
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bypassing the scaling relations in oxygen electrocatalysis with geometry-adaptive catalysts
Ritums
Cepitis
 a,
Vladislav
Ivaništšev
*a,
Jan
Rossmeisl
b and
Nadezda
Kongi
*a
a,
Vladislav
Ivaništšev
*a,
Jan
Rossmeisl
b and
Nadezda
Kongi
*a
aInstitute of Chemistry, University of Tartu, Ravila 14a, 50411 Tartu, Estonia. E-mail: vladislav.ivanistsev@ut.ee; nadezda.kongi@ut.ee
bDepartment of Chemistry, Center for High Entropy Alloy Catalysis, University of Copenhagen, Universitetsparken 5, 2100 Copenhagen, Denmark
First published on 8th March 2024
Abstract
This communication introduces the concept of geometry-adaptive electrocatalysis, where a catalyst adjusts its geometry during the reaction. A model system of metal–nitrogen–carbon (M–N–C) catalysts – the dual-atom site 2Co–N4 of variable curvature – proves the concept from the first principles. Density functional theory calculations show how cycling the curvature effect with a geometry adaptation bypasses the scaling relations. Thus, in theory, geometry-adaptive electrocatalysis offers a promising direction to address the current stagnation in the experimentally measured overpotential for oxygen evolution and reduction reactions. It also indicates the possibility of discovering the ideal oxygen electrocatalyst.
Introduction
The “holy grail” of electrocatalysis is the ideal catalyst concept – catalysts that accelerate “dream reactions”, such as oxygen evolution and reduction reactions (OER and ORR), using only the energy set by their thermodynamic potential. While the quest for better catalysts motivates much of electrocatalytic research, the ultimate goal of this journey is the discovery of the ideal catalysts. Such catalysts promise 100% efficient OER and ORR for sustainable economic development.The journey towards the ideal catalysts is theoretically and experimentally challenging due to a fundamental constraint – linear scaling relationships – tying adsorption energy values to the chemical nature, valence, and coordination of reaction intermediates.1,2 In oxygen electrocatalysis, the scaling relation dictates the relative adsorption energies of two key intermediates, OH and OOH.3,4 It is worth noting that in electrocatalysis theory, the adsorption energy (ΔG) serves as the primary descriptor.5 Ideally, the difference between ΔG for OH and OOH intermediates should be 2.46 eV.5 Yet, in computational practice, the OH–OOH scaling relation stands as: ΔGOOH ≈ ΔGOH + 3.20 eV.6 This implies that the OH and OOH intermediates cannot have the ideal ΔG difference.7 Furthermore, the deviation from the reaction thermodynamic potential on the ΔGOH scale follows the Sabatier principle to form activity volcanoes shown in Fig. 1(a).7 Specifically, the solid magenta line in Fig. 1(a) highlights the apex of the “associative mechanism” volcano defined by the OH–OOH scaling relation. The predictions from Fig. 1(a) align with experimental potential values in Fig. 1(b), where they converge towards the theoretical limit.
 | ||
| Fig. 1 (a) Overpotential volcano projected on the ΔGOH description. Triangles denote the DFT-calculated overpotentials for single-site M–N–C catalysts,8 whereas circles represent those for dual-site M–N–C with curvature.9 Dashed and dotted lines highlight the apex of these overpotential volcanoes. (b) Timeline with outstanding, experimentally measured ORR and OER potentials for both platinum-group metals and metal–carbon–nitrogen (M–N–C) catalysts. The selected potentials correspond to a current density of 10 mA cm−2 for OER and 3 mA cm−2 for ORR. Data obtained from ref. 10–33. | ||
Approaching the theoretical limit in oxygen electrocatalysis calls for creativity in exploring novel catalysts and mechanisms.34 Historically, platinum group metal catalysts have showcased superior activity, yet they raise valid concerns regarding sustainability and cost.35 Metal- and nitrogen-doped carbon (M–N–C) materials, known for their single-atom sites (M–Nx), have become a more practical alternative thanks to their high catalytic activity.36 Still, efforts to enhance the catalytic performance of both platinum group metal and M–N–C catalysts consistently encounter the constraint of the OH–OOH scaling relation, as illustrated in Fig. 1(b).
To implement the ideal catalyst, it is essential to find a way to circumvent the OH–OOH scaling relation.37 One strategy is to sidestep the associative mechanism (involving OOH) by introducing an adjacent second active site.38 In this scenario, electrocatalysis can proceed via a dissociative mechanism, substituting the OOH intermediate with O on one site and OH on the other.10,39 In competition between the associative and dissociative mechanisms, only a few model catalysts, like Fe2N6, Co2N6, and FeCoN6, favour the dissociative mechanism.40,41 The preference depends on the active site metal and coordination.42 Specifically for the 2Co–N4 model, the dissociative mechanism is preferred. This results in the OH–O/OH scaling marked with a dot-dashed green line in Fig. 1(a).9 The “dissociative mechanism” volcano suggests a feasible improvement in the ORR as well as enhanced bifunctionality, which is the difference between ORR and OER potentials for a given material. Another intriguing strategy involves altering the catalyst's geometry, as demonstrated by the geometry adaptation of Pt nanoparticles to vary the intermediates' adsorption energy.43
Combining these two strategies leads us to a handy model system that favours the dissociative pathway and can adapt its geometry through curving, as illustrated in the insets of Fig. 2(a). This system is denoted below as 2Co–N4, emphasising its nature as an M–N–C dual-atom site catalyst. A key advantage of the 2Co–N4 model is the linear dependence of energy on the squared curvature, i.e. inverse pore radius as shown in Fig. 2(a). In contrast, other geometry-adaptive catalysts, such as Pt nanoparticles, have a more complex energy–geometry relationship.44 As detailed below, linear dependence facilitates the modulation of individual adsorption energies, thereby circumventing the scaling relations in a highly controlled manner.
