption of fossil-based energy, concerns that the traditional methods of propylene production cannot meet the growing demand for propylene in the petrochemical industry have been raised. Therefore, developing alternative propylene production processes independent of fossil fuels is of great importance. Several potential processes, including the Fischer–Tropsch process, the methanol–olefin process, and the propane dehydrogenation (PDH) reaction, have been proposed.3–5PDH is attracting attention as a highly promising alternative for future propylene production needs. PDH presents multiple advantages and is the best candidate for replacing conventional processes. First, light alkanes exist in abundance owing to the development of shale gas extraction methods. Thus, propane is inexpensive to use as a reactant. Second, the PDH reaction is simple and produces few side products. In addition, the hydrogen generated as a by-product along with propylene is a valuable resource that can be further exploited for other purposes. Aromatic compounds and coke are produced through sequential reactions.6,7 Considering this reaction mechanism, the development of catalysts that can enhance the selectivity of dehydrogenation and suppress sequential reactions is required. Several metals, including Pt, Ga and Cr, have been studied as active species in PDH.8–15 Pt is the most commonly used PDH catalyst; however, it is a precious metal and expensive to use. Development of alternative abundant and inexpensive catalysts may achieve a sustainable and low-cost PDH process.
A shift from using expensive precious metal-based catalysts to the development of inexpensive and non-toxic transition metal-based catalysts, such as those bearing Zn, Fe, and Co, has recently been observed.9 In particular, research indicates that Co-based catalysts are effective in the PDH reaction.2,16–18 However, the catalytic activity of these transition-metal-based catalysts requires further improvement. The low activity of these metal-based catalysts in the PDH reaction often originates from a decrease in the amount of active transition metal species following their sintering or reduction during the PDH reaction. In addition, sintered or reduced transition metal species promote undesirable side and sequential reactions, such as cracking and carbon deposition, and forming carbon nanotubes.19 Thus, an effective strategy is required to improve the stability of active transition metal species during the PDH reaction.
Inspired by our previous and related works on core–shell structured zeolite catalysts,20–31 we confined Co species within core–shell silicalite-1 crystals by coating Co-loaded silicalite-1 with a silicalite-1 layer to overcome the above drawbacks of transition-metal-based catalysts as shown in Scheme 1. We then investigated the effects of the core–shell structure on propane conversion, propylene yield, and propane selectivity in the PDH reaction. Co species are aggregated and carbon nanotubes (CNT) are formed during PDH reaction in the case of conventional Co-loaded silicalite-1. Due to these, conventional Co-loaded silicalite-1 shows poor catalytic activity for PDH reaction. We expect to obtain Co species that remain stable during the PDH reaction owing to the presence of silicalite-1 layers which can geometrically confine Co species, leading to preventing the aggregation of Co species and the formation of CNT. Silicalite-1 is a 10-membered-ring MFI-type zeolite that allows the sufficient diffusion of propane. Because the support does not contain Al which induces acidity, sequential reactions involving acidic sites can be suppressed. Co-loaded silicalite-1 shows poor catalytic activity for PDH reaction. We expect to obtain Co species that remain stable during the PDH reaction owing to the presence of silicalite-1 layers which can geometrically confine Co species, leading to preventing the aggregation of Co species and the formation of CNT. Silicalite-1 is a 10-membered-ring MFI-type zeolite that allows the sufficient diffusion of propane. Because the support does not contain Al which induces acidity, sequential reactions involving acidic sites can be suppressed.
 | ||
| Scheme 1 Schematic representation of core–shell method. | ||
2. Experimental
2.1 Materials
Tetraethylorthosilicate (TEOS, Wako Pure Chemical Co.), fumed silica (Aldrich), 10 wt% tetrapropylammonium hydroxide (TPAOH, Wako Pure Chemical Co.), Co(NO3)2·6H2O (Wako Pure Chemical Co.), ethanol (EtOH, Wako Pure Chemical Co.) and deionized water were used without further purification to prepare the catalysts.2.2 Catalyst preparation
Silicalite-1 was prepared using the hydrothermal synthesis method described in our previous study.32 The precursor solution was stirred at room temperature for 24 h. The molar ratio of the precursor solution was 1 SiO2 (TEOS)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.25 TPAOH:60 H2O. The precursor solution was then autoclaved at 453 K for 24 h. The solid product was collected by centrifugation and washed several times with deionized water. The wet powder was dried at 363 K and calcined in air at 823 K for 5 h. As a comparison, we performed a one-pot synthesis of Co-contained silicalite-1 by just adding Co(NO3)2·6H2O in the precursor. The sample was donated as “one-pot S–Co”.
