
Introducing carbon dots to NiFe LDH via a mild coprecipitation–aging method to construct a heterojunction for effective oxygen evolution†
Zi-Ye
Liu
,
Qian-Yu
Wang
and
Ji-Ming
Hu
 *
*
Department of Chemistry, Zhejiang University, Hangzhou 310027, P. R. China. E-mail: kejmhu@zju.edu.cn
First published on 14th December 2023
Abstract
Modified NiFe-layered double hydroxide (NiFe LDH) materials with a high oxygen evolution reaction (OER) performance make it possible to replace noble metal catalysts for widespread applications. Herein, we prepare a layered carbon dot (CD) composite catalyst (denoted as NiFe LDH@CDs) by a one-step coprecipitation method without heating or hydrothermal treatment. Due to the numerous functional groups of CDs, there were strong electron interactions between the two components. The CDs promoted rapid charge transfers and accelerated OER kinetics. Moreover, through binding with the CDs, the NiFe LDH@CDs formed a heterojunction structure, which could efficiently suppress photoelectron–hole recombination. Based on the beneficial factors, the Tafel slope of NiFe LDH@CDs-200 was 44.77 mV dec−1, which further decreased to 38.44 mV dec−1 under light even with a low content of CDs.
1. Introduction
The oxygen evolution reaction (OER), a half reaction in the water electrolysis process, is an indispensable part of the hydrogen energy system.1,2 However, the sluggish four-electron coupling proton-transfer process of the OER retards the counter half-reaction.3 Therefore, a well-designed catalyst is the most efficient way to accelerate electron transfer and remove the barriers of the kinetics. Metal-based OER catalysts can be divided into two categories: noble metal catalysts, such as Ir-, Ru-, and Rh-based catalysts and non-noble metal catalysts.4–6 The undesirably high cost of noble metal catalysts limits their widespread application despite their excellent catalytic activity.7 For non-noble metal catalysts, 3d-metal compounds have been considered as great OER catalysts in the past decades, especially nickel compounds.8–10 However, their poor electrical conductivity is a major limitation to their further application. Fortunately, this problem can be easily solved by combining them with other well conductive substrates,11–13 and if some of them possess a photoelectric response, such as semiconductors, the solar energy can be used to replace a part of the power consumption for a more efficient and more environmental friendly water-splitting process.14 Unfortunately, most catalysts, such as Co3O4 and Ni(OH)2, do not display any light responses to match this assumption, while TiO2, BiVO4, and other materials with excellent photoelectric conversion fail to display good electrocatalytic activity.Layered double hydroxides (LDHs) can well meet these demands. The formula of LDH is [M2+1 − xM3+x(OH)2]x+(An−)x/n·mH2O with a structure of two-dimensional (2D) positively charged laminates with exchangeable anions in the interlayers to maintain the charge balance.15 The adjustable metal ratio and huge specific surface area make the performance of LDHs flexible, facilitating their application in corrosion prevention,16 capacitors,17 photodegradation,18 electrocatalysis,19 and other processes. Ni-based LDHs show greater OER performances in alkaline media than other LDHs and have been commonly used in water catalysis.20 For example, Fe-doped NiCo-LDH nanosheets with a high specific surface area prepared by Ni–Co glycerate spheres as a template exhibited good performance for the OER process.21 Among Ni-based LDHs, NiFe LDH is particularly highly valued for its low cost and favorable OER catalytic capacity, and the cooperation of two metal ions creates more possibility for specific applications.22 However, the low conductivity of NiFe LDH leads to a more sluggish charge-transfer process.23 The other crucial problem of NiFe LDH is its weak photocatalytic capability owing to the fast recombination of photogenerated electrons and holes.24 A conductive matrix, such as graphene25,26 or carbon nanotubes,27 is commonly chosen for the modification of NiFe LDH to improve its conductivity and to facilitate the charge transfer of the composite electrocatalysts. Meanwhile, a heterojunction structure is often designed to efficiently improve the photocatalytic performance of NiFe LDH electrocatalysts via promoting the separation of photogenerated carriers.28 For example, constructing heterojunctions by loading CdS/CdTe quantum dots onto sulfurized NiFe LDH can achieve efficient photocatalytic performance for hydrogen evolution.29 Carbon dots (CDs) have a high electric conductivity and much smaller size compared to graphene and other carbon nanomaterials, which provides a larger surface area to form strong interactions with other components. For these reasons, CDs have been widely used as a charge-transfer promoter for a variety of electrocatalysts.30–32 The difference in energy level between CDs and other substrates may meet the conditions of facilitating the photogenerated electron–hole separation.
Usually, the synthesis of such heterogenous catalysts requires strong external forces to construct a special morphology or structure. The commonly used method is hydrothermal treatment, which was recently employed by Tang et al. to prepare a carbon quantum dots/NiFe LDH complex.33 However, hydrothermal treatment needs to maintain a high temperature for a long time in the oven, and the yield is not high due to the low concentration of the material. The long-time synthesis increases the energy consumption and runs counter to the purpose of designing catalysts.
Herein, we developed a feasible method to prepare NiFe LDH@CDs at room temperature for the first time. The numerous functional groups enable the CDs to build chemical bonds with NiFe LDH easily. The coprecipitation–aging process creates the formation of strong interactions between the two compounds. Due to the combination between CDs and NiFe LDH, the electrocatalytic activity of NiFe LDH@CDs is greatly enhanced. Moreover, the heterojunction material formed by these two structures can further improve the OER activity under light conditions.
