
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polyoxometalate (POM) boosting the light-harvesting ability of graphitic carbon nitride for efficient photocatalytic hydrogen production†
Simon Yves
Djoko T.
 a,
Estella
Njoyim T.
b,
Anh Dung
Nguyen
a,
Jin
Yang
c,
Hüseyin
Küçükkeçeci
c,
Edith Mawunya
Kutorglo
a,
Babu
Radhakrishnan
d,
Klaus
Schwarzburg
d,
Shahana
Huseyinova
a,
Prasenjit
Das
c,
Minoo
Tasbihi
a,
Michael
Schwarze
a,
Arne
Thomas
c and
Reinhard
Schomäcker
*a
a,
Estella
Njoyim T.
b,
Anh Dung
Nguyen
a,
Jin
Yang
c,
Hüseyin
Küçükkeçeci
c,
Edith Mawunya
Kutorglo
a,
Babu
Radhakrishnan
d,
Klaus
Schwarzburg
d,
Shahana
Huseyinova
a,
Prasenjit
Das
c,
Minoo
Tasbihi
a,
Michael
Schwarze
a,
Arne
Thomas
c and
Reinhard
Schomäcker
*a
aDepartment of Chemistry, Multiphase Reaction Engineering, Technische Universität Berlin, TC8, Straße des 17. Juni 124, 10623 Berlin, Germany. E-mail: schomaecker@tu-berlin.de
bHigher Teacher Training College, Department of Chemistry, University of Bamenda, P.O.Box 39 Bambili, Cameroon
cDepartment of Chemistry, Functional materials, Technische Universität Berlin, BA216 Hardenbergstr. 40, 10623 Berlin, Germany
dHelmholtz-Zentrum Berlin für Materialien und Energie, Hahn-Meitner-Platz 1, 14109 Berlin, Germany
First published on 23rd October 2023
Abstract
Phosphomolybdic acid (PMA) was used to achieve simultaneously P-doping and heterojunction construction of graphitic carbon nitride (gCN). P-gCN/PMAx composites were obtained via post-thermal annealing of bulk gCN/PMAx and characterized in detail using different techniques including XRD, XPS, SEM, and DRS. As an in situ hard templating agent and doping source, the loaded PMA helps to provide porous structured materials and reinforced electronic properties whereby many transitions are enhanced resulting in the improvement of light-harvesting. The electronic properties of gCN are improved strongly with the increased amount of PMA loaded, but the crystallinity becomes worse and the material becomes much more amorphous and disordered. The materials were investigated for the hydrogen evolution reaction (HER) showing the highest H2 evolution performance of 625 μmol g−1 h−1 for the P-gCN/PMA1.5 sample, which is almost 4 times higher than that of gCN with 167 μmol g−1 h−1. The P-gCN/PMA1.5 sample was investigated under long-term irradiation and in recycling tests indicating the good photocatalytic stability of this material. The apparent quantum efficiency (AQE) exhibited by P-gCN/PMA1.5 (0.7%) is 7 times higher than the AQE measured exhibited by gCN. This is evidence of the boosting effect exhibited by the loading of PMA onto the surface of gCN (0.1%). Thus, this study aims to offer a novel strategy to improve the activity of gCN by applying polyoxometalates as modifiers for better light harvesting.
1. Introduction
Environmental and energy issues are two major challenges nowadays. Energy needs are unavoidable to perpetuate sustainable development, but it is very important to use energy resources in strict compliance with environmental protection rules.1 Among existing energy resources, solar energy is one of the most reliable and clean energy sources because it is abundant, inexhaustible, environmentally friendly, and available worldwide.2,3 Solar energy does not need to be transported like fossil fuels, it only needs to be transformed and stored. Therefore, converting solar energy into storable and transportable chemical fuels remains a challenging issue. Based on plants' photosynthesis,3 hydrogen4 has been identified as a fuel of the future that could help to end the world's dependence on fossil fuels and allow for the transition to net-zero carbon emissions.2,5Light-driven photocatalytic water splitting has emerged and is now becoming a promising technology by which solar light harvesting could be enhanced and turned into hydrogen fuel avoiding CO2 pollution.6–8 The development of efficient photocatalysts exhibiting high light-absorption ability that improve the hydrogen evolution reaction (HER) through photocatalytic water splitting remains one of the most challenging aspects in this research field. Besides inorganic metallic semiconductors (e.g. TiO2, CdS, MoP) which have often been used so far to drive photochemical water splitting, metal-free polymeric semiconductors have been explored for the photocatalytic evolution of H2 or O2 from water with higher performance as compared to TiO2.9–11 The best known one is graphitic carbon nitride (gCN).12–14 This material has a lower bandgap energy (2.7 eV), and its catalytic properties and photocatalytic activity can be tuned by synthesis. The photocatalytic efficiency of pristine gCN still suffers from the fast recombination of its photogenerated electron/hole pairs, which directly limits the light-harvesting efficiency leading to a sluggish HER performance.14–16 Thus, providing a new modified gCN with improved photocatalytic activity would be a big achievement. To date, various strategies including metallic or non-metallic molecular dopants, creating fine-tuned nano-structural motifs by controlling morphologies (nanosheets, nanoparticles, nanotubes, etc.), coupling with other semiconductors, and construction of heterojunction structures, have been established to enhance the photocatalytic activity of g-CN.17,18 Due to its easier procedure, doping methods with metallic or non-metallic ionic dopants are frequently used. This is because they lead to increased conductivity due to higher concentrations of charge carriers.19 As a result, this improves the light absorption ability of gCN and, when combined with structural templating methods, the photocatalytic properties are strongly enhanced.
This study presents a new improvement strategy by combining three single steps in one to obtain a new structurally doped gCN exhibiting higher activity under visible light absorption for the HER from water splitting. It describes how a composite catalyst material is involved in a coupling strategy with gCN to obtain a final structured doped gCN material. Here, a polyoxometalate (POM) is used as a coupling material to improve the photocatalytic activity of gCN. This work focuses on highlighting a simple, new, efficient, and green strategy that consists of the incorporation of phosphomolybdic acid (PMA) into gCN giving further rise to the formation of novel nano-structured P-doped CN together with hetero-junction construction. This promising strategy might serve as a guideline and using other POMs, novel gCN-based nanomaterials with high solar-to-energy efficiency can be designed.
Polyoxometalates constitute a well-defined subset of metal–oxygen cluster anions with enormous structural characteristics and an unmatched range of physical and chemical properties. As a result, POMs can make a significant contribution to the material development for catalysis and photochemical reactions.20,21 It is reported that POMs are not only strong photo-oxidizing compounds but also strong Brønsted acids. Therefore, they can be easily applied in photochemical redox reactions under solar irradiation and they can also help to improve the conductivity of other materials.20,21 So far, there is little attention on the use of POMs as a coupling material in the synthesis of gCN-based composites. Thus, this manuscript aims also to offer a novel strategy by which POMs of the Keggin type could be used to improve the activity of gCN by retarding the fast recombination of electron/hole pairs. PMA is very interesting as an example of POM of the Keggin type suggesting that by incorporating it into 2D structured gCN results in structured P-doped gCN with better hydrogen evolution performance compared to gCN.