 | ||
| Fig. 2 (a) Dependence of OH and O/OH intermediate adsorption energy on curvature. The inset shows the states 1O/OH and 2OH depicting the adsorption of intermediates on the 2Co–N4 model with selected curvature. (b) Top-view counter-map of the overpotential volcano for oxygen electrocatalysis. Grey lines represent overpotential contours. Coloured dots correspond to the states O/OH (pink – 21.2 Å), 2OH (blue – 4.5 Å), and a geometry-adaptation between them (pink–blue gradient). | ||
Results
Adsorption free energies
The linear dependence in Fig. 2(a) gives rise to a new scaling relation for the dissociative mechanism: ΔGO/OH = 2.93ΔGOH + 0.70 eV,9 which constrains the predicted bifunctional overpotential to 0.62 V, surpassing the theoretical limit of 0.74 V for the associative mechanism.7,8For rigid catalysts, it can be generalised that any scaling relation results in a linear path on the corresponding activity volcano. Accordingly, each ΔGOH–ΔGO/OH point aligns within its uncertainty (see Model, methods, and FAIR data below) with the shaded line on the volcano's top view in Fig. 2(b). At a first glance, this linear scaling appears to corroborate numerous similar ΔGOOHvs. ΔGOH plots from existing literature,2,6–8,10,45 where each point signifies a catalyst with a distinct chemical composition. At a second glance, the 2Co–N4 model stands out. Unlike other scaling relations, in Fig. 2(b), all points share the same chemical composition of the 2Co–N4 model. Still, the variable curvature produces a continuous range of ΔGO/OH–ΔGOH points. Thus, the transition between points requires geometry adaptation of the 2Co–N4 model, while in previous studies this would signify an alchemical transformation. This unique characteristic leads to a compelling question: “Can geometry adaptation between states of various curvature pave a nonlinear path to the apex of the activity volcano?” Answering this hypothetical question leads to the concept of geometry-adaptive electrocatalysis with a promise to bypass the scaling relations.
Geometry-adaptive electrocatalysis
Bypassing the scaling relations requires two states. For reaching the ideal catalyst these states correspond to the ideal adsorption energies of 1.23 and 3.69 eV for OH and O/OH intermediates, respectively. In geometry-adaptive electrocatalysis on the 2Co–N4 model, these two states are possible and marked as green dashed lines in Fig. 2(a). For verification, we selected states 1O/OH and 2OH in Fig. 2(a) that closely match the ideal adsorption energies. The resulting overpotential is below 0.1 V for the geometry adaptation between these states, which is visualised as an adjoining half-filled marker to the starred ideal catalyst in Fig. 2(b).Such analysis focuses solely on thermodynamics and neglects barriers due to the reaction kinetics, surface coverage, reagents diffusion, the double layer structure, and ohmic resistance. Still, as the thermodynamics is the main limitation, it is inspiring to show that in principle, a thermodynamic overpotential of 0 V is achievable:
• Because, unlike rigid catalysts, geometry-adaptive catalysts can bypass the scaling relations;
• Specifically, through fine-tuning of the 2Co–N4 model;
• Generally, via analogous dynamic geometry-adaptation.
To draw an analogy from our findings, we have the following. While the quest for new rigid catalysts might be compared to climbing straight paths on the activity volcano, geometry-adaptive electrocatalysis offers a bypass. It is like entering a tunnel at one state, moving directly to the central axis of the volcano, elevating to its apex, then returning, and finally emerging at another state. Fig. 2(b) illustrates the geometry-adaptive path with green dotted lines on the top-view counter-map of the overpotential volcano for oxygen electrocatalysis. That path throughout the volcano is the essence of geometry-adaptive electrocatalysis.
The corresponding chemical cycle, including four combinations of intermediates and curvatures, is depicted in Fig. 3(a). This electrocatalytic cycle unites two reaction paths on the energy diagram in Fig. 3(b). Each path, on its own, suggests a modest catalyst, even though they incorporate states 1O/OH and 2OH with ideal adsorption energies. For instance, on the lower pink path, a significant kinetic barrier for the dissociation of O2 hinders both OER and ORR.9 Yet, uniting these paths into a single cycle allows us to achieve almost ideal overpotential at the cost of curving the model.
 | ||
| Fig. 3 (a) Chemical cycle for the geometry-adaptation between states 1O/OH, 1OH/OH, 2OH and 2* for the O and OH intermediates adsorption at the 2Co–N4 model of selected curvatures (1 and 2). Progressing clockwise leads to OER, whereas the counterclockwise direction results in ORR. (b) Free energy diagram at 1.23 V distinguishing between electrochemical steps (same colour) and mechanical steps (change in colour). Approximate barrier for dissociation step via proton-coupled electron transfer is shown schematically based on estimations for OOH dissociation.9 | ||
In our DFT calculations, the 2Co–N4 model is artificially curved under an assumption that, in reality, such geometry adaptation can be induced by external stimuli like electromagnetic radiation, temperature, electromagnetic fields, or pressure.46 With the employed DFT methods, we can not assess whether the substantial energy cost of 2.6 eV, required to curve the model from 21 Å to 4.5 Å, is reversible or includes additional overheads. Nevertheless, based on the results presented above and the discussion given below, we anticipate geometry adaptation to emerge as a new strategy for bypassing the scaling relations.