2. Experimental
2.1 Chemical materials
Citric acid (C6H8O7, 99.5%) and dicyandiamide (C2H2N4, 99%) were purchased from Aladdin. Nickel(II) nitrate hexahydrate (Ni(NO3)2·6H2O, 98%), iron(III) nitrate nonahydrate (Fe(NO3)3·9H2O, 98%), sodium hydroxide (NaOH, 96%), potassium hydroxide (KOH, 85%), sodium carbonate (Na2CO3, 99.8%), and ethanol (C2H6O, A. R.) were purchased from Sinopharm Chemical Reagent Co. Ltd. Nafion solution (5 wt% in alcohol and water) was purchased from DuPont.2.2 Preparation of the carbon dots
First, 15 mmol of citric acid and 15 mmol of dicyandiamide were added to 12 mL of deionized water and stirred for 15 min. The solution was then transferred into a 50 mL PPL-lined stainless-steel autoclave and heated at 180 °C for 6 h. The collected solution was filtered by a 0.22 μm pore filter to remove large aggregates. Afterward, the filtrate was washed with ethanol to remove unreacted species. The resulting fallout was completely dehydrated at 70 °C for 6 h in a vacuum drying oven. Finally, the solid pieces were ground quickly to obtain a dark green powder of CDs.2.3 Synthesis of NiFe LDH and NiFe LDH@CDs-50/100/200
First, 4 mmol Fe(NO3)3·9H2O and 8 mmol Ni(NO3)2·6H2O were dissolved in 10 mL of deionized water and used as the salt solution. Next, 28 mmol NaOH and 9 mmol Na2CO3 were dissolved in 15 mL of deionized water and used as the lye. The salt solution and lye were mixed dropwise under magnetic stirring while maintaining the pH above 13 and the suspension was aged for 12 h at room temperature. The product was collected by centrifugation and washed three times with ethanol and water alternately. Then, NiFe LDH was dried in an oven at 50 °C for 4 h. The salt solution and lye were prepared by the same method. The salt solution and lye were mixed dropwise under magnetic stirring. After dropping had finished, 50/100/200/300 mg of CDs was added into the suspension and the mixture was aged for 12 h. The NiFe LDH@CDs-50/100/200/300 samples were collected in the same way as the NiFe LDH. A schematic description of the preparation process is shown in Fig. 1. | ||
| Fig. 1 The preparation of NiFe LDH@CDs by a coprecipitation–ageing process. | ||
2.4 Characterization
The morphology were captured via field emission scanning electron microscopy (SEM, SU8010, Hitachi) and transmission electron microscopy (TEM, HT7700, Hitachi). The microstructure of the lamella and distribution of elements were investigated by high-resolution transmission electron microscopy (HRTEM, 2100F, Jeol) with energy dispersive X-ray spectroscopy (EDS, OXFORD 80T, Oxford Instruments Plc.). X-ray diffraction patterns were collected by X-ray diffraction (XRD, X'Pert3 Powder, PANalytical) using Cu Kα radiation. The chemical bonds of LDH@CDs were analyzed by Fourier transform infrared spectroscopy (FTIR, Nicolet iS10, ThermoFisher Scientific). The UV-vis absorption of the prepared materials and energy gap of the LDH and CDs were evaluated by UV-vis diffuse reflectance spectroscopy (TU-1901, PERSEE). The fluorescence intensity was obtained by PL spectroscopy (PL, FLS980, Edinburgh Instruments. EI.). The contents of CDs were calculated by thermogravimetry (TG, TG 209 F1 Libra, NETZSCH) and elemental analysis (EA, Vario Micro, Elementar Analysensysteme GmbH). X-ray photoelectron spectroscopy (XPS, Thermo ESCALAB 250Xi, ThermoFisher Scientific) was used to analyze the element information of the prepared materials.2.5 Electrochemical measurements
OER electrochemical tests were carried out in 1 M KOH solution at room temperature with a three-electrode system. SCE (saturated calomel electrode) with a salt bridge was used as the reference electrode, carbon rod was used as the counter electrode, and a glassy carbon (GC) loading catalyst was used as the working electrode. First, 5 mg of sample was added into 770 μL deionized water, 200 μL isopropanol, and 30 μL Nafion mixture. The mixture was dispersed under sonication for 1 h to obtain a homogeneous ink. Next, 15 μL catalyst ink was dropped on the polished glassy carbon electrode, and the prepared electrodes were dried in the oven at 50 °C for 30 min. Additionally, the photo-electrodes were prepared with a similar way. Here, 50 μL catalyst ink was added to the FTO glassy electrode (active area of 1 × 1 cm2) to prepare the electrode, and the electrodes were dried in the oven at 50 °C for 30 min. The measured potentials were converted to potentials of the reversible hydrogen electrode using the formula: E(vs. RHE) = E(vs. SCE) + 0.241 + 0.0591 × pH. Linear sweep voltammetry (LSV) curves were obtained at a scan rate of 2 mV s−1 with 95% iR-compensation. Tafel plots were converted from LSV results by the Tafel equation (η = a + b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logj, where η is the overpotential, j is the current density, and b is the Tafel slope). Electrochemical impedance spectra were recorded at 0.45 V (vs. SCE) with an AC signal amplitude of 10 mV. The test frequency ranged from 100 k Hz to 0.1 Hz. The stability of the catalysts was proved by chronopotentiometry at a current density of 10 mA cm−2. Mott–Schottky curves were evaluated at a frequency of 2 k Hz with a sweeping applied voltage from −1.4 V to 0.4 V (vs. SCE). The electrochemical active surface area was measured by cyclic voltammetry in the range of −0.15 to −0.05 V (vs. SCE). The process for the photoelectric measurements was the same as for the electrochemical tests, with the only difference being the working electrodes. The light source for the photoelectric measurements was a Xe lamp. The photocurrent density was evaluated by chronoamperometry with intermittent light.
logj, where η is the overpotential, j is the current density, and b is the Tafel slope). Electrochemical impedance spectra were recorded at 0.45 V (vs. SCE) with an AC signal amplitude of 10 mV. The test frequency ranged from 100 k Hz to 0.1 Hz. The stability of the catalysts was proved by chronopotentiometry at a current density of 10 mA cm−2. Mott–Schottky curves were evaluated at a frequency of 2 k Hz with a sweeping applied voltage from −1.4 V to 0.4 V (vs. SCE). The electrochemical active surface area was measured by cyclic voltammetry in the range of −0.15 to −0.05 V (vs. SCE). The process for the photoelectric measurements was the same as for the electrochemical tests, with the only difference being the working electrodes. The light source for the photoelectric measurements was a Xe lamp. The photocurrent density was evaluated by chronoamperometry with intermittent light.