:0.25 TPAOH:60 H2O. The precursor solution was then autoclaved at 453 K for 24 h. The solid product was collected by centrifugation and washed several times with deionized water. The wet powder was dried at 363 K and calcined in air at 823 K for 5 h. As a comparison, we performed a one-pot synthesis of Co-contained silicalite-1 by just adding Co(NO3)2·6H2O in the precursor. The sample was donated as “one-pot S–Co”.
Next, Co was loaded onto the prepared silicalite-1 using the impregnation method described in our previous works.33,34 Briefly, silicalite-1 was immersed in an aqueous solution of Co(NO3)2·6H2O, after which the mixture was dried at 363 K. The dried powder was calcined in air at 823 K for 5 h. We prepared Co-loaded silicalite-1 with different amounts of Co (1–20 wt%) using the impregnation method described above. The Co-loaded silicalite-1 samples were denoted as “S–Co (x)”, where x indicates the mass ratio of Co in the starting materials.
The Co-loaded silicalite-1 and one-pot synthesized Co-containing silicalite-1 were coated with silicalite-1 as described in our previous studies.32,35 A precursor solution containing a molar ratio of 1 SiO2 (fumed silica):0.08 TPAOH:16 EtOH:240 H2O was stirred at room temperature for 1 h. Co-loaded silicalite-1 was added to the precursor, and the mixture was autoclaved with rotation at 453 K for 24 h. The solid product was collected, washed with deionized water, and dried at 363 K. Finally, calcination was performed in air at 823 K for 5 h. The obtained core–shell catalysts were denoted as “CS–S–Co (x),” where x indicates the mass ratio of Co.
2.3 Characterization
The crystal structures of the samples were investigated via X-ray diffraction (XRD) measurements using Cu-Kα radiation on a PANalytical X'Pert PRO MPD diffractometer. Chemical compositions were determined using energy-dispersive X-ray (EDX) analysis. The morphologies and particle sizes of the prepared catalysts were observed using a Hitachi H800 transmission electron microscope (TEM). N2 adsorption isotherms were measured at 77 K using a BELSORP MINI X instrument (MicrotracBEL).2.4 Assessment of catalytic performance
PDH reactions over the zeolite catalysts were carried out in a fixed-bed reactor at atmospheric pressure, as described in referring to our previous works.36,37 The catalyst (0.05 g) was loaded into a quartz tube (i.d., 4 mm), and the temperature was increased to 873 K under a He flow. A feed gas of 10 vol% propane and He (balance) was supplied at a total flow rate of 10 cm3 min−1. The product stream was analyzed online using a Shimadzu GC-2025 gas chromatograph equipped with a flame-ionization detector. The conversion of propane and yield of propylene were calculated using the following equations. In addition, assuming an ideal dehydrogenation reaction in which only propylene and hydrogen are produced from propane, the volume rare of change was calculated based on the composition of the feed gas: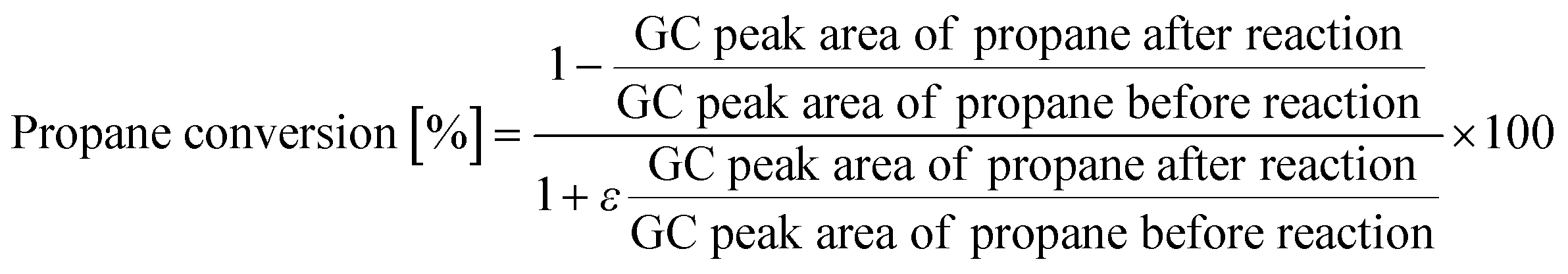