Pareto analysis
Reflections on the substantial curving energy prompted us to extend the analysis and consider the overpotential for all pairwise combinations of the curved models. The expanded analysis reveals a Pareto front, as depicted in Fig. 4(a), which visualises the trade-off between mechanical and electrochemical costs in geometry-adaptive oxygen electrocatalysis. Namely, for the 2Co–N4 model, improving its catalytic activity in terms of overpotential comes at the expense of increasing curvature energy. The slope of the Pareto front is influenced by the energy required to curve the 2Co–N4 model; as previously noted, the optimal overpotential is realised with a curvature energy of 2.6 eV. The Pareto analysis shows that, even for the 2Co–N4 model, there are promising candidates with reasonable curving energy and improved overpotential. | ||
| Fig. 4 (a) Pareto plot illustrating the trade-off between bifunctional overpotential (ηORR + ηOER) and curvature energy. (b) Approximate dependence of overall efficiency (of H2 production in an electrolyser and its oxidation in a fuel cell) on the bifunctional overpotential. | ||
To put the Pareto front in perspective, let us turn the reader's attention to the approximate dependence of the overall efficiency of H2 production in an electrolyzer, followed by its oxidation in a fuel cell, on the bifunctional overpotential in Fig. 4(b). One can see that even a modest curving energy of 0.5 eV can reduce overpotential by 0.2 V, which translates to a near doubling of the energy efficiency compared to the state-of-the-art rigid catalysts (from ∼25% to ∼40%). Therefore, even modest geometry-adaptive catalysts could significantly boost the efficiency of oxygen electrocatalysis. This enhancement would benefit devices that rely on the OER and ORR, such as hydrogen electrolysers, fuel cells, and certain metal-ion batteries. As a result, these electrochemical devices could become more potent competitors to existing technologies, including internal combustion engines and traditional metal-ion batteries.
Discussion
Comparison of catalysts
In the diverse landscape of electrocatalysts, a systematic classification emerges on the basis of two criteria – structural geometry adaptability and definiteness of the active sites. The former criterion divides catalysts into rigid and geometry-adaptive. As the name suggests, rigid catalysts retain their structure throughout electrocatalytic reactions. In contrast, geometry-adaptive catalysts tend to alter their geometry to optimise performance. The latter criterion categorises catalysts as either well-defined or stochastic. Well-defined catalysts possess active sites with precise atomic surroundings, whereas stochastic ones exhibit a degree of randomness in the atomic arrangement surrounding the active site.Examples of well-known model systems corresponding to real catalysts are:
• Well-defined rigid platinum group metals and oxides are widely used in electrocatalysis due to their exceptional electrocatalytic activity controlled through geometry and composition.47,48 Their structures, especially active sites, are well-defined and remain rigid during electrocatalytic reactions.
• Stochastic and rigid high-entropy alloys (HEAs) have a random atomic arrangement formed by mixing multiple principal elements.49 Their stochastic nature means that the surrounding atoms of a given active site are random, providing a high degree of variability in electrocatalytic activity.
• Well-defined geometry-adaptive Fe–Cu dual-atom site in cytochrome c oxidase (CcO) follows the dissociative mechanism while reducing oxygen to water.50,51 The difference in Fe–Cu distance in reduced and oxidised forms of the enzyme can be viewed as geometry adaptation.
• Stochastic geometry-adaptive M–N–C catalysts with dual-atom sites,52 exemplified by the 2Co–N4 model (in this work), uniquely combine design with inherent randomness in electrocatalysis. Their intrinsic curvature can facilitate geometry adaptation. This adaptability and random atom site distribution define their distinct catalytic character.
Together, these catalysts represent a spectrum of properties, from the randomness of HEAs to the precision of enzymes, each offering unique advantages and challenges in electrocatalysis. M–N–C materials with 2Co–N4 dual-atom sites emerge as a unique blend in this classification. These materials are inherently curved, relatively flexible, and have a stochastic distribution of catalytic sites.53–56 Each dual-atom site can potentially adopt geometry during reaction independently of others under external stimuli.46 However, mastering such geometry adaptation control (like in enzyme cofactors) is an open challenge. The presented 2Co–N4 model is just a first step in unravelling geometry-adaptive mechanisms in artificial catalysts.
Perspective on circumventing the scaling relations
Let us put the challenge of implementing geometry-adaptive electrocatalysis into a broader context of circumventing scaling relations. There are five general strategies:1. Tuning by varying the OH adsorption energy;57–59
2. Breaking by stabilising OOH relative to OH;1,7,60
3. Switching from the associative to the dissociative mechanism;61–65
4. Pushing by destabilising O relative to OH;9,61
5. Bypassing by completely decoupling the adsorption energies.
While tuning and breaking the OH–OOH scaling relation are well-established in current state-of-the-art,1,7,34,57–60,66 switching mechanisms represent the cutting edge of research, with confirmed experiments.62,64,65 To our knowledge, pushing the OH–O/OH scaling relation is computationally conceptualised,9,61 and awaits experimental verification in direct relation to research on switching. In this work, we propose the geometry adaptation concept as a candidate for the bypassing strategy.
The rhetorical question “How can this adaptation occur at a frequency matching catalytic turnover?” inspires us to look closer at the CcO enzyme. Regarding the turnover frequency, CcO surpasses all known artificial catalysts by orders of magnitude.38 The active site of CcO favours the dissociative mechanism and undergoes structural changes during catalysis.67–70 Therefore, we speculate that CcO follows geometry-adaptive catalysis and bypasses scaling relations. Due to the trade-off between costs for geometry adaptation and overpotential, CcO is not the ideal catalyst, yet it shows a record low compromise overpotential of 0.19 V.71
Although CcO biocatalysis, including switching configurations and pumping protons, is a marvel of complexity, it also highlights the limitations inherent in biological systems. Unlike biocatalysis, artificial electrocatalysis is not bound by the constraints of biological evolution and can utilise a broader range of chemical elements and create more diverse and exotic structures; it can also apply effects that are rare or non-existent in biological systems, such as electromagnetic fields, extreme pressures or temperatures. Moreover, human engineering can often surpass natural processes in terms of efficiency, scalability, and specificity.