3. Results and discussion
The abundant OH− of the lye precipitates, Fe3+ and Ni2+ simultaneously, and the CO32− of the lye served as buffers to avoid rapid pH changes caused by the addition of the salt solution, and stabilized the structure of LDH through the electrostatic interactions between the electropositive layers and negative ions, which made the LDH easier to construct.34 The cooperation between the carbonate and hydroxyl groups ensured the facile formation of NiFe LDH, and promoted the good crystal structure formation at room temperature even without hydrothermal treatment.The X-ray diffraction (Fig. S1a†) results illustrated that the diffraction peak of CDs was similar to the (002) crystallographic plane of graphene. The Fourier transform infrared spectrum (FTIR, Fig. S1b†) showed that the carbon dots synthesized by hydrothermal approach35 were rich in hydroxyl, amino, carboxyl, and amide groups. The abundant functional groups of the CDs facilitated the formation of chemical bonds with metal atoms. Therefore, the CDs enables the coordination with NiFe LDH. In addition, the adequate aging time created enough chance for the formation of chemical bonds between the CDs and NiFe LDH.
The scanning electron microscopy (SEM) images show that NiFe LDH was a multilayer petaloid structure with a smooth surface (Fig. 2a). Compared with NiFe LDH, the surface of NiFe LDH@CDs-200 (Fig. 2b) was covered with a number of particles whose grain diameter was ∼3 nm, and the diameter of these particles well matched with that shown in the transmission electron microscopy (TEM) image of CDs (Fig. S2†). High-resolution transmission electron microscopy (HRTEM) was used to more intuitively illustrate the differences between the NiFe LDH and NiFe LDH@CDs (Fig. 2c and d). Here, the layers of NiFe LDH could be seen to be flat while the surface of NiFe LDH@CDs-200 was rough with convex surfaces. Furthermore, the lattice fringes of NiFe LDH@CDs-200 (Fig. 2e) indicated that there were two phases of the crystallographic planes in NiFe LDH@CDs-200, among which the fringes with a spacing of 0.527 nm were the (003) lattice plane of NiFe LDH, and the 0.241 nm spacing fringes corresponded to the (002) lattice plane of CDs. The different lattice fringes proved the bulges on the surface were CDs rather than NiFe LDH itself. Meanwhile, the surface and bulk phase of NiFe LDH were surrounded by C signals in the EDS mapping of NiFe LDH@CDs-200 (Fig. 2f). These results were mutually consistent with the lattice fringe image, both of which verified the distribution of CDs on the LDH surfaces, while pure NiFe LDH had a weak signal for C (Fig. S3 and S4†). The signal intensity and mass percent of C in NiFe LDH (Fig. S4†) were lower than that of NiFe LDH@CDs-200, which also indicated that the CDs had been successfully loaded on NiFe LDH.
 | ||
| Fig. 2 SEM (a and b) and HRTEM (c and d) images of NiFe LDH (a and c) and NiFe LDH@CDs-200 (b and d). (e) The high magnification HRTEM image of NiFe LDH@CDs-200. (f) The EDS mapping results of NiFe LDH@CDs-200. | ||
The X-ray diffraction patterns (XRD, Fig. 3a) showed that the diffraction peaks of NiFe LDH appeared at 11.5°, 23°, 34°, 38°, 58°, and 61°, corresponding to the (003), (006), (012), (015), (110), and (113) planes, respectively.36 Even though the CDs were added in the aging process, the crystal structure of LDH was not crushed. Similar diffraction patterns of NiFe LDH@CDs proved the feasibility of the synthesis method. Thermogravimetry analysis (Fig. 3b) allowed estimating the loading capacity of CDs, which was about 1–3 wt%. This result agreed well with the elemental analysis (Table S1†). The FTIR spectra showed that the absorption peak (Fig. 3c) near ∼3500 cm−1 corresponded to the O–H bonds of LDH, while the peak at ∼1370 cm−1 was assigned to the intercalated NO3− in the LDH, and the peaks at 800–600 cm−1 corresponded to M–O bonds.37 A peak at ∼1560 cm−1 appeared in the NiFe LDH@CDs, which was due to the existence of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching vibration from the CDs (Fig. S5†). The red-shift of the Ni–O peak with increasing the content of CDs in NiFe LDH@CDs (Fig. 3d) indicated that chemical interactions might exist between these two different materials. The formation of chemical bonds between LDH and CDs via the formation of metal–oxygen bonds, such as COO–M, decreased the electron cloud density of Ni and caused the red-shift.
N stretching vibration from the CDs (Fig. S5†). The red-shift of the Ni–O peak with increasing the content of CDs in NiFe LDH@CDs (Fig. 3d) indicated that chemical interactions might exist between these two different materials. The formation of chemical bonds between LDH and CDs via the formation of metal–oxygen bonds, such as COO–M, decreased the electron cloud density of Ni and caused the red-shift.