2. Experimental section
2.1 Synthesis of P-gCN/PMA
The synthesis of P-doped gCN consisted of three steps as shown in Fig. 1. At first, 2 g of gCN was mixed with a corresponding amount of PMA in a Teflon-lined stainless-steel autoclave using methanol and water in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, and the mixture was heated up to 180 ° C for 13–17 hours to obtain a yellow-to-brownish precipitate. Secondly, the precipitate was collected, carefully washed with methanol and water, and dried overnight at 60 ° C. The dried material was ground and labeled as bulk gCN/PMAx. For comparison, composites named gCN/PMAx using different ratios of gCN and PMA have been synthesized using the same synthesis procedure with x being 0.1, 0.2, 0.3, 0.4, 0.5, 1, 1.5, 2, and 2.5 g of PMA. The third and last step involves post-annealing of gCN/PMAx (450 °C, 2 h, HR = 5 C min−1), which leads directly to the P-doped gCN material labeled as P-gCN/PMAx. This annealing process results in the removal of the templating (molding) matrix and creates structural defects. A detailed experimental procedure of the synthesis is well described in section 1 of the ESI.†
:1 ratio, and the mixture was heated up to 180 ° C for 13–17 hours to obtain a yellow-to-brownish precipitate. Secondly, the precipitate was collected, carefully washed with methanol and water, and dried overnight at 60 ° C. The dried material was ground and labeled as bulk gCN/PMAx. For comparison, composites named gCN/PMAx using different ratios of gCN and PMA have been synthesized using the same synthesis procedure with x being 0.1, 0.2, 0.3, 0.4, 0.5, 1, 1.5, 2, and 2.5 g of PMA. The third and last step involves post-annealing of gCN/PMAx (450 °C, 2 h, HR = 5 C min−1), which leads directly to the P-doped gCN material labeled as P-gCN/PMAx. This annealing process results in the removal of the templating (molding) matrix and creates structural defects. A detailed experimental procedure of the synthesis is well described in section 1 of the ESI.†
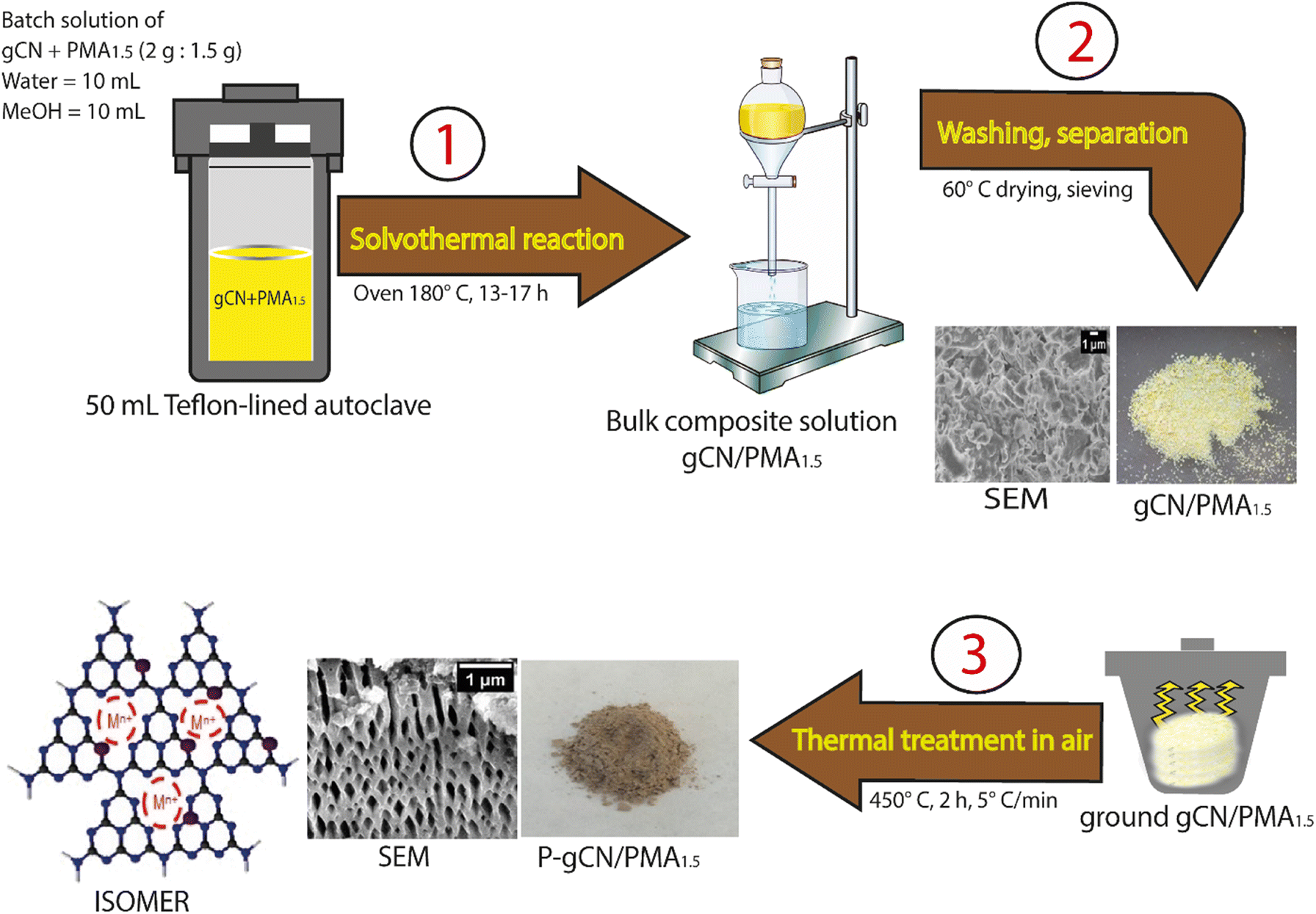 | ||
| Fig. 1 Schematic illustration of the synthesis pathway. | ||
2.2 Catalyst characterization
Scanning electron microscopy (SEM) images were collected from an InLens secondary electron detector (SE) with the help of a GeminiSEM-500 higher resolution scanning electron microscope (HR-SEM) from ZEISS company. N2 sorption measurements were performed on a volumetric sorption instrument (Autosorb-iQ-MP). Using the N2 adsorption isotherms, the specific surface area, pore size, and volume distribution of samples were investigated by Brunauer–Emmett–Teller and Barrett–Joyner–Halenda (BET/BJH) methods on a Quantachrome QuadraSorb S.I instrument at 77 K in nitrogen adsorption/desorption experiments. The optical properties of the samples have been investigated by solid-state UV-vis diffuse reflectance spectroscopy (DRS) with the help of a UV-vis spectrophotometer (Varian Cary 300) with a Ø 60 mm integrating sphere and BaSO4 as a reference standard. XPS measurements were carried out in an ion-pumped chamber (evacuated to 10–9 Torr) of a photoelectron spectrometer (Thermo Scientific) equipped with a Hemispheric 180° dual-focus analyzer with a 128-channel detector and with a focused X-ray source (Al Kα, hν = 1360 eV). The data were collected with an X-ray spot size of 400 μm, after performing 20 scans for the survey, 50 scans for the specific regions, and 100 scans for the valence band (VB) regions. Powder X-ray diffraction data were collected on a Bruker D8 Advance diffractometer in reflection geometry operating with a Cu Kα anode (λ = 1.54178 Å) operating at 40 kV and 40 mA. Samples were ground and mounted as loose powders onto a Si sample holder. PXRD patterns were collected from 2 to 80 2θ degrees with a step size of 0.02 degrees and an exposure time of 2 seconds per step. The Fourier transform infrared spectroscopy (FTIR) analyses of the samples were carried out on a Varian 640IR spectrometer equipped with an ATR cell. The separation efficiency and the re-combination of photogenerated electrons and holes were investigated by photoluminescence (PL) spectroscopy whose spectra of the samples were obtained using a Varian Cary Eclipse fluorometer with an excitation wavelength of 420 nm. An EG&G Princeton Applied Research potentiostat (Model 273 A) coupled with a WACOM Class AAA solar simulator (AM1.5, 100 mW cm−2) was used to perform the photo-electrochemical measurements (PEC) using a custom Teflon cell in a three-electrode configuration.222.3 Hydrogen evolution reaction
Hydrogen evolution reactions were carried out in a homemade side-irradiation quartz glass reactor having a total volume of Vo = 35 mL, which was connected to a thermostat that was set to 25 ° C for the water-circulating cooling system. In practice, 10 mg of the P-gCN/PMAx sample was transferred to the reactor and dispersed by 1 min sonication in 18 mL of a mixture of water and triethanolamine (used as a sacrificial agent) in a volume ratio of 16:2 mL (H2O/TEOA). Then 10 μL of H2PtCl6 aqueous solution (8 wt%) was added to the reactor as the source of the Pt co-catalyst. After sealing the reactor with rubber stoppers, the dispersion was flushed/degassed with argon for 15–20 minutes to remove air and to ensure that the reaction system was maintained under anaerobic conditions before irradiation. The irradiation was carried out with a 300 W Xe arc lamp through a 395 nm cut-off filter aiming to remain near visible light conditions. After the desired irradiation time, a gas sample was taken from the headspace, and H2 was quantified using a GC (Agilent 7820A) equipped with a thermal conductivity detector. The detailed experimental procedure is described in the ESI† together with the laboratory setup for the photocatalytic HER (Fig. S16†). The solar to hydrogen efficiency (STH) was calculated following an experimental procedure described in the ESI.†
3. Results and discussion
3.1 Physicochemical characterization of composite materials
 | ||
| Fig. 2 SEM images of P-gCN/PMAx composite materials: P-gCN/PMA0.1 (a), P-gCN/PMA0.2 (b), P-gCN/PMA0.3 (c), P-gCN/PMA0.4 (d), P-gCN/PMA0.5 (e), P-gCN/PMA1 (f), P-gCN/PMA1.5 (g), P-gCN/PMA2 (h), and P-gCN/PMA2.5 (i). | ||
As shown in Fig. 2a, the sample P-gCN/PMA0.1 consists of highly crystalline nanorods with multi-walled porous arrays and regular shapes obtained by self-stacking of numerous layered nanosheets. The formation of porous nanosheets can most likely be ascribed to the in situ hard-templating effect from where gas bubbles are taken out during the thermal treatment.23 As a result of post-thermal annealing, some molecules are taken out of the network leaving behind an ordered porous structure formed by rearrangement and hard weaving between atoms of layered nanosheets. The gradual introduction of PMA further contributes to a great improvement in crystallinity and geometric structure. This is shown in Fig. 2a–d, in which it could be observed that the more PMA in the precursor composite (gCN/PMAx), the more layered the porous nanosheets in the final material (P-gCN/PMAx). These nanosheets are highly self-stacked after rearrangement, forming extremely closed-packed 3D nanorods P-gCN/PMAx arrays with irregular shapes and less visible pores due to the crowding created during the stacking of many nanosheet layers. However, the crystallinity is being improved with the increase of PMA in the bulk solution, and P-gCN/PMA0.5 is found to be the most crystalline