For inspiration, consider the ferroelectric model, which demonstrates high-frequency switching between states during reaction.72 Besides, alternative models and mechanisms for geometry adaptation could sustain a higher frequency. For example, using rotational degrees of freedom in nanotubes and buckyballs (see Fig. 5) offers a less energy-intensive means of adjusting the active site geometry. In addition, controlling geometry using cis–trans photoisomerisation could induce localised curving around active sites in M–N–C-like materials.73 The possibilities are only limited by the laws of nature and human creativity. As a source of ideas, we recommend a recent review by Wang et al. with examples of geometry adaptations in electrocatalysts, highlighting many opportunities for further advancements in this field.74
 | ||
| Fig. 5 Visualisation of M–N–C single- and dual-atom site models within a graphene layer, nanotubes, and buckyballs. The dissociative oxygen electrocatalysis on these sites can be controlled by curving and rotating. | ||
Further considerations
This study builds upon the emphasis on the geometric effect in electrocatalysis, recently recognised as an independent and significant factor.75 On the one hand, models focusing on the geometric effect for single-atom site catalysts (in particular with variable curvature) help reach the apex of the “associative mechanism” activity volcano in Fig. 1(a). One of the pioneering works, by Xie et al. in 2018,76 demonstrated an energy–curvature relationship (like in Fig. 2) employing a model similar to 2Co–N4. More recent works on M–N–C- and metal phthalocyanine-decorated carbon nanotubes, as well as curved graphene, have predicted enhanced performance attributed to the curvature-induced strain effect.77–79 The strain effect was known for a long time and nowadays is used in modelling catalysts for oxygen evolution and reduction reactions,80,81 as it alters the OH–OOH scaling relation.82On the other hand, models of curved in-pore, slit-pore, biphenylene, difullerenes, dimicrocycles, and diporphyrins catalysts have been proposed for dual-atom site catalysts.9,10,39,61,83,84 All referred models, firstly, favour the dissociative mechanism and, secondly, can adjust geometry for specific intermediates. Therefore, such models can be considered candidates for geometry-adaptive oxygen electrocatalysis.
Notably, Wan et al. overviewed the fundamentals of electronic and geometric effects by comparing metal with dual-atom site catalysts to conclude: “the direct way to achieve this (higher intrinsic catalytic activity) is to design catalysts that go through a direct dissociative mechanism, bypassing the OOH intermediate”.10 Also highlighted are the main complication beyond thermodynamic analysis – the kinetic barriers.85,86 Specifically, the dissociative mechanism has a notable kinetic barrier for the O2 reduction via a proton-coupled electron transfer to O and OH intermediates.87 Using the Brønsted–Evans–Polanyi relation and data from the literature,9,10 we approximated the kinetic barrier as schematically shown in Fig. 3(b). Due to the near-optimum inter-site distance in the state 2, the barrier for both O2 and OOH dissociation is low. In particular, the geometry-adaptive mechanism in the 2Co–N4 model is adjustable by curving. To fully understand the kinetic aspects of proton-coupled electron transfer during dissociation, DFT-based molecular dynamics is essential,87,88 which, while beyond the scope of this study, would further reinforce the concept of geometry-adaptive electrocatalysis.
Conclusions
We have explored an approach to elevate the efficiency of oxygen electrocatalysis using a geometry-adaptive model system and density functional theory methods. We show that geometry adaptation helps to bypass the scaling relations by adjusting the geometry of the model system and adapting the geometry during the catalytic reaction.The introduced model system is a dual-atom site 2Co–N4 with a unique geometric effect – a linear dependence between geometry and the adsorption energy of reaction intermediates. In the example of the 2Co–N4 model, we highlight the difference between novel geometry-adaptive and classical rigid catalysts. Namely, the geometry adaptation during reactions can bypass the fundamental limitation due to scaling relations.
Our computational modelling of the 2Co–N4 model revealed several key insights:
• The geometric effect allows reaching ideal adsorption energies for reaction intermediates.
• Geometry adaption during oxygen evolution/reduction reactions allows to reach the ideal potential for oxygen electrocatalysis.
• The direction for improving geometry-adaptive catalysts towards optimal catalysts is given by the Pareto front – a trade-off between costs for geometry adaptation and overpotential (deviation from the thermodynamic potential).
With these insights, we hope to inspire further research on geometry-adaptive catalysts to discover the true ideal oxygen electrocatalysts – elevating to the apex of the activity volcano – and address the current lack of significant progress in the experimentally observed activities.