 | ||
| Fig. 3 The XRD pattern (a), thermogravimetric analysis (b), FTIR spectra (c and d), UV-vis diffuse reflection spectra (e) and PL spectra (f) of NiFe LDH, NiFe LDH@CDs-50, NiFe LDH@CDs-100, and NiFe LDH@CDs-200. | ||
The optical responses of NiFe LDH and NiFe LDH@CDs were investigated by UV-visible diffuse reflectance spectroscopy (UV-vis, Fig. 3e). The absorption edge of NiFe LDH was red-shifted with the addition of the CDs, suggesting that the CDs enabled an expansion of the optical response range of NiFe LDH.38 To further understand the migration of photogenerated carriers, we used Tauc plots and the XPS valence band spectra (VB-XPS) to calculate the valence band, conduction band, and band gap. According to the UV-vis analysis, the band gaps of the CDs and NiFe LDH were calculated to be 2.29 and 2.73 eV, respectively (Fig. S6†). The XPS valence band spectra (VB-XPS) results (Fig. S7†) showed that the valence band energies of NiFe LDH and CDs were 1.61 and 2.60 eV, respectively. The conduction band energies of NiFe LDH and CDs were calculated to be −1.12 and 0.31 eV by using the formula: ECB = EVB − Eg (vs. Fermi level). The moderate gap band (Eg,NiFe LDH = 2.73 eV) indicated that NiFe LDH had a visible photoelectric response. Furthermore, the difference in the energy levels between the CDs and LDH layer provided a foundation to construct the structure of a heterojunction. The existence of the heterojunction structure could be proved by the photoluminescence spectra (PL). The PL spectra (Fig. 4d) showed that NiFe LDH exhibited two emission peaks at ∼450 and ∼515 nm. However, the intensity decreased apparently, especially for the peak at ∼515 nm after incorporating the strongly fluorescent CDs. The attenuation of the PL intensity proved that the photogenerated electron–hole pairs were effectively suppressed thanks to the structure of the heterojunction.39
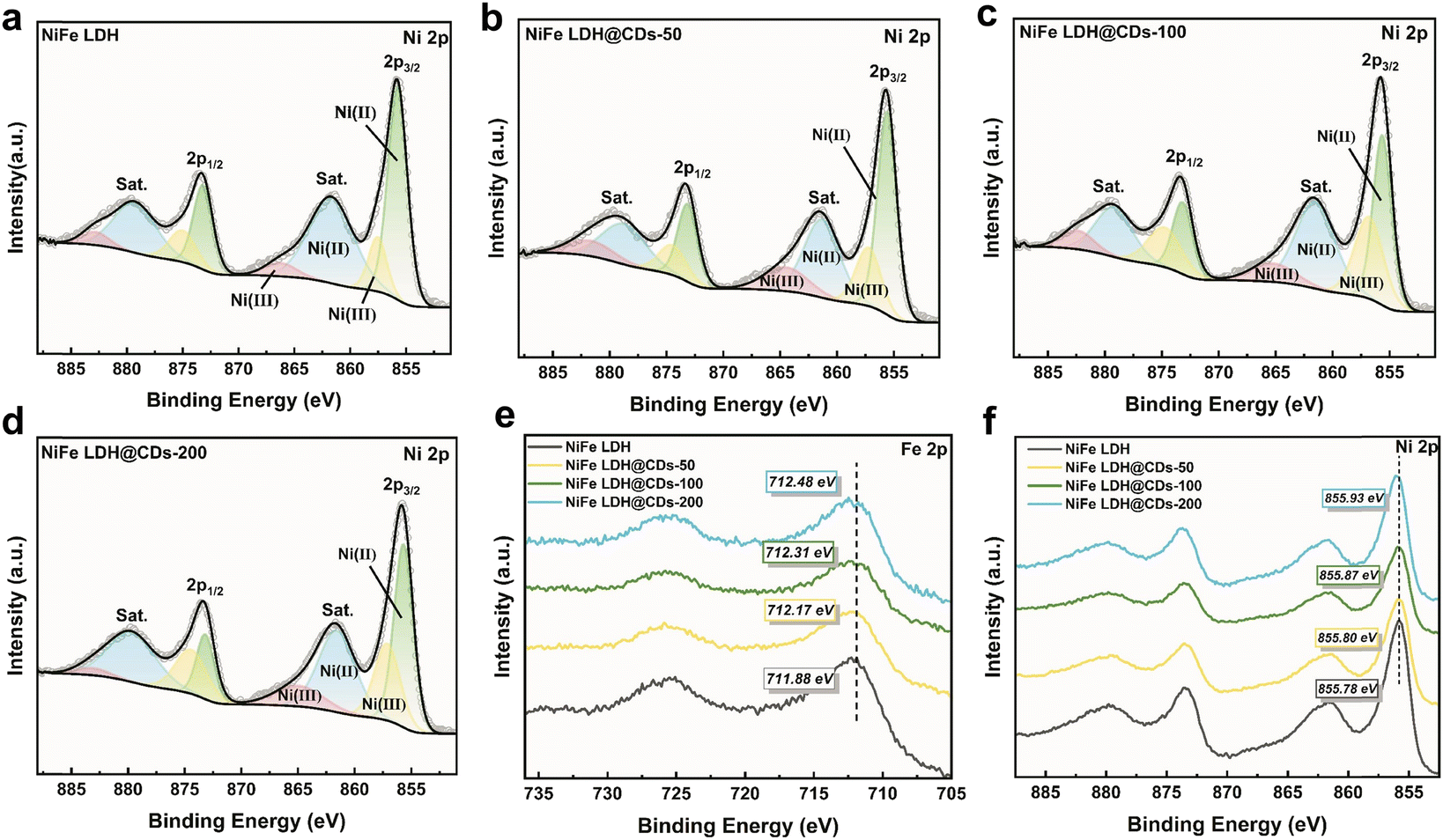 | ||
| Fig. 4 The XPS spectra of Ni 2p (a–d) and their decompositions for NiFe LDH (a), NiFe LDH@CDs-50 (b), NiFe LDH@CDs-100 (c), and NiFe LDH@CDs-200 (d). (e) The XPS spectra of Fe 2p. (f) An overview of the XPS spectra of Ni 2p. | ||
The X-ray photoelectron spectra were used to evaluate the binding energies of Fe 2p and Ni 2p. According to the fitting results, Fe only existed in the Fe3+ state (Fig. S8†), which originated from the reagents of iron(III) nitrate. Ni could be assigned to two forms with the states: Ni2+ and Ni3+, and the content of Ni(III) consecutively increased with addition of the CDs content in the NiFe LDH@CDs, with the value varying from 18.2% in NiFe LDH to 29.0%, 34.7%, and 37.5% in NiFe-LDH@CDs-50, -100, and -200, respectively (Fig. 4a–d). The higher content of Ni(III) indicated that NiFe LDH could easily transform into the NiOOH phase, while the quick formation of Ni(III) compounds is beneficial for the OER,40 and therefore the increase in CDs content would be expected to enhance the OER electrocatalytic activity of NiFe LDH. Compared with NiFe LDH, a negative shift in binding energy was both observed on Ni (Fig. 4e) and Fe (Fig. 4f) in the NiFe LDH@CDs. These shifts indicated that there were electronic interactions between the NiFe LDH and CDs, and also demonstrated the existence of electron transport from NiFe LDH to CDs.41 This result was consistent with the red-shift in the FTIR absorption peaks (Fig. 3d).