material. From P-gCN/PMA1 (Fig. 2f), the crystallinity becomes worse and the material becomes much more amorphous and disordered when the bulk composite solution is saturated with PMA. Moreover, the 3D nanorod arrays are gradually lost as shown in Fig. 2g–i. For additional comparison, Fig. S1 and S2 in the ESI† show the SEM images of bulk precursors gCN/PMAx and gCN, respectively. The physical aspect of gCN/PMAx looks almost the same whatever the saturation level of the bulk material with PMA. From TEM images (Fig. 3a), gCN (TEM 1) exhibits a geometrical shape that is less defined and less oriented than P-gCN/PMA1.5 (TEM 2). As compared to gCN, the loading of PMA during the solvothermal synthesis increases of course the FWHM of their corresponding XRD patterns in the final P-gCN/PMAx material resulting in a slightly smaller crystalline size but provides a well-defined 3D polymeric shape with a structure well oriented like nanorods. In Fig. 3b, the elemental mapping of sample P-gCN/PMA1.5 allows us to suggest that the P atoms are localized within the structure at areas nearby or into the electronic cloud of C and Mo atoms, suggesting a replacement of some C atoms or a doping connection with some of them. Additional information could be obtained from Fig. S23 to S28.†
 | ||
| Fig. 3 a) TEM images of gCN (TEM 1) and P-gCN/PMA1.5 (TEM 2) and b) elemental mapping of P-gCN/PMA1.5. | ||
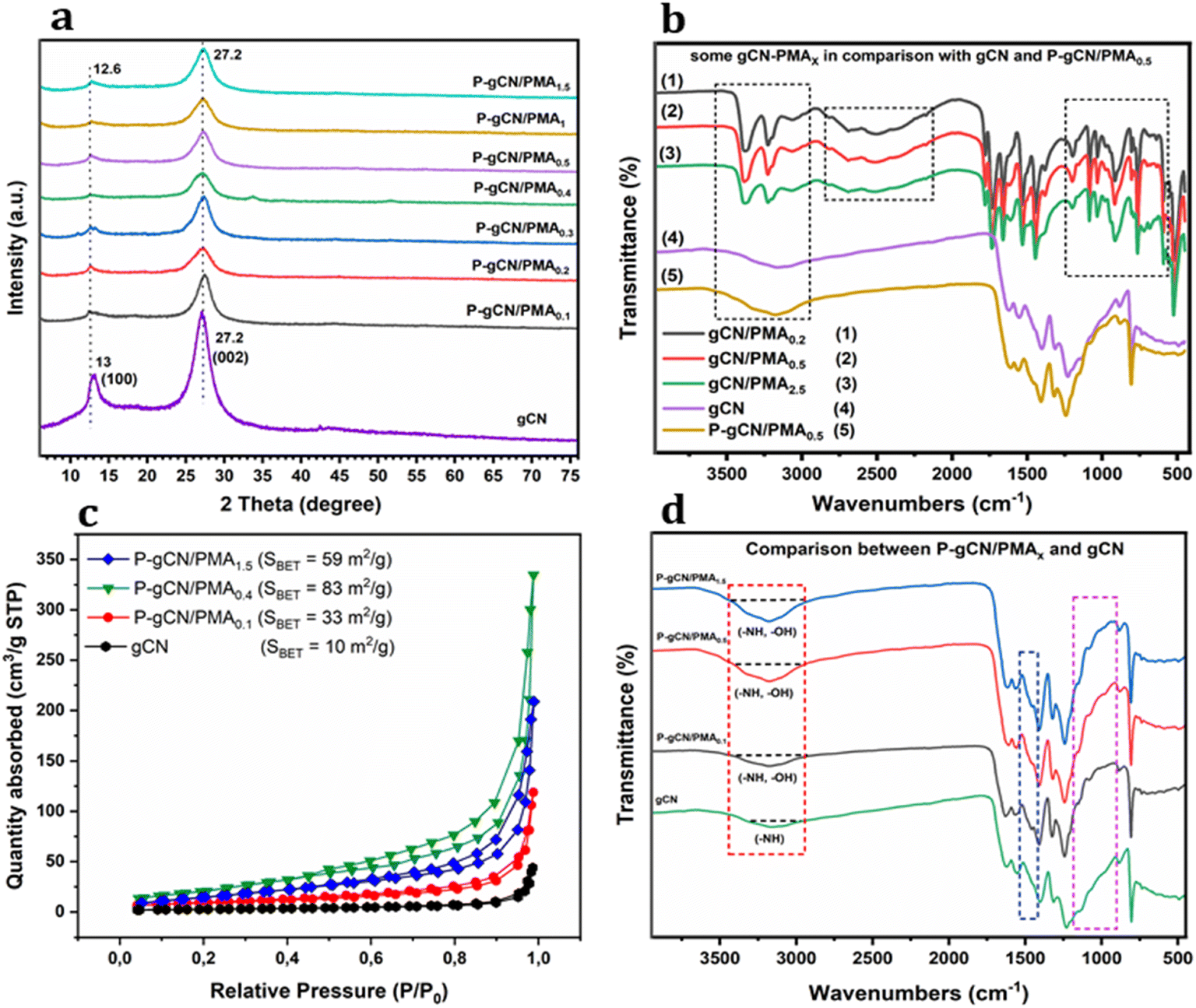 | ||
| Fig. 4 XRD patterns (a), FT-IR spectra (b and d), and N2 adsorption–desorption isotherms (c) of selected samples. | ||
The gCN XRD spectrum exhibits diffraction peaks at 2θ = 13° (1 0 0) and 27.2° (0 0 2) with the highest intensity implying its highest crystallinity among others. The (1 0 0) peak at 13° is assigned to the in-plane trigonal nitrogen linkage of tri-s-triazine motifs and the (0 0 2) peak at 27.2° corresponds to the stacking layer of conjugated aromatic rings.24–26 For P-gCN/PMAx samples (x = 0.1–1.5 g), the peaks at 27.2° of pure gCN are observed clearly but the one initially at 13° shifts a little to 12.6° which indicated that the loading with PMA maintains the basic structure of gCN. The slight shift of peak (1 0 0) from 13° to 12.6° means that the stacking distance between gCN layers is decreasing.33,43 These two peaks become less significant with increasing PMA concentration suggesting that the MO atom and some clusters are filled in the in-plane cavities of gCN.27–29 In addition, when the batch mixture is saturated with PMA during the solvothermal synthesis (x > 1.5 g), the XRD patterns of the corresponding P-gCN/PMAx come closer to that of PMA-450° (Fig. S3 in the ESI†). The increase of PMA concentration along with solvothermal synthesis does not affect the XRD patterns of all gCN/PMAx bulk composites (Fig. S4 in the ESI†), indicating that the loading equilibrium might be achieved so far for x < 1.5 g. Moreover, the diffraction peaks related to MO nanoparticles are not exhibited in P-gCN/PMAx XRD diffractograms (x < 1.5 g), which indicates that MO atoms are not chemically bonded to the gCN backbone but form a coordination bond with it.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CO) or to phosphate group atoms –P–NCO. During the post-thermal annealing of these bulk precursors, CO2 molecules are taken out from this group of atoms leaving behind a porous structure followed by rearrangement which replaces some C atoms with P atoms, hence the P-doping effect.
CO) or to phosphate group atoms –P–NCO. During the post-thermal annealing of these bulk precursors, CO2 molecules are taken out from this group of atoms leaving behind a porous structure followed by rearrangement which replaces some C atoms with P atoms, hence the P-doping effect.
After proceeding with the post-thermal treatment of gCN/PMAx at 450°, the P-gCN/PMAx composites (x = 0.1 to 1.5) are obtained and their IR spectra are almost similar to those of gCN only with small differences (Fig. 4d). The post-thermal treatment helps to finalize the intercalation process and to recover the polymeric backbone of gCN by taking some oxide molecules out of the network leaving behind an ordered porous structure. As depicted in Fig. 4b, P-gCN/PMAx exhibits a characteristic peak in its FTIR spectra around 810 cm−1 assigned to the bending vibration modes of tri-s-triazine units of the gCN.30–32 A series of peaks in the range from 1200 and 1700 cm−1 are assigned to the stretching vibration modes of the aromatic CN from heterocyclic units.33,34 The most interesting observation in this range is the appearance of several strong and wrinkled peaks showing the vibrations of the metal–NC2 moiety resulting from the intercalation of metal ions. These wrinkled peaks are exactly observed in the range from 800–1200 cm−1 and 1450–1600 cm−1, and might correspond to the bond vibrations of P–O and Mo–O.26,27
 | ||
| Fig. 5 UV-vis diffuse absorption spectra (a), band gap energy of P-gCN/PMA1.5 (b), transient photocurrent responses (c), XPS valence band spectra (d), steady-state PL spectra, (e) and time-resolved transient PL decay spectra (f). | ||
Fig. 5b shows the plots of 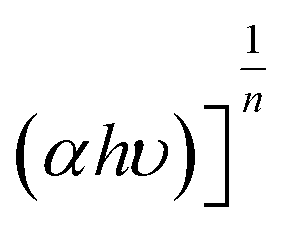 versus hυ with n = 0.5 for indirect transitions. Based on the existing literature, photocatalysts with narrow bandgap energies are highly desired in the photocatalytic water splitting process to ease photoactivation and retain a moderately large overpotential level. This leads to an improvement in the separation of photogenerated electrons and inhibition of the electron–hole pair recombination, maintaining them for more redox reactions. Therefore, to achieve a narrow band gap, raising the VB-XPS position of the photocatalyst is the best option since it results in the lowering of its CB making it as negative as possible compared to the reduction potential of H+ to H2 (0 V vs. NHE at pH 0).38 In this work, we achieved the VB-XPS position raising only by increasing the amount of PMA resulting in the CB lowering. As illustrated in Fig. 5d and S9,† the increase of PMA in the bulk synthesis solution helps in the further post-thermal step to lower both the CB and VB position and reduce the bandgap energy as compared to gCN by providing additional optical and electronic properties to the final corresponding catalyst. The P-gCN/PMA1.5 sample, having its CB positions closer to the reduction potential of H+ to H2, is suspected to be the more photoactive material somewhat confirmed from the experimental part by its photocatalytic H2 evolution performances. The band energy decreased from 3.02 eV for P-gCN/PMA0.1 to 2.46 and 2.70 eV for P-gCN/PMA1 and P-gCN/PMA1.5 respectively changing the mode into indirect transition (Fig. S10 in the ESI†). This can be explained by the fact that, when increasing PMA in the solvothermal step, we provide enough P atoms, which in turn possess the required electronic energy to be engaged in the P-doping mechanism in competition with the O-doping mechanism. The O2P orbital becomes less dominant as a result; we assume that the cooperation between P-doping and O-doping gives rise to possible in situ heterojunction constructions in the final material. This results in the improvement of photogenerated charge separation efficiency and effectively reduces the recombination efficiency of the photogenerated electrons and holes.19 The change in transition mode from the direct to indirect band gap is proof that the P-doping effect needs to be achieved at a higher amount of PMA loaded.