| ε = 0.1 (Volume rate of change) |
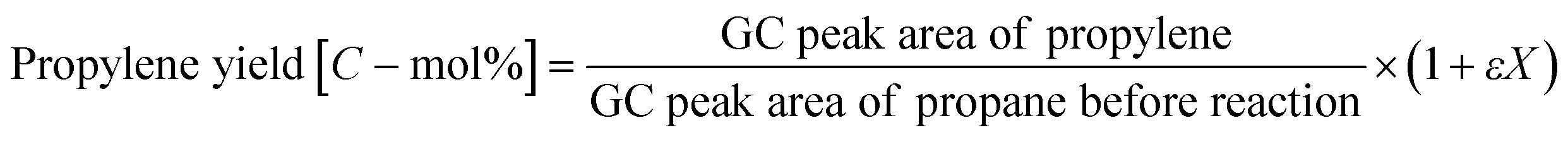
| X = (Propane conversion)/100 |
The amount of carbon deposition was analyzed by Thermogravimetric Analysis (TGA) under an air atmosphere using DTG-60 (Shimadzu). The weight loss from 523 to 1073 K was assigned to the combustion of carbon. We did not confirm the significant mass gain derived from the oxidation of Co species. The regeneration for the best sample was performed under an air atmosphere at 873 K for 2 h. The heating rate was 5 K min−1.
3. Results and discussion
The XRD patterns of silicalite-1, S–Co (1), and CS–S–Co (1) catalysts are shown in Fig. 1. Silicalite-1 displayed specific peaks attributable to MFI-type zeolite, indicating that the prepared sample possessed a highly pure MFI-type crystal structure. These peaks were maintained after Co impregnation, thus suggesting that the crystal structure did not undergo significant decomposition. Moreover, clear peaks derived from Co species were not observed, likely because of the high dispersion of Co species on the silicalite-1 support. We then investigated the XRD patterns of the Co-loaded silicalite-1 samples. Silicalite-1 loaded with low amounts of Co did not show peaks that could be derived from Co species, whereas those loaded with high amounts of Co exhibited peaks attributable to Co3O4, as shown in Fig. S1(a).†38 These results indicate that the aggregation of Co species occurs with increasing amounts of Co, as often observed in previous studies.39 Next, we focused on the silicalite-1 coating. All silicalite-1 coated samples showed specific peaks attributable to MFI-type zeolite, as shown in Fig. 1 and S1(b),† and the peak intensity increased after silicalite-1 coating. These findings imply that highly crystalline silicalite-1 was formed after silicalite-1 coating. | ||
| Fig. 1 XRD patterns of silicalite-1, S–Co (1), and CS–S–Co (1). | ||
We analyzed the compositions of the catalysts by EDX analysis. The Si/Co ratio decreased with an increasing amount of Co in the aqueous solution during impregnation, as shown in Table S1.† By contrast, the Si/Co ratio increased after silicalite-1 coating, thus implying the successful formation of an additional silicalite-1 layer. Next, we performed SEM to investigate the morphological changes in the catalysts during their modification. As shown in Fig. 2, no significant changes were observed in the SEM image of S–Co (1). Similar results were obtained when the amount of Co was varied, as shown in Fig. S2.† These results indicate that microscale changes in morphology do not occur and that the silicalite-1 structure is maintained after Co impregnation, in agreement with the results of the XRD measurements. This is in accord with the results of XRD measurements. After silicalite-1 coating, the particle size increased, as shown in Fig. 2 and S2.† The results of XRD measurements and EDX analysis indicated that the silicalite-1 shell layers grew epitaxially.