Linear sweep voltammetry (LSV) was next used to investigate the OER performances of the prepared materials in 1 M KOH electrolyte in a three-electrode system (Fig. 5a). As shown from the LSV curves, the pure CDs were nonactive for OER, but when they were incorporated with NiFe LDH, they remarkably enhanced the OER catalytic activity of NiFe LDH. Such an improving effect became more obvious when the CDs content increased, till the content of CDs excessed 200 mg. The results show that the OER performance of NiFe LDH@CDs-300 was worse than that of CDs-200 (Fig. S9a and b†). It is supposed that the amount of catalytic sites on the catalyst surface may decrease under an excessive amount of CDs due to the coverage of CDs, even though the photo-response of NiFe LDH@CDs-300 was also enhanced. So CDs-200 was suggested as the optimal value.
 | ||
| Fig. 5 The OER LSV curves (a) and Tafel plots (b) of NiFe LDH, NiFe LDH@CDs-50, NiFe LDH@CDs-100, and NiFe LDH@CDs-200 under light on or off conditions in 1 M KOH with 95% iR-compensation. | ||
The overpotential at the current density of 10 mA cm−2 (denoted as η10) decreased from 360 mV on NiFe LDH to 279 mV on NiFe LDH@CDs-200, and the Tafel slope decreased from 126.75 mV dec−1 to 44.37 mV dec−1 (Fig. 5b). The Tafel slope of NiFe LDH@CDs-200 was quite small in the various nickel-based LDH catalysts (Table S2†). Interaction between the CDs and NiFe LDH was the main reason for the rapid charge transfer from the catalytic centers, and the combination of CDs accelerated the electron-transfer process to different sheets in bulk NiFe LDH. After applying light irradiation, the OER polarization curves of NiFe LDH and NiFe LDH@CDs composites were shifted to a lower potential range with the decrease in the overpotentials and Tafel slopes. Moreover, the positive effect of light irradiation was more obvious on the CDs-incorporated samples. For instance, η10 and η25 of NiFe LDH@CDs-200 decreased from 279 and 303 mV before to 273 mV and 291 mV after illumination. Also, the Tafel slopes decreased from 44.77 mV dec−1 to 38.44 mV dec−1. Although the overpotential of NiFe LDH@CDs-200 may not be as good as the other Ni-based LDH catalysts, its Tafel slope was quite small (Table S2†).33,41–50 A lower Tafel slope means that the overpotential does not increase rapidly with the current density, which indicated that NiFe LDH@CDs-200 was comparable to other catalysts at high current density. It is supposed that the CDs work like a ‘electron transportation bridge’ to suppress the recombination of photogenerated electron–hole pairs, reflected in the lower overpotential and Tafel slope.
Electrochemical impedance spectroscopy (EIS) tests showed there were two semicircles in the complex planes (Fig. 6a), corresponding to physical electric impedance between the glassy carbon substrate and catalyst membrane in the higher-frequency range, and electrochemical impedance of the catalyst/electrolyte interface in the lower-frequency range. The impedance data thus could be fitted to the equivalent circuit shown in Fig. 6a, where Rs is the solution resistance, Rif is the resistance of the catalyst membrane in parallel with the capacitance (Cif), which represents the membrane of the catalysts attached on the surface of the GC electrodes, and Rct is the charger-transfer resistance between the catalysts and electrolyte in parallel with the double-layer capacitance (Cdl).51 To obtain more detailed results, a constant phase angle element (CPE) was used for the fitting of the capacitance. The fitting results (Table S3†) showed that the Rct of NiFe LDH decreased after being incorporated with the CDs, indicating that the CDs could facilitate charge transfer of the biphasic interface and weaken the kinetic hindrance. This resulted in the greater OER performance. The Rif of NiFe LDH also decreased simultaneously after being composited with the CDs because of their favorable conductivities. Cyclic voltammetry tests were next used to evaluate the electrochemical active area (Fig. S10†). The working electrodes were prepared by the same processes, but they had different Cdl values. The Cdl of NiFe LDH@CDs-200 was 152 μF cm−2, much higher than that of pure NiFe LDH (Fig. 6b). This indicated that NiFe LDH@CDs had a larger electrochemical active surface area (ECSA) and that NiFe LDH@CDs-200 could expose more active sites for the oxidation of water.