Model, methods, and FAIR data
The 2Co–N4 model is an M–N–C dual-atom site catalyst of a constant composition and variable curvature. Two distinct geometry states of the model, nearly planar and curved, are depicted in Fig. 2(a).DFT calculations for intermediates adsorbed on the 2Co–N4 model followed standard practices, as referenced in previous works.61,89 Spin-polarised DFT calculations were performed with the GPAW 22.8 and ASE 3.22.1 software using the revised Perdew–Burke–Ernzerhof (RPBE) functional.90–92 For optimisation and energy calculations, 3 × 1 × 1 k-point sampling, grid spacing of 0.18 Å was used. After curving, a 5 Å vacuum layer was added in all non-periodic directions. Only edge carbon atoms were constrained to define the curvature, while all other atoms, including intermediates, were fully optimised below 0.1 eV Å−1. The dissociative mechanism was the focus of this study:
| 2* + O2 + (H+ + e−) → *O + *OH | (1) |
| *O + *OH + (H+ + e−) → *OH + *OH | (2) |
| *O + * + (H+ + e−) → *OH + * | (3) |
| *OH + * + (H+ + e−) → 2* + H2O | (4) |
The free energy (G) of each intermediate was calculated as:
| G = E + ZPE + TS + S | (5) |
 | (6) |
| ΔGOH/OH = GOH/OH − G* + GH2 − 2GH2O − 2eU | (7) |
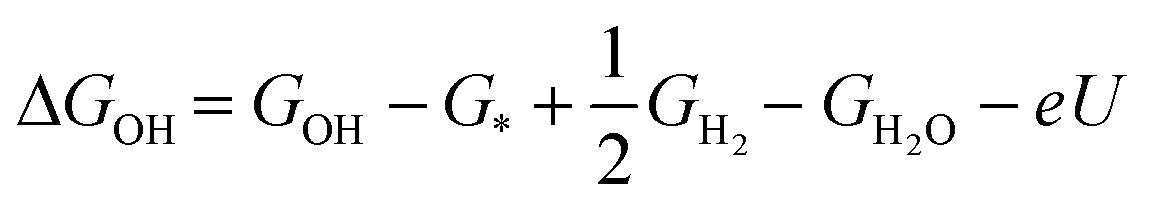 | (8) |
 with e being the elementary charge.5
with e being the elementary charge.5
The OER and ORR overpotential values were evaluated in terms of energy differences between reaction stages in Fig. 3:
| ηORR = 1.23 V − min(ΔG1−4)/e, ηOER = max(ΔG1−4)/e − 1.23 V | (9) |
FAIR data on the modelled structures, total energy values, and analysis scripts are accessible on the webpages https://chem.ku.dk/research_sections/nanochem/theoretical-electrocatalysis/ and https://nano.ku.dk/english/research/theoretical-electrocatalysis/katladb/geometry-adaptive-catalysis/.
Author contributions
R. Cepitis: conceptualisation, data curation, formal analysis, investigation, methodology, writing – original draft; V. Ivaništšev: conceptualisation, software, supervision, validation, visualisation, writing – review & editing; J. Rossmeisl: conceptualisation, resources, validation, visualisation; N. Kongi: supervision, funding acquisition, writing – review & editing, project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
V. I. and J. R. acknowledge the Danish National Research Foundation Centers of Excellence, The Center for High Entropy Alloys Catalysis (Project DNRF149), and the Independent Research Fund Denmark, grant no. 0217-00014B. V. I. receives funding from the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement no. 101031656. This research was also supported by the Estonian Research Council grant PSG250 and by the Estonian Ministry of Education and Research (TK210).Notes and references
- L. Li, K. Yuan and Y. Chen, Acc. Mater. Res., 2022, 3, 584–596 CrossRef CAS.
- M. M. Montemore and J. W. Medlin, Catal. Sci. Technol., 2014, 4, 3748–3761 RSC.
- I. C. Man, H.-Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS.
- V. Viswanathan, H. A. Hansen, J. Rossmeisl and J. K. Nørskov, ACS Catal., 2012, 2, 1654–1660 CrossRef CAS.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- S. Divanis, T. Kutlusoy, I. M. Ingmer Boye, I. C. Man and J. Rossmeisl, Chem. Sci., 2020, 11, 2943–2950 RSC.
- M. Busch, N. B. Halck, U. I. Kramm, S. Siahrostami, P. Krtil and J. Rossmeisl, Nano Energy, 2016, 29, 126–135 CrossRef CAS.
- F. Calle-Vallejo, J. I. Martínez and J. Rossmeisl, Phys. Chem. Chem. Phys., 2011, 13, 15639 RSC.
- R. Cepitis, N. Kongi, J. Rossmeisl and V. Ivaništšev, ACS Energy Lett., 2023, 8, 1330–1335 CrossRef CAS PubMed.
- H. Wan, A. W. Jensen, M. Escudero-Escribano and J. Rossmeisl, ACS Catal., 2020, 10, 5979–5989 CrossRef CAS.
- H. Tian, A. Song, P. Zhang, K. Sun, J. Wang, B. Sun, Q. Fan, G. Shao, C. Chen, H. Liu, Y. Li and G. Wang, Adv. Mater., 2023, 35, 2210714 CrossRef CAS PubMed.
- W. Yang, X. Liu, X. Yue, J. Jia and S. Guo, J. Am. Chem. Soc., 2015, 137, 1436–1439 CrossRef CAS PubMed.
- P. Yin, T. Yao, Y. Wu, L. Zheng, Y. Lin, W. Liu, H. Ju, J. Zhu, X. Hong, Z. Deng, G. Zhou, S. Wei and Y. Li, Angew. Chem., Int. Ed., 2016, 55, 10800–10805 CrossRef CAS PubMed.
- Y. Chen, S. Ji, Y. Wang, J. Dong, W. Chen, Z. Li, R. Shen, L. Zheng, Z. Zhuang, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2017, 56, 6937–6941 CrossRef CAS PubMed.
- R. Jiang, L. Li, T. Sheng, G. Hu, Y. Chen and L. Wang, J. Am. Chem. Soc., 2018, 140, 11594–11598 CrossRef CAS PubMed.
- C. Zhu, Q. Shi, B. Z. Xu, S. Fu, G. Wan, C. Yang, S. Yao, J. Song, H. Zhou, D. Du, S. P. Beckman, D. Su and Y. Lin, Adv. Energy Mater., 2018, 8, 1801956 CrossRef.
- J. Han, X. Meng, L. Lu, J. Bian, Z. Li and C. Sun, Adv. Funct. Mater., 2019, 29, 1808872 CrossRef CAS.