 | ||
| Fig. 2 SEM images of (a) silicalite-1, (b) S–Co (1), and (c) CS–S–Co (1). | ||
We performed TEM observations to investigate the morphology of the catalysts at the nanoscale level. Comparisons of the TEM images of pristine silicalite-1 and Co-loaded specimens (Fig. 3 and S3†) revealed the presence of Co nanoparticles on the silicalite-1 support after Co impregnation. The Co nanoparticles aggregated, and their sizes increased with increasing Co content, which agrees with the XRD results. TEM images of the silicalite-1 coated samples confirmed that the silicalite-1 shell layers grew epitaxially and that Co nanoparticles were encapsulated in the silicalite-1 shell layers, as shown in Fig. 3 and S3.† Porosity is important for the application of these samples as catalysts in PDH reactions. Thus, we measured the N2 adsorption isotherms of all samples (Fig. S4†). The adsorption volumes of the specimens decreased with increasing amounts of Co during impregnation. These decreases were due to the mass gain of the nonporous Co nanoparticles. After silicalite-1 coating, the adsorption volumes of the specimens increased, which could be attributed to the increase in the mass ratio of porous silicalite-1. No significant decrease was observed after silicalite-1 coating, thus confirming that the silicalite-1 shell layers grew epitaxially without blocking the zeolitic micropores on the interfaces.
 | ||
| Fig. 3 TEM images of (a) silicalite-1, (b) S–Co (1), and (c) CS–S–Co (1). | ||
We measured UV-vis spectra to investigate the chemical state of Co species. The following two peaks are generally known to suggest the introduction of Co species for Co-loaded silicalite-1. The adsorption bands at 330–480 nm and approximately 720 nm are ascribed to CoOx oligomer and Co3O4, respectively.40–43 As shown in Fig. 4, S–Co (1) and CS–S–Co (1) were dominated by the peak which are assigned to CoOx oligomer, and showed the modest peak which are assigned to Co3O4. For the catalysts of other loading amounts, as shown in Fig. S5,† only CoOx oligomer was detected at low loading amount, and Co3O4 was the majority at high loading amount. The formation of bulk Co3O4 at a higher loading amount is reasonable because of the consistence with the XRD result and TEM images.
 | ||
| Fig. 4 UV-vis spectra of silicalite-1, S–Co (1) and CS–S–Co (1). | ||
We conducted the PDH reaction over S–Co (1) and CS–S–Co (1). CS–S–Co (1) exhibited much higher propane conversion and propylene yields on all time-on-stream conditions investigated as shown in Fig. 5(a) and (b). These improvements in propane conversion and propylene yields were observed for all samples after silicalite-1 coating, regardless of the amount of Co impregnation (Fig. S6†). The deactivation rates were calculated referring to a previous report.44 The deactivation rates decreased after silicalite-1 coating for the almost series (Table S2†). Among the samples investigated, CS–S–Co (1) showed the best performance in the PDH reaction despite its low Co content. We prepared physically mixed S–Co (1) and silicalite-1 to verify the effect of the location of silicalite-1 on the PDH reaction. The amount of pristine silicalite-1 in the physically mixed catalyst was identical to the mass gain of SiO2 in CS–S–Co (1). The physically mixed catalyst showed slightly inferior catalytic performance in the PDH reaction compared with S–Co (1), as shown in Fig. 5(a) and (c), likely because of the mass gain of pristine silicalite-1. In other words, pristine silicalite-1 is inert and does not contribute to the reaction. The physically mixed catalyst also showed much poorer catalytic performance in the PDH reaction than CS–S–Co (1), as shown in Fig. 5(b) and (c), thus implying that the silicalite-1 shell layers on S–Co (1) play an important role in improving propane conversion and propylene yields.