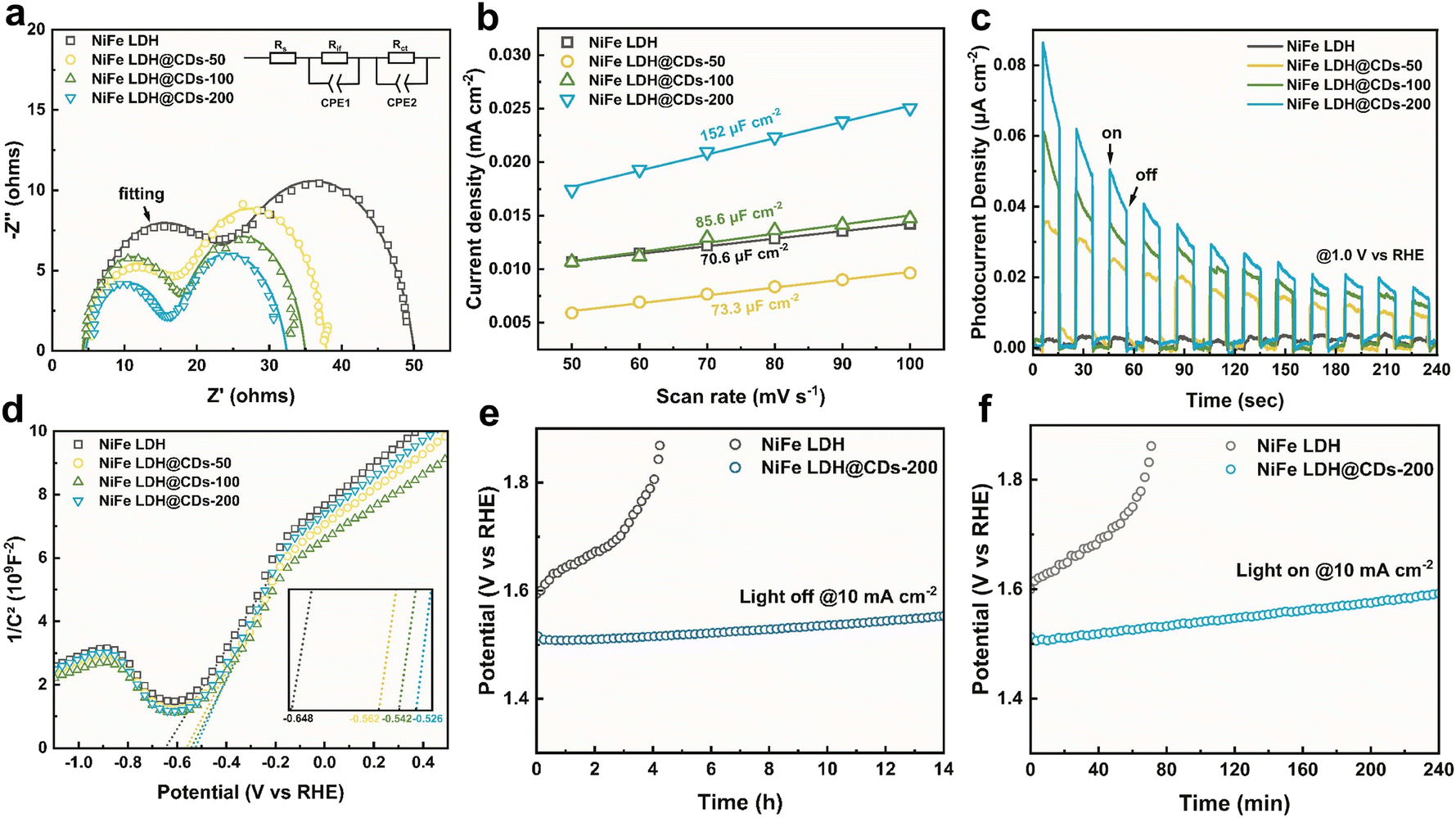 | ||
| Fig. 6 (a) Nyquist plots, (b) double-layer capacitance estimated by CV at various scan rates. (c) The photocurrent density and (d) Mott–Schottky curves of NiFe LDH, NiFe LDH@CDs-50, NiFe LDH@CDs-100, and NiFe LDH@CDs-200. Chronopotentiometry responses at 10 mA cm−2 on NiFe LDH and NiFe LDH@CDs-200 without irritation (e) and under the Xe lamp (f). | ||
The photocurrent density (at 1.0 V vs. RHE) was used to evaluate the light responses of NiFe LDH@CDs (Fig. 6c). Pure NiFe LDH only showed tiny responses under intermittent illumination. In contrast, the addition of CDs greatly improved the photoelectric response of the LDH. The Mott–Schottky plots (Fig. 6d) showed that the flat band potentials (Efb) of NiFe LDH and NiFe LDH@CDs-50/100/200 were −0.648, −0.598, −0.542, and −0.526 V (vs. RHE), respectively. The positive shift of Efb indicated that the bending of the band edges decreased, which was attributed to the faster interface charge transfer between the catalyst and electrolyte.52 It is worth noting that a more positive Efb is favorable for transferring photogenerated holes to the surfaces and separating electron–hole pairs.38 To evaluate the stability of the composites, we used chronopotentiometry to monitor the potential change at 10 mA cm−2 (Fig. 6e and f). It was illustrated that LDH@CDs-200 still maintained good catalytic activity when working for 14 h at 10 mA cm−2, while NiFe LDH was gradually deactivated under the same condition for 4 h accompanied with a rapidly increased potential. Under the Xe lamp illumination, NiFe LDH@CDs-200 still maintained better activity than NiFe LDH. After the OER process, the morphology of NiFe LDH@CDs-200 did not change much, and it maintained the NiFe LDH phase as well (Fig. S11†). The favorable stability of LDH@CDs was due to the promotion of the conductivity, which avoided forfeiting the catalytic activity caused by the destruction of the NiFe LDH layers structure caused by charge accumulation.
The possible reaction mechanism is proposed based on the results of the EIS, XPS, Mott–Schottky curves, and Tauc plots and is illustrated in Scheme 1. Here, before the occurrence of the OER, OH− is absorbed on the surfaces of catalyst. When applying a sufficient overpotential, the NiFe LDH@CDs offer nickel and iron atoms as the active centers. The nickel and iron sites transform the absorbed OH− into intermediates such as O*, OOH*, and OH*.53 Then the electrons obtained from the active sites are transferred to CDs bound on the NiFe LDH (Scheme 1a). The electrons cross through the interbedded CDs to reach the inner layers and enter the circuit through the interface between the electrode and catalyst under the electric field. Due to the favorable conductivity of the CDs, NiFe LDH@CDs shows better electrocatalytic performance. The rapid conduction of electrons effectively alleviates the charge accumulation of NiFe LDH, thus greatly improving the stability. Furthermore, water splitting and oxidation by photogenerated holes occurs simultaneously under the light. When visible light is irradiated on the composite surfaces, both the CDs and NiFe LDH can generate photogenerated electron–hole pairs (Scheme 1b). The heterojunction structure ensures that the photoelectrons migrate to the lower energy band. The electrons of NiFe LDH excited by the Xe lamp transit from the valence band (1.62 V vs. RHE) to the conduction band (−1.21 V vs. RHE). Then photoelectrons are rapidly transferred to the conduction band of CDs (0.31 V vs. RHE). Due to the separation of the charge, the photogenerated holes derived from CDs and NiFe LDH enable the oxidation of OH− or H2O.