- Z. Yang, Y. Wang, M. Zhu, Z. Li, W. Chen, W. Wei, T. Yuan, Y. Qu, Q. Xu, C. Zhao, X. Wang, P. Li, Y. Li, Y. Wu and Y. Li, ACS Catal., 2019, 9, 2158–2163 CrossRef CAS.
- K. Chen, K. Liu, P. An, H. Li, Y. Lin, J. Hu, C. Jia, J. Fu, H. Li, H. Liu, Z. Lin, W. Li, J. Li, Y.-R. Lu, T.-S. Chan, N. Zhang and M. Liu, Nat. Commun., 2020, 11, 4173 CrossRef CAS PubMed.
- H. Shang, X. Zhou, J. Dong, A. Li, X. Zhao, Q. Liu, Y. Lin, J. Pei, Z. Li, Z. Jiang, D. Zhou, L. Zheng, Y. Wang, J. Zhou, Z. Yang, R. Cao, R. Sarangi, T. Sun, X. Yang, X. Zheng, W. Yan, Z. Zhuang, J. Li, W. Chen, D. Wang, J. Zhang and Y. Li, Nat. Commun., 2020, 11, 3049 CrossRef CAS PubMed.
- Y. Chen, R. Gao, S. Ji, H. Li, K. Tang, P. Jiang, H. Hu, Z. Zhang, H. Hao, Q. Qu, X. Liang, W. Chen, J. Dong, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 3212–3221 CrossRef CAS PubMed.
- X. Xie, L. Peng, H. Yang, G. I. N. Waterhouse, L. Shang and T. Zhang, Adv. Mater., 2021, 33, 2101038 CrossRef CAS PubMed.
- J. Han, H. Bao, J.-Q. Wang, L. Zheng, S. Sun, Z. L. Wang and C. Sun, Appl. Catal., B, 2021, 280, 119411 CrossRef CAS.
- A. Han, X. Wang, K. Tang, Z. Zhang, C. Ye, K. Kong, H. Hu, L. Zheng, P. Jiang, C. Zhao, Q. Zhang, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 19262–19271 CrossRef CAS PubMed.
- H. Karimi-Maleh, C. Karaman, O. Karaman, F. Karimi, Y. Vasseghian, L. Fu, M. Baghayeri, J. Rouhi, P. Senthil Kumar, P.-L. Show, S. Rajendran, A. L. Sanati and A. Mirabi, J. Nanostruct. Chem., 2022, 12, 429–439 CrossRef CAS.
- T. Cui, Y.-P. Wang, T. Ye, J. Wu, Z. Chen, J. Li, Y. Lei, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2022, 61, e202115219 CrossRef CAS PubMed.
- J. Woo, J. S. Lim, T. Lim, D. S. Baek, J. H. Kim, J. H. Lee, H. Y. Jeong, C. H. Choi and S. H. Joo, EES Catal., 2023, 1, 62–73 RSC.
- R. Li and D. Wang, Nano Res., 2022, 15, 6888–6923 CrossRef CAS.
- M.-T. Chen, Z.-X. Huang, X. Ye, L. Zhang, J.-J. Feng and A.-J. Wang, J. Colloid Interface Sci., 2023, 637, 216–224 CrossRef CAS PubMed.
- A. Bala Musa, M. Tabish, A. Kumar, M. Selvaraj, M. Abubaker Khan, B. M. Al-Shehri, M. Arif, M. Asim Mushtaq, S. Ibraheem, Y. Slimani, S. Ajmal, T. Anh Nguyen and G. Yasin, Chem. Eng. J., 2023, 451, 138684 CrossRef CAS.
- H. Chang, Y.-F. Guo, X. Liu, P.-F. Wang, Y. Xie and T.-F. Yi, Appl. Catal., B, 2023, 327, 122469 CrossRef CAS.
- J. Huo, X. Cao, Y. Tian, L. Li, J. Qu, Y. Xie, X. Nie, Y. Zhao, J. Zhang and H. Liu, Nanoscale, 2023, 15, 5448–5457 RSC.
- T. Tang, Y. Wang, J. Han, Q. Zhang, X. Bai, X. Niu, Z. Wang and J. Guan, Chin. J. Catal., 2023, 46, 48–55 CrossRef CAS.
- J. Pérez-Ramírez and N. López, Nat. Catal., 2019, 2, 971–976 CrossRef.
- S. Macchi, I. Denmark, T. Le, M. Forson, M. Bashiru, A. Jalihal and N. Siraj, Electrochemistry, 2022, 3, 1–27 CAS.
- Y. He, S. Liu, C. Priest, Q. Shi and G. Wu, Chem. Soc. Rev., 2020, 49, 3484–3524 RSC.
- A. Vojvodic and J. K. Nørskov, Natl. Sci. Rev., 2015, 2, 140–143 CrossRef CAS.
- A. Pedersen, J. Barrio, A. Li, R. Jervis, D. J. L. Brett, M. M. Titirici and I. E. L. Stephens, Adv. Energy Mater., 2022, 12, 2102715 CrossRef CAS.
- K. L. Svane, H. A. Hansen and T. Vegge, J. Catal., 2021, 393, 230–237 CrossRef CAS.
- P. Lv, W. Lv, D. Wu, G. Tang, X. Yan, Z. Lu and D. Ma, Phys. Rev. Appl., 2023, 19, 054094 CrossRef CAS.
- J. Liu, H. Xu, J. Zhu and D. Cheng, JACS Au, 2023, 3, 3031–3044 CrossRef CAS PubMed.
- M. Li, R. Lu, Y. Mao, Z. Hu and Z. Wang, J. Phys. Chem. C, 2024, 128, 1964–1970 CrossRef CAS.