versus hυ with n = 0.5 for indirect transitions. Based on the existing literature, photocatalysts with narrow bandgap energies are highly desired in the photocatalytic water splitting process to ease photoactivation and retain a moderately large overpotential level. This leads to an improvement in the separation of photogenerated electrons and inhibition of the electron–hole pair recombination, maintaining them for more redox reactions. Therefore, to achieve a narrow band gap, raising the VB-XPS position of the photocatalyst is the best option since it results in the lowering of its CB making it as negative as possible compared to the reduction potential of H+ to H2 (0 V vs. NHE at pH 0).38 In this work, we achieved the VB-XPS position raising only by increasing the amount of PMA resulting in the CB lowering. As illustrated in Fig. 5d and S9,† the increase of PMA in the bulk synthesis solution helps in the further post-thermal step to lower both the CB and VB position and reduce the bandgap energy as compared to gCN by providing additional optical and electronic properties to the final corresponding catalyst. The P-gCN/PMA1.5 sample, having its CB positions closer to the reduction potential of H+ to H2, is suspected to be the more photoactive material somewhat confirmed from the experimental part by its photocatalytic H2 evolution performances. The band energy decreased from 3.02 eV for P-gCN/PMA0.1 to 2.46 and 2.70 eV for P-gCN/PMA1 and P-gCN/PMA1.5 respectively changing the mode into indirect transition (Fig. S10 in the ESI†). This can be explained by the fact that, when increasing PMA in the solvothermal step, we provide enough P atoms, which in turn possess the required electronic energy to be engaged in the P-doping mechanism in competition with the O-doping mechanism. The O2P orbital becomes less dominant as a result; we assume that the cooperation between P-doping and O-doping gives rise to possible in situ heterojunction constructions in the final material. This results in the improvement of photogenerated charge separation efficiency and effectively reduces the recombination efficiency of the photogenerated electrons and holes.19 The change in transition mode from the direct to indirect band gap is proof that the P-doping effect needs to be achieved at a higher amount of PMA loaded.
To obtain better insight into the electronic behavior of the final photocatalyst, the following discussions are based on the P-gCN/PMA1.5 sample compared with the bulk gCN/PMA1.5 composite precursor and gCN framework as reference materials. As mentioned in Fig. 6c, (Fig. S13 in the ESI†) the high-resolution C 1s spectrum of the P-gCN/PMA1.5 sample mainly revealed two wide peaks as well as the C1s spectrum of gCN (Fig. 6a) which are attributed to different types of chemical interactions of carbons within the whole molecule. These peaks can be further deconvoluted into main associated peaks. The C 1s peaks of P-gCN/PMA1.5 (Fig. 6c) at 284.5 and 287.6 eV which are similar to that of gCN (Fig. 6a) correspond to surface adventitious carbon atoms (C–C) and sp2-hybridized C atoms (NC–N) bonded to N (which might probably also be bonded to P atoms) in an aromatic ring respectively. Similar observations have been already described.39 Furthermore, the C1s peak located at 288.1 eV could be ascribed to sp2-hybridized C atoms ((C)3–N) in the aromatic ring attached to the N group (–NHO or –NPH). The same peak appears at 288.6 eV in the C 1s spectrum of gCN but results in the sp2-hybridization of the C atom in the aromatic ring ((C)3–N) attached to –NH2 groups.40,41 The other C 1s peaks centered at 285.9 and 293 eV are indexed to oxygen-containing carbonaceous bonds corresponding mainly to C–O and –COO groups respectively. This might come from water molecules adsorbed, oxidation, or acidification of the milieu by POMs in the previous solvothermal step of the synthesis. The C 1s XPS spectrum of the gCN/PMA1.5 precursor depicted in Fig. 6b (Fig. S12 in the ESI†) unveiled a peak at 287.8 eV and a shoulder peak at 289.2 eV revealing strong oxidized carbon bonds by intermediates molecules which might be probably ascribed to MoOx, P2Ox or POx molecules. These molecules are responsible for the upshift in the binding energy and further, the post-thermal treatment of this bulk precursor leaves behind a well-modified gCN called P-gCN/PMA1.5. The change in the chemical state of P-gCN/PMA1.5 is due to the phosphorus doping element and a new coordination environment in which Moy+ metal ions are involved in the formation of strong electronic interactions, evidencing a charge transfer from MoOx to gCN. Similar observations have already been mentioned by previous works when using other metal oxides like TiO2, and Bi2O3 to improve the photocatalytic activity of gCN.42–44
 | ||
| Fig. 6 High-resolution deconvoluted XPS spectra of C 1s (a–c), N 1s (d–f), O 1s (g), Mo 3d (h), and P 2p (i) for the P-gCN/PMA1.5 catalyst in comparison with high-resolution deconvoluted XPS spectra of gCN/PMA1.5 and gCN. | ||
The N 1s XPS spectra of gCN (Fig. 6d) can be fitted into four characteristic peaks, whereas the deconvolution of the N 1s XPS spectrum of P-gCN/PMA1.5 exhibited five fitted peaks (Fig. 6f). This observation confirms the introduction of new electronic transition levels in which the electronic cloud of nitrogen is much in demand suggesting the reaction site where the heterojunction phenomenon might probably occur as well. N 1s deconvoluted XPS spectra of gCN (Fig. 6d) show peaks centered at 397.9, 398, 400, and 430.6 eV which are ascribed to sp2-hybridized N from the heterocycle bonded to carbon atoms (CN–N), tertiary N in the form of N(–C)3, uncondensed amino functional groups (NH2 or NH), and charging effects represented by a wide π→π* excitation band, respectively. These observations have been already reported in the literature.23,24 Similar peaks are reflected in the N 1s XPS spectra of P-gCN/PMA1.5 (Fig. 6f) but shifts of these peaks are also noticed and centered at 397.9, 398.2, 399.7, and 403.8 eV corresponding to the above-mentioned functional groups with a little change of their electronic environments. The N 1s BE at 398.2 eV of P-gCN/PMA1.5 is slightly different from that of gCN confirming that the gCN framework in the P-gCN/PMA1.5 is kept; the triazine unit remains the building block that drives the polycondensation growthof the polymer chains providing the final chemical structure of the synthesized materials. Other peaks (399.7 eV, 400.8 eV) exhibited by P-gCN/PMA1.5 have higher BE than their corresponding XPS peaks exhibited in gCN (398 eV, 400 eV) confirming the presence of new electronic environments that influence the sp3-hybridized N suggesting the presence of P-bonded atoms from which tertiary C atoms and amino functional groups (quaternary N) might be substituted giving rise to the P-doping effect or bonded giving rise to NPH or N2P bonds. Moreover, a new BE is exhibited in the N 1s XPS of P-gCN/PMA1.5 with a peak fitted at 403.8 eV that could presumably be attributed to sp2-hybridized N (from the aromatic ring) bonded by coordination to Mo containing species (MoOx, MoyOx, MoOH, etc.) or sp3-hybridized N bonded to O containing trace species (POx, P2Ox, etc.). The N 1s XPS spectra of the gCN/PMA1.5 precursor depicted in Fig. 6e (Fig. S12 in the ESI†) confirm that after the solvothermal preparation, the gCN matrix is completely broken and lost so that only the post-thermal treatment can help to resurrect this backbone.
It is well known from the literature that the XPS survey spectrum of gCN does not exhibit oxygen atoms except the ones coming from water molecules adsorbed on the surface.24 But regarding the high-resolution O 1s XPS spectra of P-gCN/PMA1.5 depicted in Fig. 6g (Fig. S13 in the ESI†), the O peak intensity allows us to demonstrate that the occurrence of O does not only come from water molecule adsorption but mostly from the clustering molecule PMA (POMs) used in this synthesis as a doping and coupling reagent (starting molecule). Looking at O 1s deconvolution XPS spectra depicted in Fig. 6g, the sample P-gCN/PMA1.5 exhibits four oxygen characteristic peaks located at 525, 527.7, 528, and 529 eV which are identified as metal oxides (Mo(IV)), metal hydroxyl groups (Mo(IV)) from lattice oxygen, surface adsorbed oxygen (from water molecules), and phosphate oxide PO4−, respectively, which is closer to something already observed in the literature.26,27 However, these O 1s peaks exhibit a different BE than somewhat already observed in many Mo and P-modified gCN compounds.26,45,46 These differences could be explained by an electronic environment strongly influenced by Mo oxides and P oxides. The stronger O 1s peak at 529 eV in P-gCN/PMA1.5 could be attributed to the interstitial doping phenomenon caused by possible PO4− and the larger peak at 528 eV indicates the occurrence of more OH groups in the lattice. The up-shift (0.3 eV) for tertiary nitrogen peaks of P-gCN/PMA1.5 (399.7 eV) compared to the reference value (gCN-399.4 eV) is explained by the strong interactions of N atoms with interstitial doped PO4− and the possibility of coordination bonds with metal oxides and metal hydroxides.