 | ||
| Fig. 5 Results of PDH reaction over (a) S–Co (1), (b) CS–S–Co (1), and (c) physically-mixed catalyst with S–Co (1) and silicalite-1. | ||
We obtained TEM images of the spent catalysts to obtain further insights into the improvement in propane conversion and propylene yields in the PDH reaction. Aggregated Co nanoparticles and carbon nanotubes are observed in the TEM image of spent S–Co (1), as shown in Fig. 6(a). This phenomenon is often observed in hydrocarbon transformation reactions involving transition metals.45–47 The aggregation of Co species and the formation of carbon nanotubes lead to the poor catalytic performance of S–Co (1) in the PDH reaction. Meanwhile, there are no aggregated Co nanoparticles and carbon nanotubes in the TEM image of spent CS–S–Co (1) as shown in Fig. 6(b). For all other samples with different amounts of Co, the presence of silicalite-1 layers suppressed the formation of aggregated Co nanoparticles and carbon nanotubes (Fig. S7†), although some aggregated Co nanoparticles and carbon nanotubes were present in spent CS–S–Co (x) samples with x = 3, 5, 10, and 20. It was confirmed that the weight loss of carbon nanotubes in the period from 573 to 1073 K by thermogravimetric analysis as shown in Fig. S8.†48 The mass loss of CS–S–Co (1) was much smaller than that of S–Co (1). This result indicates that the improvements endowed by the silicalite-1 coating originate from the suppression of the formation of aggregated Co nanoparticles and carbon nanotubes during the PDH reaction. Therefore, CS–S–Co (1), which hardly showed aggregated Co nanoparticles or carbon nanotubes after the PDH reaction, exhibited the best performance in the PDH reaction. Regarding the improvement at 10 min by silicalite-1 coating, a possible reason is that Co species were aggregated during the heating process under inert gas before the reaction and the initial reaction period of up to 10 min for uncoated samples, while significant aggregation of Co species did not occur for silicalite-1 coated samples during those processes, leading to higher activities of silicalite-1 coated samples even at 10 min. Table S3† presents the results of the PDH reaction tests over Co-based catalysts previously reported. The CS–S–Co (1) synthesized in this study is superior to any of the samples listed in the table in terms of propane conversion and propylene yield. In general, as conversion and yields increase, selectivity tends to decrease, but our results suggest that selectivity can be maintained using our core–shell catalysts even at high conversion.
 | ||
| Fig. 6 TEM images of spent (a) S–Co (1), (b) CS–S–Co (1), and (c) physically-mixed catalyst with S–Co (1) and silicalite-1. | ||
We obtained a TEM image of the spent physical mixture of S–Co (1) and silicalite-1. As shown in Fig. 6(c), we observed aggregated Co nanoparticles and carbon nanotubes, similar to that observed in the TEM image of spent S–Co (1). This result indicates that silicalite-1 shell layers are important for improving propane conversion and propylene yields in PDH reactions. Thus, the suppression of the formation of aggregated Co nanoparticles and carbon nanotubes during the PDH reaction led to improvements in propane conversion and propylene yields. Another concern is that the different chemical states of Co species formed after silicalite-1 coating and the Co species after silicalite-1 coating may show higher activity than the Co species before silicalite-1 coating. One possible Co species is Co2+ interacting with the zeolite framework. We synthesized silicalite-1 with the Co species (one-pot S–Co) by one-pot hydrothermal synthesis method. According to UV-vis spectra of the sample, the Co species was formed as shown in Fig. S9.† The catalytic activity of one-pot S–Co was lower than that of Co-impregnated silicalite-1 (S–Co (1)) as shown in Fig. 5(a) and S10(a).† This result indicates that extra framework Co species are better active sites for PDH reaction than Co species Co2+ interacting with the zeolite framework. In addition, we performed silicalite-1 coating for one-pot S–Co. The silicalite-1 coated one-pot S–Co showed better catalytic activity before silicalite-1 coating as shown in Fig. S10(b).