 | ||
| Scheme 1 (a) Possible electrocatalytic reaction processes of LDH@CDs. (b) Schematic diagram of the LDH@CDs photoelectrocatalytic reaction mechanism. | ||
4. Conclusion
In summary, we devised a one-step coprecipitation–aging method to synthesize endoplasmic reticulum-liked NiFe LDH@CDs. The chemical binding and electrostatic attraction between CDs and NiFe LDH ensured strong interactions between the two materials. The CDs could rapidly transport the electrons obtained from the active sites, thus greatly reducing the charge-transfer resistance. Moreover, the combination of CDs and NiFe LDH formed a heterojunction structure, which could effectively suppress the recombination of photoelectrons and holes. Therefore, both components of the NiFe LDH@CDs could be excited to generate holes to oxidize water under the illumination. This work briefly illustrates that CDs can enable the promotion of the OER electrocatalytic capacity of NiFe LDH and act as a co-photocatalyst with NiFe LDH to split water. This simple coprecipitation–aging process may be used as a feasible strategy for the industrial preparation of highly efficient LDH-based catalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Natural Science Foundation of China (No. 52071283) and Zhejiang Provincial Natural Science Foundation of China (No. Z24E010002) and Zhejiang Province High-level Talent Supporting Program (2022R52001).References
- I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev. Chem., 2017, 1, 0003 CrossRef CAS
.
- B. Chen, H. Miao, R. Hu, M. Yin, X. Wu, S. Sun, Q. Wang, S. Li and J. Yuan, J. Alloys Compd., 2021, 852, 157012 CrossRef CAS
- S. Sultan, M. Ha, D. Y. Kim, J. N. Tiwari, C. W. Myung, A. Meena, T. J. Shin, K. H. Chae and K. S. Kim, Nat. Commun., 2019, 10, 5195 CrossRef PubMed
- L. She, G. Zhao, T. Ma, J. Chen, W. Sun and H. Pan, Adv. Funct. Mater., 2022, 32, 2108465 CrossRef CAS
- S. S. Narwade, S. M. Mali, V. S. Sapner and B. R. Sathe, ACS Appl. Nano Mater., 2020, 3, 12288–12296 CrossRef CAS
- D. F. Abbott, R. K. Pittkowski, K. Macounová, R. Nebel, E. Marelli, E. Fabbri, I. E. Castelli, P. Krtil and T. J. Schmidt, ACS Appl. Mater. Interfaces, 2019, 11, 37748–37760 CrossRef CAS PubMed
- Y. Kumar, E. Kibena-Poldsepp, J. Kozlova, M. Rahn, A. Treshchalov, A. Kikas, V. Kisand, J. Aruvali, A. Tamm, J. C. Douglin, S. J. Folkman, I. Gelmetti, F. A. Garces-Pineda, J. R. Galan-Mascaros, D. R. Dekel and K. Tammeveski, ACS Appl. Mater. Interfaces, 2021, 13, 41507–41516 CrossRef CAS PubMed
- P. Bhanja, Y. Kim, B. Paul, Y. V. Kaneti, A. A. Alothman, A. Bhaumik and Y. Yamauchi, Chem. Eng. J., 2021, 405, 126803 CrossRef CAS
- P. Bhanja, Y. Kim, B. Paul, J. Lin, S. M. Alshehri, T. Ahamad, Y. V. Kaneti, A. Bhaumik and Y. Yamauch, ChemCatChem, 2020, 12, 2091–2096 CrossRef CAS
- Y. Guo, C. Zhang, J. Zhang, K. Dastafkan, K. Wang, C. Zhao and Z. Shi, ACS Sustainable Chem. Eng., 2021, 9, 2047–2056 CrossRef CAS
- H. Jung, A. Karmakar, A. Adhikari, R. Patel and S. Kundu, Sustainable Energy Fuels, 2022, 6, 640–663 RSC
- G. Wu, A. Santandreu, W. Kellogg, S. Gupta, O. Ogoke, H. Zhang, H.-L. Wang and L. Dai, Nano Energy, 2016, 29, 83–110 CrossRef CAS
- Y. Tang, R. Liu, S. Liu, B. Zheng, Y. Lu, R. Fu, D. Wu, M. Zhang and M. Rong, Carbon, 2019, 141, 704–711 CrossRef CAS
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed
- D. Wang, Q. Li, C. Han, Q. Lu, Z. Xing and X. Yang, Nat. Commun., 2019, 10, 3899 CrossRef PubMed
- M. Salzano de Luna, G. G. Buonocore, C. Giuliani, E. Messina, G. Di Carlo, M. Lavorgna, L. Ambrosio and G. M. Ingo, Angew. Chem., Int. Ed., 2018, 57, 7380–7384 CrossRef CAS PubMed
- H. Liang, J. Lin, H. Jia, S. Chen, J. Qi, J. Cao, T. Lin, W. Fei and J. Feng, J. Mater. Chem. A, 2018, 6, 15040–15046 RSC
- H. Hu, J. Liu, Z. Xu, L. Zhang, B. Cheng and W. Ho, Appl. Surf. Sci., 2019, 478, 981–990 CrossRef CAS
- M. Liu, K. A. Min, B. Han and L. Y. S. Lee, Adv. Energy Mater., 2021, 11, 2101281 CrossRef CAS
- H. You, D. Wu, D. Si, M. Cao, F. Sun, H. Zhang, H. Wang, T. F. Liu and R. Cao, J. Am. Chem. Soc., 2022, 21, 9245–9263 Search PubMed