The XPS deconvolution spectrum of the Mo 3d core-level indicates four exhibited peaks as depicted in Fig. 6h. These exhibited peaks are described as two doublets because of the low spin–orbit splitting (3.2 eV) among Mo 3d core excitation levels (3/2 and 5/2) as already mentioned in the literature.27,28 As displayed in this figure, the first XPS Mo 3d doublet centered at 231 and 234 eV is identified as Mo 3d5/2 and Mo 3d3/2 of the metallic Mo3+ cation respectively that can be attributed to a possible formation of MoP species (even in very low concentration) and the former is considered as one of the active sites toward the efficient HER.27,28 The second XPS Mo 3d doublet located at 231.7 and 234.8 eV might be ascribed to Mo4+ connected with oxygen vacancies (oxide or hydroxide) for Mo 3d5/2 and Mo 3d3/2 respectively. In other words, Mo4+ can originate from surface-oxidized Mo species.27,46
With a surface composition of 0.61% wt (ICP-OES, Table S3 in the ESI†), P is successfully loaded in the gCN units. This is confirmed by the survey XPS spectra of P-gCN/PMA1.5 (Fig. S14 in the ESI†) even though not visible, this is identified in the P 2p high-resolution XPS spectra (Fig. 6i). The P 2p XPS deconvolution spectrum exhibited only one doublet peak centered at 132.3 and 133.2 eV for P 2p3/2 and P 2p1/2 respectively, confirming that P atoms are mainly involved in only two bonding relationships with another specific atom in the lattice. This doublet peak could be ascribed to phosphorus (metal phosphate) bonded to oxygen (P–O) and phosphorus bonded to nitrogen (P–N bonds) with a high oxidation state (+4).26,28 As reported in the literature, this observation suggests that phosphorus is interstitially loaded into the gCN mostly in the form of PO4− by substituting some C atoms in CN heterocycles.26 In other words, this result suggests that some P atoms are bonding to tertiary N atoms in the form of PO4−. The absence of C–P and the presence of a P–N signal normally suggest that there is a direct replacement of C atoms by P atoms in the tri-s-triazine framework of gCN.47 But this is difficult to argue if we consider PO4− species as a doping agent group. However, traces of P2O5 could also be an indicator of the phosphorus source in the lattice.28 Moreover, the XPS spectral pattern of Mo 3d shows many more important shearings at BE values lower than 134 eV. This helps us to validate the hypothesis that there would probably be weak Mo–P (metal phosphide) and P–C interactions.28
Considering the previously discussed data in addition to discussions in the ESI,† the experimental energy band structures corresponding to each sample were drawn. It could be easily observed that the gradual introduction of PMA into 2D gCN has little change in the VBM of samples although their VBM is lower than the VBM of the gCN framework. This situation implies that the incorporation of PMA into 2D gCN followed by post-thermal treatment results also in the lowering of its activation energy which helps to ease the photon absorption and increase its light absorption ability.26 The VBM shift (ΔVB) of −0.11 eV for the P-gCN/PMA1.5 sample suggests a stronger oxidation of photogenerated holes (Fig. 7). However, one can observe that the gradual incorporation of PMA into 2D gCN has a significant change in the CBM which gradually decreases with the increased fraction of PMA. Concerning the CBM of gCN, the CBM of P-gCN/PMA1.5 shifts by ΔCB = −0.28 eV near the hydrogen reduction potential (EVB (H+/H2) = −3.85 eV at pH = 10.7) and this shift results in the narrowing of the band gap making this material much more favorable for proton reduction. The lowering of the CB leads to an appropriate reduction of the band gap energy that matches suitably with the redox potential of water. Thus, P-gCN/PMA1.5 having the lowest CB value is revealed to possess the strongest reduction ability favorable for photocatalytic hydrogen production from water. Regarding the narrowing of the band gap energy, the easy recombination reactions of photogenerated species (electrons and holes) were strongly expected as already reported in the literature.48,49 But, regarding the incredible photocatalytic efficiency of synthesized samples that revealed fewer recombination reactions, and considering the decreasing of the band gap energy, it is believed that there is a mid-gap energy state and defects states induced by the P-doping effect and formed within the band gap that leads to heterojunction construction between 2D gCN and 2D MoyOx (from in situ PMA decomposition).50,51 These mid-gap states together with the heterojunction phenomenon facilitate the separation of carriers and the electron transfer and hence limit the recombination process of electrons and holes.19,38,49 The lowest CB is observed in the P-gCN/PMA1.5 sample making it more available to establish indirect transition with the intermediate mid-gap states and that results in the increase of lifetime species and the reduction of the recombination reaction. As mid-gap states are suggested to enable mediation in the excitation of electrons from the valence band edge to the conduction band edge, one can assume that a type-II heterojunction mechanism is more favorable than the direct Z-scheme for highly efficient water splitting regarding the band structure of the P-gCN/PMA1.5 sample.38,52
 | ||
| Fig. 7 Electronic band structures of selected samples. | ||
To investigate the transfer dynamics of the charge carriers under irradiation and the underlying mechanism explaining this emission quenching, time-resolved fluorescence decay spectra were recorded as shown in Fig. 5f. By fitting the TRPL decay spectra to a 3rd-order exponential function using the origin software, samples exhibited three distinct radiative lifetimes. Then, the average percentage of each sample was calculated from the fitted graph (Table 1), and their corresponding exciton average lifetime was calculated using the following equation:

| Samples | Components | Lifetimes (ns) | Relative percentage A2 (%) | X 2 | Average lifetime (ns) |
|---|---|---|---|---|---|
| gCN | τ 1 | 1.26 | 71.83 | 0.999 | 3.37 |
| τ 2 | 4.76 | 34.82 | |||
| τ 3 | 20.24 | 6.12 | |||
| P-gCN/PMA1.5 | τ 1 | 8.62 | 4.00 | 0.998 | 4.72 |
| τ 2 | 3.00 | 21.37 | |||
| τ 3 | 34.54 | 0.59 |
3.2 Application in photocatalytic H2 production
To estimate the light-harvesting ability of the P-gCN/PMAx samples, their activities have been evaluated through photocatalytic tests for hydrogen production (Fig. S16†). Hence, the intrinsic photocatalytic activities of the as-prepared samples were evaluated by monitoring the H2 evolution reaction (HER) via the water-splitting reaction under visible light irradiation. Fig. 8a displays hydrogen production using different P-gCN/PMAx samples. The increasing amount of PMA in the bulk solution during the solvothermal synthesis does not only affect the electronic structure of the final material but also affects the photocatalytic performance. The relative order of the photocatalytic HER activity is P-gCN/PMA0.1 < P-gCN/PMA0.2 < P-gCN/PMA0.3 < P-gCN/PMA0.4 < P-gCN/PMA0.5 < P-gCN/PMA1 < P-gCN/PMA1.5 > P-gCN/PMA2 > P-gCN/PMA2.5 which might be attributed to both the P-doping effect and heterojunction construction.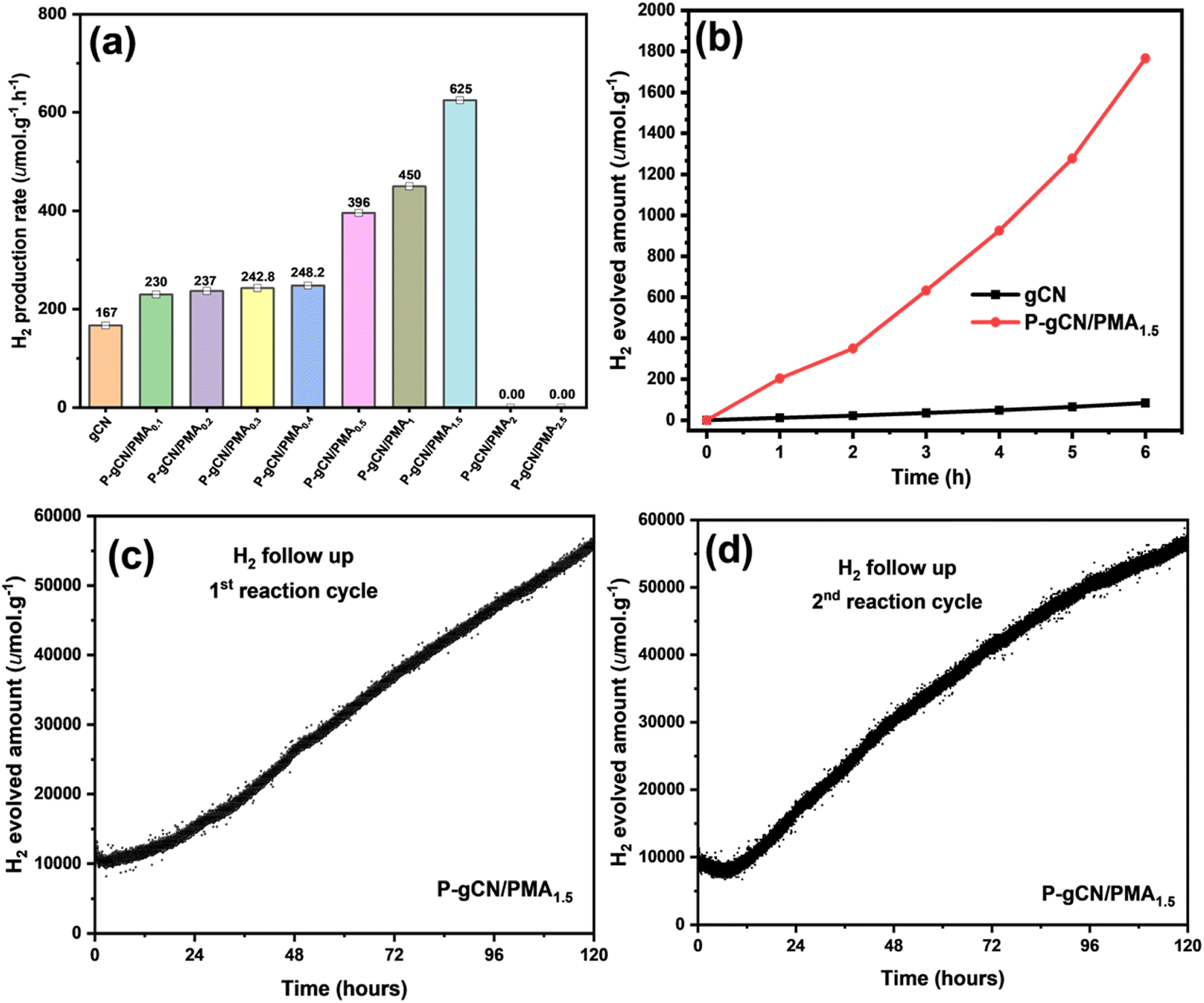 | ||