† Thus, silicalite-1 is an effective method for improving the catalytic activity regardless of the chemical state of Co species. Moreover, the propylene selectivity was calculated from the results of PDH reaction as shown in Table S4.† There was a clear difference between S–Co (1) and CS–S–Co (1), and the coating improved the propylene selectivity. However, the coating was less effective when the loading amount was higher than 3 wt%. The reason for this is suggested to be that the silicalite-1 layer does not adequately cover the active species if the loading amount is too large. To quantitatively confirm the suppression of sintering by the silicalite-1 coating, we observed the particle size of Co particles in TEM images of the catalyst after the reaction. As shown in Table S5,† the particle size tended to be smaller in CS–S–Co (x) than in S–Co (x), and it was quantitatively confirmed that the coating was effective in preventing the aggregation of Co particles. The amount of carbon deposition was quantitatively analyzed by TGA as shown in Table S6.† The amount of carbon deposition decreased after silicalite-1 coating except for S–Co (20) series. Up to x = 10, silicalite-1 shell prevents the formation of carbon physically. As for the S–Co (20) series, this may be because too aggregated Co particles in S–Co (20) did not promote the carbon deposition while CS–S–Co (20) had highly dispersed Co species like S–Co (10) which can produce carbon due to silicalite-1 shell. In summary, Co species were geometrically confined in the silicalite-1 crystals owing to the presence of silicalite-1 layers. The confined Co species were difficult to sinter and could not induce the subsequent reactions necessary to form carbon nanotubes during the PDH reaction, leading to the high catalytic activity of CS–S–Co (x) in the PDH reaction. Finally, we investigated the reusability of CS–S–Co (1). The spent CS–S–Co (1) was calcined under an oxygen atmosphere at 873 K for 2 h. The refreshed catalyst was applied for the PDH reaction under the same reaction condition. As shown in Fig. S11,† the catalyst performed as well as fresh catalyst in terms of propane conversion and propylene yield after oxygen treatment.
4. Conclusions
We prepared Co confined within core–shell silicalite-1 zeolite crystals by coating Co-loaded silicalite-1 with silicalite-1 layers. The overgrowth of silicalite-1 layers was confirmed by various characterization techniques. The performance of the catalysts in the PDH reaction improved after silicalite-1 coating. This improvement was due to the suppression of the formation of aggregated Co nanoparticles and carbon nanotubes owing to the presence of silicalite-1 layers.Author contributions
Shohei Kubota: conceptualization, investigation, visualization, writing – original draft. Tomoka Sumi: methodology, writing – review & editing. Haruna Kitamura: methodology, writing – review & editing. Koji Miyake: conceptualization, supervision, visualization, methodology, writing – review, and editing. Yoshiaki Uchida: writing – review & editing. Norikazu Nishiyama: supervision, resources, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A part of the present experiments was carried out by using a facility in the Research Center for Ultra-High Voltage Electron Microscopy, Osaka University.Notes and references
- S. Chen, X. Chang, G. Sun, T. Zhang, Y. Xu, Y. Wang, C. Pei and J. Gong, Chem. Soc. Rev., 2021, 50, 3315–3354 RSC.
- Z. Bian, N. Dewangan, Z. Wang, S. Pati, S. Xi, A. Borgna, H. Kus and S. Kawi, ACS Appl. Nano Mater., 2021, 4, 1112–1125 CrossRef CAS.
- M. Martino, E. Meloni, G. Festa and V. Palma, Catalysts, 2021, 11, 1070 CrossRef CAS.
- B. M. Weckhuysen and R. A. Schoonheydt, Catal. Today, 1999, 51, 223–232 CrossRef CAS.
- W. Won, K. S. Lee, S. Lee and C. Jung, Comput. Chem. Eng., 2010, 34, 508–517 CrossRef CAS.
- M. Miyamoto, K. Mabuchi, J. Kamada, Y. Hirota, Y. Oumi, N. Nishiyama and S. Uemiya, J. Porous Mater., 2015, 22, 769–778 CrossRef CAS.
- L. H. Nguyen, T. Vazhnova, S. T. Kolaczkowski and D. B. Lukyanov, Chem. Eng. Sci., 2006, 61, 5881–5894 CrossRef CAS.
- N. S. Gnep, J. Y. Doyemet, A. M. Seco, F. R. Ribeiro and M. Guisnet, Appl. Catal., 1987, 35, 93–108 CrossRef CAS.