- B. Zandkarimi and A. N. Alexandrova, J. Phys. Chem. Lett., 2019, 10, 460–467 CrossRef PubMed.
- J. Munarriz, Z. Zhang, P. Sautet and A. N. Alexandrova, ACS Catal., 2022, 12, 14517–14526 CrossRef CAS.
- O. Piqué, F. Illas and F. Calle-Vallejo, Phys. Chem. Chem. Phys., 2020, 22, 6797–6803 RSC.
- J. Park, A. Adhikary and H. R. Moon, Coord. Chem. Rev., 2023, 497, 215402 CrossRef CAS.
- J. Greeley, I. E. L. Stephens, A. S. Bondarenko, T. P. Johansson, H. A. Hansen, T. F. Jaramillo, J. Rossmeisl, I. Chorkendorff and J. K. Nørskov, Nat. Chem., 2009, 1, 552–556 CrossRef CAS PubMed.
- Q. Shi, C. Zhu, D. Du and Y. Lin, Chem. Soc. Rev., 2019, 48, 3181–3192 RSC.
- T. Löffler, A. Ludwig, J. Rossmeisl and W. Schuhmann, Angew. Chem., Int. Ed., 2021, 60, 26894–26903 CrossRef PubMed.
- S. Yoshikawa and A. Shimada, Chem. Rev., 2015, 115, 1936–1989 CrossRef CAS PubMed.
- I. Ishigami, R. G. Sierra, Z. Su, A. Peck, C. Wang, F. Poitevin, S. Lisova, B. Hayes, F. R. Moss, S. Boutet, R. E. Sublett, C. H. Yoon, S.-R. Yeh and D. L. Rousseau, Nat. Commun., 2023, 14, 5752 CrossRef CAS PubMed.
- J. Wang, C.-X. Zhao, J.-N. Liu, Y.-W. Song, J.-Q. Huang and B.-Q. Li, Nano Energy, 2022, 104, 107927 CrossRef CAS.
- X. Gong, J. Zhu, J. Li, R. Gao, Q. Zhou, Z. Zhang, H. Dou, L. Zhao, X. Sui, J. Cai, Y. Zhang, B. Liu, Y. Hu, A. Yu, S.-h. Sun, Z. Wang and Z. Chen, Adv. Funct. Mater., 2021, 31, 2008085 CrossRef CAS.
- H. Xu, D. Wang, P. Yang, L. Du, X. Lu, R. Li, L. Liu, J. Zhang and M. An, Appl. Catal., B, 2022, 305, 121040 CrossRef CAS.
- J. Y. Jung, S. Kim, J.-G. Kim, M. J. Kim, K.-S. Lee, Y.-E. Sung, P. Kim, S. J. Yoo, H.-K. Lim and N. D. Kim, Nano Energy, 2022, 97, 107206 CrossRef CAS.
- Y. Liu, Z. Chen, Z. Li, N. Zhao, Y. Xie, Y. Du, J. Xuan, D. Xiong, J. Zhou, L. Cai and Y. Yang, Nano Energy, 2022, 99, 107325 CrossRef CAS.
- M. Escudero-Escribano, P. Malacrida, M. H. Hansen, U. G. Vej-Hansen, A. Velázquez-Palenzuela, V. Tripkovic, J. Schiøtz, J. Rossmeisl, I. E. L. Stephens and I. Chorkendorff, Science, 2016, 352, 73–76 CrossRef CAS PubMed.
- L. Banko, O. A. Krysiak, J. K. Pedersen, B. Xiao, A. Savan, T. Löffler, S. Baha, J. Rossmeisl, W. Schuhmann and A. Ludwig, Adv. Energy Mater., 2022, 12, 2103312 CrossRef CAS.
- Z.-J. Zhao, S. Liu, S. Zha, D. Cheng, F. Studt, G. Henkelman and J. Gong, Nat. Rev. Mater., 2019, 4, 792–804 CrossRef.
- T. Sours, A. Patel, J. Nørskov, S. Siahrostami and A. Kulkarni, J. Phys. Chem. Lett., 2020, 11, 10029–10036 CrossRef CAS PubMed.
- H. Wan, T. M. Østergaard, L. Arnarson and J. Rossmeisl, ACS Sustainable Chem. Eng., 2019, 7, 611–617 CrossRef CAS.
- R. Gao, J. Wang, Z.-F. Huang, R. Zhang, W. Wang, L. Pan, J. Zhang, W. Zhu, X. Zhang, C. Shi, J. Lim and J.-J. Zou, Nat. Energy, 2021, 6, 614–623 CrossRef CAS.
- F. Liu, R. Gao, C. Shi, L. Pan, Z.-F. Huang, X. Zhang and J.-J. Zou, J. Am. Chem. Soc., 2023, 128(5), 1964–1970 CrossRef PubMed.
- H. Xia, R. Pang, X. Dong, Q. Liu, J. Chen, E. Wang and J. Li, J. Am. Chem. Soc., 2023, 145, 25695–25704 CrossRef CAS PubMed.
- Y. Xie, X. Chen, K. Sun, J. Zhang, W.-H. Lai, H. Liu and G. Wang, Angew. Chem., Int. Ed., 2023, 62, e202301833 CrossRef CAS PubMed.
- Z.-F. Huang, J. Song, S. Dou, X. Li, J. Wang and X. Wang, Matter, 2019, 1, 1494–1518 CrossRef.
- J. Liu, C. Hiser and S. Ferguson-Miller, Biochem. Soc. Trans., 2017, 45, 1087–1095 CrossRef CAS PubMed.