| Fig. 8 Photocatalytic sample screening through light-driven HER performance (a), time course of the HER using P-gCN/PMA1.5 and gCN under simulated light irradiation (b), and long-time photocatalytic reaction cycles with HER stability test (c and d). | ||
The optimal photocatalytic performance is obtained for the P-gCN/PMA1.5 sample that exhibits a strong hydrogen production with a rate of 625 μmol g−1 h−1, which is almost 4 times higher than that of the gCN backbone (167 μmol g−1 h−1). This is because the addition of 1.5 g of PMA to 2 g of gCN provides the required amount of P atoms for a successful P-doping effect during the post-thermal phase. As a result, the doping of carbon atoms in the graphitic carbon nitride framework not only enhances light absorption but also inhibits the recombination of charge carriers which consequently results in the modification of the electronic structure and therefore improvement of the photocatalytic performance. Such a P-doping effect has already been reported in the literature.24,28Fig. 8b shows the monitoring of H2 evolved, related to P-gCN/PMA1.5 photocatalytic activity over time. As soon as the light irradiation is switched on, the HER starts immediately but the H2 hardly evolves during the first 2 hours of irradiation. That is simply because, during the first 2 hours of irradiation, the in situ photodeposition of Pt (from H2PtCl6 as the precursor) is taking place, thus, the catalytic system is not yet completely ready to start the HER. Beyond 2 hours of irradiation, Pt photodeposition is finalized and the catalyst is active for the HER. Then, from the 3rd hour of irradiation, the HER is constant and the total amount of H2 increased continuously with an evolved amount of 1800 μmol g−1 after 6 hours of light irradiation. The total amount of H2 evolved using gCN under similar reaction conditions was only 97 μmol g−1 after 6 hours of irradiation. As the gCN performances are being improved by elements from the PMA precursor, P-gCN/PMA1.5 is considered a bi-component catalyst and exhibits the highest performance compared to catalysts from the same category (Table 2 and Fig. S29†). Looking at performances exhibited by multi-component catalysts (Table 2), the performances of P-gCN/PMA1.5 could be better improved with the addition of a 3rd precursor during the synthesis, somewhat will be investigated in the future. The stability of the catalyst was also supported by the XRD patterns of gCN and P-gCN/PMA1.5 before and after the photocatalytic HER provided in Fig. S31.†
| Catalysts | Number of components | Sacrificial agents TEOA (%) | H2 evolution rate (umol g−1 h−1) | Ref. |
|---|---|---|---|---|
| NiS/CdS/g-C3N4 | 3 | 20 | 3015 | 6 |
| Holey ultrathin g-C3N4 | 1 | 12 | 2860 | 55 |
| NiCo-MOF/ZIF | 2 | 15 | 4170 | 8 |
| Co@NC/g-C3N4-2 | 2 | 10 | 161 | 56 |
| C3N4–Pd–Cu2O | 2 | 10 | 32.5 | 57 |
| 4NiL/CN | 2 | 10 | 303.3 | 25 |
| 4Nil/NiOx/CN | 3 | 10 | 524.1 | 25 |
| Protonated imine-linked COFs | 2 | 10 | 20.7 | 58 |
| W-TEOS-CN | 3 | 10 | 326 | 16 |
| W-SNP-CN | 3 | 10 | 286 | 16 |
| P-gCN/PMA1.5 | 2 | 10 | 625 | This work |
As depicted in Fig. S17a,† the change of catalyst concentration has no significant effect on the photocatalytic HER performances both for gCN and P-gCN/PMA1.5. This confirms that the HER performances do not depend too much on the contact surface of light with the material catalyst but that they mainly depend on the electronic structure of the catalyst and the availability of reaction sites. The increase in temperature has a significant effect on both catalysts for HER performances. The temperature of 50 °C is regarded as the ideal temperature for optimal HER performances and might be appropriate for large-scale applications (Fig. S17b†).
Another operation mode that has been carried out to evaluate the performance range of the as-prepared P-gCN/PMA1.5 was the photocatalytic recycling test. The experimental procedure is described in detail in the ESI† (Fig. S16). This is usually done to ensure the stability of the photoactivity, to show the durability of the catalyst or the extension of the catalyst performance over the irradiation time, and to check whether this material could be a candidate for a large-scale application. Often, the catalyst durability test is performed by running up to three tests of photocatalytic reaction in relatively short irradiation times (3–6 hours) with the recycled catalyst from the previous reaction batch.47,53 However, it is believed that for short irradiation times, the durability test and the stability of photoactivity would not be reliable. In this work, the photoactivity stability test was done only with two tests of photocatalytic reactions with prolonged irradiation times of 120 hours for each test (Fig. 8c and d). The amount of H2 that evolved after the second test was about 56944 μmol g−1 and was higher than the amount of H2 that evolved after the first run (55620 μmol g−1). In the second run, the reaction started with Pt already loaded onto P-gCN/PMA1.5 implying that the catalytic system is much more improved in the second run than the first one. Further, during the first run, a large fraction of Pt nanoparticles from the photoreduction process are still in solution, having an impact on the light absorption. But, during the second run, those nanoparticles are not present and the catalytic system is improved for light absorption. P-gCN/PMA1.5 not only retains its photoactivity but also maintains the photocatalytic H2 production without being deactivated after two extended runs of 120 hours each, which revealed its stability and durability making it suitable for large-scale practical applications. gCN was not stable during the first extended run and it seems to be deactivated after 48 hours of irradiation during the first test (Fig. S18 in the ESI†). Further, the apparent quantum efficiency (AQE) at 415 nm was measured to be 0.7% for the sample P-gCN/PMA1.5 which is 7 times higher than the AQE measured for gCN. This is evidence for the boosting effect through loading PMA onto gCN. Details for the calculation of the AQE are given in the ESI.†
3.3 Proposed charge transfer mechanism of redox reactions on the P-gCN/PMA1.5 surface