- N. L. W. Septiani, Y. V. Kaneti, Y. Guo, B. Yuliarto, X. Jiang, Y. Ide, N. Nugraha, H. K. Dipojono, A. Yu, Y. Sugahara, D. Golberg and Y. Yamauchi, ChemSusChem, 2020, 13, 1645–1655 CrossRef CAS PubMed
- Y. Wang, M. Qiao, Y. Li and S. Wang, Small, 2018, 14, e1800136 CrossRef PubMed
- H. Han, K. M. Kim, J. H. Ryu, H. J. Lee, J. Woo, G. Ali, K. Y. Chung, T. Kim, S. Kang, S. Choi, J. Kwon, Y.-C. Chung, S. Mhin and T. Song, Nano Energy, 2020, 75, 104945 CrossRef CAS
- A. Khataee, T. Sadeghi Rad, S. Nikzat, A. Hassani, M. H. Aslan, M. Kobya and E. Demirbaş, Chem. Eng. J., 2019, 375, 122102 CrossRef CAS
- X. Long, J. Li, S. Xiao, K. Yan, Z. Wang, H. Chen and S. Yang, Angew. Chem., Int. Ed., 2014, 53, 7584–7588 CrossRef CAS PubMed
- M. Laipan, J. Yu, R. Zhu, J. Zhu, A. T. Smith, H. He, D. O'Hare and L. Sun, Mater. Horiz., 2020, 7, 715–745 RSC
- M. Gong, Y. Li, H. Wang, Y. Liang, J. Z. Wu, J. Zhou, J. Wang, T. Regier, F. Wei and H. Dai, J. Am. Chem. Soc., 2013, 135, 8452–8455 CrossRef CAS PubMed
- D. P. Sahoo, S. Nayak, K. H. Reddy, S. Martha and K. Parida, Inorg. Chem., 2018, 57, 3840–3854 CrossRef CAS PubMed
- D. Yue, X. Qian, M. Kan, M. Ren, Y. Zhu, L. Jiang and Y. Zhao, Appl. Catal., B, 2017, 209, 155–160 CrossRef CAS
- Y. Liu, R. Ge, Y. Chen, M. Huang, R. Zhu, W. Li, Y. Liu, L. Feng and R. Che, Chem. Eng. J., 2021, 420, 127598 CrossRef CAS
- D. Wu, H. Huang, Y. Zhou, Y. Liu and Z. Kang, J. Electroanal. Chem., 2019, 855, 113617 CrossRef CAS
- L. Tian, J. Wang, K. Wang, H. Wo, X. Wang, W. Zhuang, T. Li and X. Du, Carbon, 2019, 143, 457–466 CrossRef CAS
- D. Tang, J. Liu, X. Wu, R. Liu, X. Han, Y. Han, H. Huang, Y. Liu and Z. Kang, ACS Appl. Mater. Interfaces, 2014, 6, 7918–7925 CrossRef CAS PubMed
- J. Shin, J. Guo, T. Zhao and Z. Guo, Small, 2019, 15, e1900296 CrossRef PubMed
- J. Chakraborty, S. Sengupta, S. Dasgupta, M. Chakraborty, S. Ghosh, S. Mallik, K. L. Das and D. Basu, J. Ind. Eng. Chem., 2012, 18, 2211–2216 CrossRef CAS
- C. Depège, F.-Z. El Metoui, C. Forano, A. de Roy, J. Dupuis and J.-P. Besse, Chem. Mater., 1996, 8, 952–960 CrossRef
- T. Zhao, C. Liu, T. Meng, W. Deng, L. Zheng, F. Yi, A. Gao and D. Shu, Small, 2022, 18, e2201286 CrossRef PubMed
- R. Boppella, C. H. Choi, J. Moon and D. Ha Kim, Appl. Catal., B, 2018, 239, 178–186 CrossRef CAS
- W. Kong, Z. Xing, B. Fang, Y. Cui, Z. Li and W. Zhou, Appl. Catal., B, 2022, 304, 120969 CrossRef CAS
- H.-Y. Wang, Y.-Y. Hsu, R. Chen, T.-S. Chan, H. M. Chen and B. Liu, Adv. Energy Mater., 2015, 5, 1500091 CrossRef
- Y. Lin, H. Wang, C. K. Peng, L. Bu, C. L. Chiang, K. Tian, Y. Zhao, J. Zhao, Y. G. Lin, J. M. Lee and L. Gao, Small, 2020, 16, e2002426 CrossRef PubMed
- Y. Ma, K. Wang, Y. Chen, X. Yang, S. Zhao, K. Xi, S. Xie, S. Ding and C. Xiao, Int. J. Hydrogen Energy, 2019, 44, 20085–20092 CrossRef CAS
- Q. Wen, S. Wang, R. Wang, D. Huang, J. Fang, Y. Liu and T. Zhai, Nano Res., 2023, 16, 2286–2293 CrossRef CAS
- Y. Shi, J. Li, B. Zhang, S. Lv, T. Wang and X. Liu, Appl. Surf. Sci., 2021, 565, 150506 CrossRef CAS
- Y. Yang, W.-J. Wang, Y.-B. Yang, P.-F. Guo, B. Zhu, K. Wang, W.-T. Wang, Z.-H. He and Z.-T. Liu, J. Electrochem. Soc., 2022, 169, 024503 CrossRef CAS
- H. Yang, S. Luo, Y. Bao, Y. Luo, J. Jin and J. Ma, Inorg. Chem. Front., 2017, 4, 1173–1181 RSC
- X. Hou, J. Li, J. Zheng, L. Li and W. Chu, Dalton Trans., 2022, 51, 13970–13977 RSC
- Z. Hu, D. Zhang, C. Sun, C. Song and D. Wang, Electrochim. Acta, 2021, 391, 138932 CrossRef CAS
- L. Song, X. Zhang, X. Du, S. Zhu, Y. Xu and Y. Wang, Phys. Chem. Chem. Phys., 2022, 24, 24902–24909 RSC
- Q. Dong, C. Shuai, Z. Mo, Z. Liu, G. Liu, J. Wang, Y. Chen, W. Liu, N. Liu and R. Guo, New J. Chem., 2020, 44, 17744–17752 RSC
- F. Ning, M. Shao, S. Xu, Y. Fu, R. Zhang, M. Wei, D. G. Evans and X. Duan, Energy Environ. Sci., 2016, 9, 2633–2643 RSC
- W. Li, P. Da, Y. Zhang, Y. Wang, X. Lin, X. Gong and G. Zheng, ACS Nano, 2014, 8, 11770–11777 CrossRef CAS PubMed
- C. Hu, L. Zhang and J. Gong, Energy Environ. Sci., 2019, 12, 2620–2645 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cy01621h |
| This journal is © The Royal Society of Chemistry 2024 |