- P. L. De Cola, R. Gläser and J. Weitkamp, Appl. Catal., A, 2006, 306, 85–97 CrossRef CAS.
- Y. Zhang, Y. Zhou, A. Qiu, Y. Wang, Y. Xu and P. Wu, Catal. Commun., 2006, 7, 860–866 CrossRef CAS.
- Z. Nawaz, T. Xiaoping and W. Fei, Korean J. Chem. Eng., 2009, 26, 1528–1532 CrossRef CAS.
- H. Zhu, D. H. Anjum, Q. Wang, E. Abou-Hamad, L. Emsley, H. Dong, P. Laveille, L. Li, A. K. Samal and J.-M. Basset, J. Catal., 2014, 320, 52–62 CrossRef CAS.
- S. Gómez-Quero, T. Tsoufis, P. Rudolf, M. Makkee, F. Kapteijn and G. Rothenberg, Catal. Sci. Technol., 2013, 3, 962–971 RSC.
- X. Fan, J. Li, Z. Zhao, Y. Wei, J. Liu, A. Duan and G. Jiang, RSC Adv., 2015, 5, 28305–28315 RSC.
- S. Kaneko, T. Arakawa, M. Ohshima, H. Kurokawa and H. Miura, Appl. Catal., A, 2009, 356, 80–87 CrossRef CAS.
- Y. Gao, L. Peng, J. Long, Y. Wu, Y. Dai and Y. Yang, Microporous Mesoporous Mater., 2021, 323, 111187 CrossRef CAS.
- N. Jeon, J. Oh, A. Tayal, B. Jeong, O. Seo, S. Kim, I. Chung and Y. Yun, J. Catal., 2021, 404, 1007–1016 CrossRef CAS.
- J. Long, S. Tian, S. Wei, H. Lin, G. Shi, X. Zong, Y. Yang, D. Yang, Y. Tang and Y. Dai, Appl. Surf. Sci., 2023, 614, 156238 CrossRef CAS.
- T. W. Hansen, A. T. Delariva, S. R. Challa and A. K. Datye, Acc. Chem. Res., 2013, 46, 1720–1730 CrossRef CAS PubMed.
- M. Miyamoto, T. Kamei, N. Nishiyama, Y. Egashira and K. Ueyama, Adv. Mater., 2005, 17, 1985–1988 CrossRef CAS.
- D. Van Vu, M. Miyamoto, N. Nishiyama, Y. Egashira and K. Ueyama, J. Catal., 2006, 243, 389–394 CrossRef.
- D. V. Vu, M. Miyamoto, N. Nishiyama, S. Ichikawa, Y. Egashira and K. Ueyama, Microporous Mesoporous Mater., 2008, 115, 106–112 CrossRef CAS.
- D. Van Vu, M. Miyamoto, N. Nishiyama, Y. Egashira and K. Ueyama, Catal. Lett., 2009, 127, 233–238 CrossRef CAS.
- M. Miyamoto, K. Mabuchi, J. Kamada, Y. Hirota, Y. Oumi, N. Nishiyama and S. Uemiya, J. Porous Mater., 2015, 22, 769–778 CrossRef CAS.
- K. Miyake, Y. Hirota, K. Ono, Y. Uchida and N. Nishiyama, ChemistrySelect, 2016, 1, 967–969 CrossRef CAS.
- K. Miyake, Y. Hirota, K. Ono, Y. Uchida, S. Tanaka and N. Nishiyama, J. Catal., 2016, 342, 63–66 CrossRef CAS.
- S. Yusuke, H. Yuichiro, U. Yoshiaki and N. Norikazu, Chem. Lett., 2015, 44, 477–479 CrossRef.
- T. T. Le, K. Shilpa, C. Lee, S. Han, C. Weiland, S. R. Bare, P. J. Dauenhauer and J. D. Rimer, J. Catal., 2022, 405, 664–675 CrossRef CAS.
- M. Okamoto and Y. Osafune, Microporous Mesoporous Mater., 2011, 143, 413–418 CrossRef CAS.