The photocatalytic reaction mechanism is a designed concept that helps to materialize the light-initiated reactions through which the semiconductor photocatalyst, when irradiated by light with energy equal to or greater than its band gap energy, produces oxidizing and reducing entities. Thus, for photocatalytic water splitting, the reaction mechanism requires consideration of all the active sites in the whole catalytic system that are involved in charge separation and electron transfer. This allows the prediction of the reaction path and product formation based on the electronic transitions and the reactant molecules. Considering the band structure and structural composition of P-gCN/PMA1.5, there is a non-negligible fraction of gCN that remains unchanged and is assumed to be part of the catalytic system. The modified areas of gCN are represented by the sites where the doping process and the hetero-hybridization took place, symbolizing P-gCN/PMA1.5. Therefore, a catalytic system made up of a bi-component semi-conductor originating from gCN and molybdenum oxide provided by PMA would have been considered as the better prediction matching with our input. However since the second semiconductor component is not precisely identified, its band gap determination could not be provided and hence its band structure could not be described in the redox mechanism demonstrating the heterojunction configuration as mentioned above. Therefore, the doped gCN was assumed a bi-component and considered as P-gCN/PMA1.5 catalyst material. Considering platinum and TEOA in the catalytic system, the improvement of the photocatalytic mechanism of P-gCN/PMA1.5 is shown in Fig. 9. Both P-gCN/PMA1.5 and gCN can be initiated to generate photogenerated electron–hole pairs under light irradiation, but the initiation process is much easier in the P-gCN/PMA1.5 sample. In gCN, the recombination of electron/hole pairs is the promoted reaction, which is the reason for its weak HER performance. Since the CB potential of P-gCN/PMA1.5 is negative enough (potential vs. E(ev) NHE), the photoinduced electrons on the CB could be easily transferred to the reduction site (Pt) for hydrogen production. Finally, the accumulated electrons in the CB of P-gCN/PMA1.5 can be involved in the photochemical reduction of protons to generate hydrogen. Simultaneously, the holes in the VB of P-gCN/PMA1.5 are trapped by TEOA as scavengers. Together with the midgap state (ms), the occurrence of hetero-hybridization helps to prevent the recombination of electron–hole pairs, resulting in a remarkably enhanced photocatalytic activity, 4 times higher compared to pure gCN.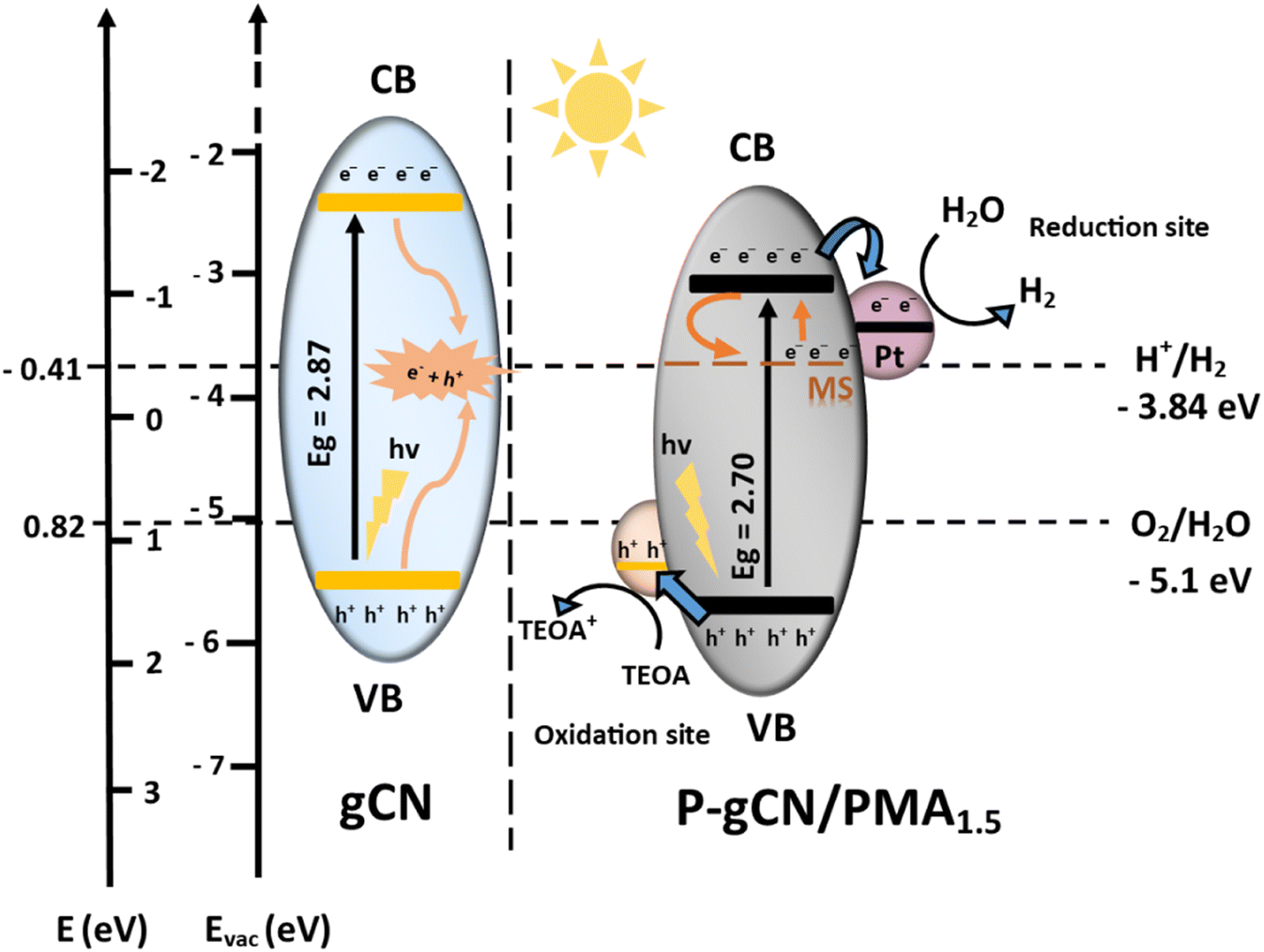 | ||
| Fig. 9 Proposed photocatalytic type II heterojunction mechanism of the as-designed catalyst material system for light-driven H2 evolution through water splitting. | ||
4. Conclusions
In summary, the development of new photocatalysts with efficient charge separation and an extended wavelength range for light absorption is the key in photocatalytic water splitting to enhance the sunlight harvesting process and convert it into a storable energy form. However, it is challenging for a single photocatalyst such as gCN to simultaneously exhibit all the required properties. To boost the photocatalytic activity, the structure of gCN was modified by introducing the phosphorus dopant element and heterojunction construction together with defect structures. This has been achieved by adding phosphomolybdic acid (PMA). The results revealed that there is a competition between the P-doping phenomenon and the heterojunction construction during the synthesis. At lower concentrations of PMA, the P-doping effect has a weak impact on the final structure. With 1.5 g of PMA loaded in the solvothermal step (gCN/PMA1.5), the obtained P-gCN/PMA1.5 catalyst exhibits the highest P-doping effect and hydrogen production through photocatalytic water splitting. PMA assumes three functions during the synthesis: a) as an in situ templating agent, it helps to provide porous structured materials; b) as a doping source, it helps to provide new and reinforced electronic properties whereby many transitions are enhanced resulting in the improvement of light-harvesting; and c) by the heterojunction configuration, PMA helps to introduce bridges between HOMO and LUMO levels to optimize the charge carrier transport. All these properties lead to a boost in the photocatalytic hydrogen production compared to pure gCN, and the H2 evolution rate increased by a factor of four to 625 μmol g−1 h−1 (gCN: 167 μmol g−1 h−1) with a stable photoactivity over 2 reaction cycles of 120 hours, making it valuable for large-scale applications. By using POMs as a new class of materials, this work provides a new concept for the design and fabrication of highly efficient catalysts that could be used for sustainable hydrogen production through water splitting or other light-harvesting technologies, but further optimization is needed to better meet today's energy challenges.Author contributions
Conceptualization: SYDT, RS, MS. Research project – writing and designing: SYDT, RS, ENT. Methodology: SYDT. Validation: AT, RS. Synthesis and characterization: SYDT. Preliminary analysis: SYDT, JY, ENT, HK. Photocatalytic experiments: SYDT, MS, AND, EMK. Investigations – further analysis: SYDT, MT, YZ, SH, EMK, BR, KS, PD. Early discussions: SYDT, RS, MS, AT, MT, JY, EMK, AND, ENT, BR, PD. Data curation: SYDT, MS, MT, EMK. Writing – original draft: SYDT. Writing – review and editing: SYDT, EMK, JY, ENT, MS, RS. Visualisation: RS, AT. Resources: RS, AT. Funding acquisition: RS. Supervision: RS. All authors have read, checked the English grammar, contributed to the discussions, and agreed to the published version of the manuscript.Conflicts of interest
The authors declare no competing interests.Acknowledgements
The authors acknowledge support from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC 2008/1 (UniSysCat) – 390540038. Simon Yves Djoko and Hüseyin Küçükkeçeci are grateful to Einstein Center of Catalysis/Berlin International Graduate School of Natural Sciences and Engineering (EC2/BIG-NSE) for the doctoral training program and financial support at the Technische Universität Berlin (EZ-2014-227). SYD gratefully acknowledges the contribution of the Elsa Neumann Stiftung. In addition, Simon also acknowledges the contributions of the University of Dschang in Cameroon for their initial training and assistance in the early stages of this research work. We also gratefully thank Ulrich Gernert, Christoph Fahrenson, Ron Justin Simke J., and Sören Selve from the ZELMI team of TU-Berlin for their assistance with the spectroscopy measurements and all the technical staff of our laboratories especially Gabriele Vetter, Maria Unterweger, and Christina Eichenauer. We thank Dr. Heinrich Vocke from the University of Oldenburg for the PL measurements. Yasmine Ziouani is acknowledged for her technical contribution to this work.References
- H. Kent Baker and J. R. Nofsinger, Socially Responsible Finance and Investing, John Wiley & Sons, Inc. USA, 2012, pp. 1–66 Search PubMed.
- V. Balzani, A. Credi and M. Venturi, ChemSusChem, 2008, 1, 26–58 CrossRef CAS PubMed.
- S. B. Agrawal, S. Singh and M. Agrawal, Int. J. Plant, Anim. Environ. Sci., 2015, 1, 43–56 CrossRef.
- P. E. V. de Miranda, in Science and Engineering of Hydrogen-Based Energy Technologies: Hydrogen Production and Practical Applications in Energy Generation, Elsevier, 2018, pp. 1–38 Search PubMed.
- I. McConnell, G. Li and G. W. Brudvig, Chem. Biol., 2010, 17, 434–447 CrossRef CAS.
- X. Wang, H. Jiang, M. Zhu and X. Shi, Chin. Chem. Lett., 2023, 34, 107683 CrossRef CAS.
- Z. Fan, X. Guo, Z. Jin, X. Li and Y. Li, Langmuir, 2022, 38, 3244–3256 CrossRef CAS.
- T. Li, Y. Li and Z. Jin, Inorg. Chem., 2022, 61, 12809–12821 CrossRef CAS PubMed.
- C. Li, J. Yang, P. Pachfule, S. Li, M. Y. Ye, J. Schmidt and A. Thomas, Nat. Commun., 2020, 11, 4712 CrossRef CAS.
- P. Pachfule, A. Acharjya, J. Roeser, R. P. Sivasankaran, M. Y. Ye, A. Brückner, J. Schmidt and A. Thomas, Chem. Sci., 2019, 10, 8316–8322 RSC.
- L. Gu, J. Wang, Z. Zou and X. Han, J. Hazard. Mater., 2014, 268, 216–223 CrossRef CAS PubMed.
- J. Liu, T. Zhang, Z. Wang, G. Dawson and W. Chen, J. Mater. Chem., 2011, 21, 14398–14401 RSC.
- M. Schröder, K. Kailasam, J. Borgmeyer, M. Neumann, A. Thomas, R. Schomäcker and M. Schwarze, Energy Technol., 2015, 3, 1014–1017 CrossRef.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- A. Thomas, A. Fischer, F. Goettmann, M. Antonietti, J. O. Müller, R. Schlögl and J. M. Carlsson, J. Mater. Chem., 2008, 18, 4893–4908 RSC.
- K. Kailasam, A. Fischer, G. Zhang, J. Zhang, M. Schwarze, M. Schröder, X. Wang, R. Schomäcker and A. Thomas, ChemSusChem, 2015, 8, 1404–1410 CrossRef CAS.
- R. Huang, J. Wu, M. Zhang, B. Liu, Z. Zheng and D. Luo, Mater. Des., 2021, 210, 110040 CrossRef CAS.
- S. Y. T. Djoko, H. Bashiri, E. T. Njoyim, M. Arabameri, S. Djepang, A. K. Tamo, S. Laminsi, M. Tasbihi, M. Schwarze and R. Schomäcker, J. Photochem. Photobiol., A, 2020, 398, 112596 CrossRef.
- L. Jiang, X. Yuan, Y. Pan, J. Liang, G. Zeng, Z. Wu and H. Wang, Appl. Catal., B, 2017, 217, 388–406 CrossRef CAS.
- G. Marcì, E. I. García-Lõpez and L. Palmisano, Eur. J. Inorg. Chem., 2014, 21–35 CrossRef.
- N. Al-zaqri, A. Alsalme, S. F. Adil, A. Alsaleh, S. G. Alshammari, S. I. Alresayes, R. Alotaibi, M. Al-Kinany and M. R. H. Siddiqui, J. Saudi Chem. Soc., 2017, 21, 965–973 CrossRef CAS.
- S. Gahlawat, P. Schnell, R. Irani, I. Y. Ahmet, L. Choubrac, S. Fiechter, P. P. Ingole and F. F. Abdi, Sol. RRL, 2022, 6, 2200129 CrossRef CAS.
- X. Kang, Y. Kang, X. Hong, Z. Sun, C. Zhen, C. Hu, G. Liu and H. Cheng, Prog. Nat. Sci.: Mater. Int., 2018, 28, 183–188 CrossRef CAS.
- L. Zhang, Z. Jin, H. Lu, T. Lin, S. Ruan, X. S. Zhao and Y. J. Zeng, ACS Omega, 2018, 3, 15009–15017 CrossRef CAS PubMed.
- Y. X. Zhang, S. Tang, W. De Zhang and Y. X. Yu, ACS Appl. Mater. Interfaces, 2019, 11, 14986–14996 CrossRef CAS.
- B. Liu, L. Ye, R. Wang, J. Yang, Y. Zhang, R. Guan, L. Tian and X. Chen, ACS Appl. Mater. Interfaces, 2018, 10, 4001–4009 CrossRef CAS.
- N. Iqbal, A. Afzal, I. Khan, M. S. Khan and A. Qurashi, Sci. Rep., 2021, 11, 16886 CrossRef CAS.
- J. S. Li, S. Zhang, J. Q. Sha, H. Wang, M. Z. Liu, L. X. Kong and G. D. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 17140–17146 CrossRef CAS.
- Y. Li, L. Cai, Q. Huang, J. Liu, R. Tang and W. Zhou, Nanoscale Res. Lett., 2020, 15, 3246 Search PubMed.
- H. B. Fang, Y. Luo, Y. Z. Zheng, W. Ma and X. Tao, Ind. Eng. Chem. Res., 2016, 55, 4506–4514 CrossRef CAS.
- K. Maeda, D. An, R. Kuriki, D. Lu and O. Ishitani, Beilstein J. Org. Chem., 2018, 14, 1806–1812 CrossRef CAS.
- A. Naseri, M. Samadi, A. Pourjavadi, A. Z. Moshfegh and S. Ramakrishna, J. Mater. Chem. A, 2017, 5, 23406–23433 RSC.
- M. R. Gholipour, F. Béland and T. O. Do, Int. J. Chem. React. Eng., 2016, 14, 851–858 CrossRef CAS.
- D. Huang, X. Yan, M. Yan, G. Zeng, C. Zhou, J. Wan, M. Cheng and W. Xue, ACS Appl. Mater. Interfaces, 2018, 10, 21035–21055 CrossRef CAS PubMed.
- A. Fujishima, X. Zhang and D. A. Tryk, Surf. Sci. Rep., 2008, 63, 515–582 CrossRef CAS.
- Q. Lin, L. Li, S. Liang, M. Liu, J. Bi and L. Wu, Appl. Catal., B, 2015, 163, 135–142 CrossRef CAS.
- H. Zhang and A. Yu, J. Phys. Chem. C, 2014, 118, 11628–11635 CrossRef CAS.
- B. J. Ng, L. K. Putri, X. Y. Kong, Y. W. Teh, P. Pasbakhsh and S. P. Chai, Adv. Sci., 2020, 7, 1903171 CrossRef CAS PubMed.
- A. N. Shafawi, R. A. Mahmud, K. Ahmed Ali, L. K. Putri, N. I. Md Rosli and A. R. Mohamed, J. Photochem. Photobiol., A, 2020, 389, 112289 CrossRef CAS.
- X. Wang, S. Blechert and M. Antonietti, ACS Catal., 2012, 2, 1596–1606 CrossRef CAS.
- X. Zhang, J. P. Veder, S. He and S. P. Jiang, Chem. Commun., 2019, 55, 1233–1236 RSC.
- J. Lei, B. Chen, W. Lv, L. Zhou, L. Wang, Y. Liu and J. Zhang, Dalton Trans., 2019, 48, 3486–3495 RSC.
- Y. Li, S. Wu, L. Huang, H. Xu, R. Zhang, M. Qu, Q. Gao and H. Li, J. Phys. Chem. Solids, 2015, 76, 112–119 CrossRef CAS.
- R. Boppella, S. T. Kochuveedu, H. Kim, M. J. Jeong, F. M. Mota, J. H. Park and D. H. Kim, ACS Appl. Mater. Interfaces, 2017, 9, 7075–7083 CrossRef CAS.
- G. Boopathy, M. Keerthi, S. M. Chen, S. Meenakshi and M. J. Umapathy, Mater. Chem. Phys., 2021, 269, 124735 CrossRef CAS.
- Z. Huang, J. Liu, S. Zong, X. Wang, K. Chen, L. Liu and Y. Fang, J. Colloid Interface Sci., 2022, 606, 848–859 CrossRef CAS PubMed.
- J. Y. Tang, D. Yang, W. G. Zhou, R. T. Guo, W. G. Pan and C. Y. Huang, J. Catal., 2019, 370, 79–87 CrossRef CAS.
- V. Loddo, M. Bellardita, G. Camera-Roda, F. Parrino and L. Palmisano, Heterogeneous Photocatalysis: A Promising Advanced Oxidation Process, Elsevier Inc., 2018 Search PubMed.
- J. Fu, J. Yu, C. Jiang and B. Cheng, Adv. Energy Mater., 2018, 8, 1701503 CrossRef.
- B. N. Popov, in Corrosion Engineering, Elsevier, 2015, pp. 29–92 Search PubMed.
- P. Sarangapani, Y. Chu, J. Charles, G. Klimeck and T. Kubis, Phys. Rev. Appl., 2019, 12, 044045 CrossRef CAS.
- B. Y. Wu, Design of Water-Splitting Photocatalysts by First Principles Computations, PhD thesis, Massachusetts Institute of Technology, 2008 Search PubMed.
- P. Kumar, D. Laishram, R. K. Sharma, A. Vinu, J. Hu and M. G. Kibria, Chem. Mater., 2021, 33, 9012–9092 CrossRef CAS.
- F. Xu, C. R. Haughn, X. Ma, M. F. Doty and S. G. Cloutier, Appl. Phys. Lett., 2014, 104, 0511112 Search PubMed.
- Y. Li, R. Jin, Y. Xing, J. Li, S. Song, X. Liu, M. Li and R. Jin, Adv. Energy Mater., 2016, 1601273 CrossRef.
- Y. Li, S. Zhu, X. Kong, Y. Liang, Z. Li, S. Wu, C. Chang, S. Luo and Z. Cui, Environ. Res., 2021, 197, 111002 CrossRef CAS.
- W. Yin, L. Bai, Y. Zhu, S. Zhong, L. Zhao, Z. Li and S. Bai, ACS Appl. Mater. Interfaces, 2016, 8, 23133–23142 CrossRef CAS.
- J. Yang, A. Acharjya, M. Y. Ye, J. Rabeah, S. Li, Z. Kochovski, S. Youk, J. Roeser, J. Grüneberg, C. Penschke, M. Schwarze, T. Wang, Y. Lu, R. van de Krol, M. Oschatz, R. Schomäcker, P. Saalfrank and A. Thomas, Angew. Chem., 2021, 60, 19797–19803 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cy00496a |
| This journal is © The Royal Society of Chemistry 2024 |