- G. Xu and X. Zhu, Appl. Catal., B, 2021, 293, 120241 CrossRef CAS.
- A. Ghorbanpour, A. Gumidyala, L. C. Grabow, S. P. Crossley and J. D. Rimer, ACS Nano, 2015, 9, 4006–4016 CrossRef CAS PubMed.
- K. Miyake, R. Inoue, M. Nakai, Y. Hirota, Y. Uchida, S. Tanaka, M. Miyamoto and N. Nishiyama, Microporous Mesoporous Mater., 2018, 271, 156–159 CrossRef CAS.
- T. Sumi, S. Kokuryo, Y. Fujimoto, X. Li, K. Miyake, Y. Uchida and N. Nishiyama, Catal. Sci. Technol., 2022, 12, 7010–7017 RSC.
- S. Kokuryo, K. Tamura, K. Miyake, Y. Uchida, M. Miyamoto, Y. Oumi, A. Mizusawa, T. Kubo and N. Nishiyama, Catal. Sci. Technol., 2022, 12, 7270–7274 RSC.
- K. Miyake, R. Inoue, T. Miura, M. Nakai, H. Al-Jabri, Y. Hirota, Y. Uchida, S. Tanaka, M. Miyamoto, S. Inagaki, Y. Kubota, C. Y. Kong and N. Nishiyama, Microporous Mesoporous Mater., 2019, 288, 109523 CrossRef CAS.
- R. Inoue, K. Miyake, Y. Hotta, L. Xinyu, R. Yashiro, Y. Hirota, Y. Uchida, M. Miyamoto, Y. Oumi, C. Yi Kong and N. Nishiyama, Fuel, 2021, 305, 121487 CrossRef CAS.
- M. Nakai, K. Miyake, R. Inoue, K. Ono, H. A. Jabri, Y. Hirota, Y. Uchida, S. Tanaka, M. Miyamoto, Y. Oumi, C. Y. Kong and N. Nishiyama, Catal. Sci. Technol., 2019, 9, 6234–6239 RSC.
- B. Qiu, W. Guo, Z. Liang, W. Xia, S. Gao, Q. Wang, X. Yu, R. Zhao and R. Zou, RSC Adv., 2017, 7, 13340–13346 RSC.
- R. Dehghan, T. W. Hansen, J. B. Wagner, A. Holmen, E. Rytter, Ø. Borg and J. C. Walmsley, Catal. Lett., 2011, 141, 754–761 CrossRef CAS.
- L. Wu, Z. Ren, Y. He, M. Yang, Y. Yu, Y. Liu, L. Tan and Y. Tang, ACS Appl. Mater. Interfaces, 2021, 13, 48934–48948 CrossRef CAS PubMed.
- Y. Dai, J. Catal., 2019, 381, 482–492 CrossRef.
- C. Chong, S. Zhang, Z. Wang and Z.-Y. Yuan, J. Catal., 2020, 383, 77–87 CrossRef.
- S. Dzwigaj and M. Che, J. Phys. Chem. B, 2006, 110, 12490–12493 CrossRef CAS PubMed.
- J. J. H. B. Sattler, J. Ruiz-Martinez, E. Santillan-Jimenez and B. M. Weckhuysen, Chem. Rev., 2014, 114, 10613–10653 CrossRef CAS.
- A. N. Chernov, V. I. Sobolev, E. Y. Gerasimov and K. Y. Koltunov, Catalysts, 2022, 12, 1262 CrossRef CAS.
- Z. Chen, D. Y. Kim, K. Hasegawa and S. Noda, ACS Nano, 2013, 7, 6719–6728 CrossRef CAS PubMed.
- Y. Wang, L. Qiu, L. Zhang, D.-M. Tang, R. Ma, Y. Wang, B. Zhang, F. Ding, C. Liu and H.-M. Cheng, ACS Nano, 2020, 14, 16823–16831 CrossRef CAS PubMed.
- C. Paukner and K. K. K. Koziol, Sci. Rep., 2014, 4, 3903 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cy01637d |
| This journal is © The Royal Society of Chemistry 2024 |