- I. Ishigami, A. Lewis-Ballester, A. Echelmeier, G. Brehm, N. A. Zatsepin, T. D. Grant, J. D. Coe, S. Lisova, G. Nelson, S. Zhang, Z. F. Dobson, S. Boutet, R. G. Sierra, A. Batyuk, P. Fromme, R. Fromme, J. C. H. Spence, A. Ros, S.-R. Yeh and D. L. Rousseau, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 3572–3577 CrossRef CAS PubMed.
- A. Shimada, F. Hara, K. Shinzawa-Itoh, N. Kanehisa, E. Yamashita, K. Muramoto, T. Tsukihara and S. Yoshikawa, J. Biol. Chem., 2021, 297, 100967 CrossRef CAS PubMed.
- M. Wikström, R. B. Gennis and P. R. Rich, Biochim. Biophys. Acta, Bioenerg., 2023, 1864, 148933 CrossRef PubMed.
- C. H. Kjaergaard, J. Rossmeisl and J. K. Nørskov, Inorg. Chem., 2010, 49, 3567–3572 CrossRef CAS PubMed.
- S. Jung, C. Pizzolitto, P. Biasi, P. J. Dauenhauer and T. Birol, Nat. Commun., 2023, 14, 7795 CrossRef CAS PubMed.
- E. Merino and M. Ribagorda, Beilstein J. Org. Chem., 2012, 8, 1071 CrossRef CAS PubMed.
- F. Wang, L. Xie, N. Sun, T. Zhi, M. Zhang, Y. Liu, Z. Luo, L. Yi, Q. Zhao and L. Wang, Nano-Micro Lett., 2023, 16, 32 CrossRef PubMed.
- H. Wen, Z. Zhao, Z. Luo and C. Wang, ChemSusChem, 2024, e202301859 CrossRef PubMed.
- Y. Xie, Z.-W. Wang, T.-Y. Zhu, D.-J. Shu, Z.-F. Hou and K. Terakura, Carbon, 2018, 139, 129–136 CrossRef CAS.
- J. Su, C. B. Musgrave, Y. Song, L. Huang, Y. Liu, G. Li, Y. Xin, P. Xiong, M. M.-J. Li, H. Wu, M. Zhu, H. M. Chen, J. Zhang, H. Shen, B. Z. Tang, M. Robert, W. A. Goddard and R. Ye, Nat. Catal., 2023, 6, 818–828 CrossRef CAS.
- X. Li, X. Wu, Y. Zhao, Y. Lin, J. Zhao, C. Wu, H. Liu, L. Shan, L. Yang, L. Song and J. Jiang, Adv. Mater., 2023, 35, 2302467 CrossRef CAS PubMed.
- T. Kropp and M. Mavrikakis, J. Catal., 2020, 390, 67–71 CrossRef CAS.
- Z. Hou, C. Cui, Y. Li, Y. Gao, D. Zhu, Y. Gu, G. Pan, Y. Zhu and T. Zhang, Adv. Mater., 2023, 35, 2209876 CrossRef CAS PubMed.
- A. Kulkarni, S. Siahrostami, A. Patel and J. K. Nørskov, Chem. Rev., 2018, 118, 2302–2312 CrossRef CAS PubMed.
- A. Khorshidi, J. Violet, J. Hashemi and A. A. Peterson, Nat. Catal., 2018, 1, 263–268 CrossRef.
- Z. Feng, M. Fang, R. Li, B. Ma, H. Wang, H. Ding, G. Su, K. Tao, Y. Tang and X. Dai, Int. J. Hydrogen Energy, 2022, 47, 36294–36305 CrossRef CAS.
- X. Li, S. Duan, E. Sharman, Y. Zhao, L. Yang, Z. Zhuo, P. Cui, J. Jiang and Y. Luo, J. Mater. Chem. A, 2020, 8, 10193–10198 RSC.
- H. Ooka, J. Huang and K. S. Exner, Front. Energy Res., 2021, 9, 654460 CrossRef.
- H. Ooka and R. Nakamura, J. Phys. Chem. Lett., 2019, 10, 6706–6713 CrossRef CAS PubMed.
- R. E. Warburton, A. V. Soudackov and S. Hammes-Schiffer, Chem. Rev., 2022, 122, 10599–10650 CrossRef CAS PubMed.
- A. Bouzid, P. Gono and A. Pasquarello, J. Catal., 2019, 375, 135–139 CrossRef CAS.
- R. Cepitis, N. Kongi, V. Grozovski, V. Ivanistsev and E. Lust, Catalysts, 2021, 11, 1165 CrossRef CAS.
- J. Enkovaara, C. Rostgaard, J. J. Mortensen, J. Chen, M. Dułak, L. Ferrighi, J. Gavnholt, C. Glinsvad, V. Haikola, H. A. Hansen, H. H. Kristoffersen, M. Kuisma, A. H. Larsen, L. Lehtovaara, M. Ljungberg, O. Lopez-Acevedo, P. G. Moses, J. Ojanen, T. Olsen, V. Petzold, N. A. Romero, J. Stausholm-Møller, M. Strange, G. A. Tritsaris, M. Vanin, M. Walter, B. Hammer, H. Häkkinen, G. K. H. Madsen, R. M. Nieminen, J. K. Nørskov, M. Puska, T. T. Rantala, J. Schiøtz, K. S. Thygesen and K. W. Jacobsen, J. Phys.: Condens. Matter, 2010, 22, 253202 CrossRef CAS PubMed.
- A. H. Larsen, J. J. Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Dulak, J. Friis, M. N. Groves, B. Hammer, C. Hargus, E. D. Hermes, P. C. Jennings, P. B. Jensen, J. Kermode, J. R. Kitchin, E. L. Kolsbjerg, J. Kubal, K. Kaasbjerg, S. Lysgaard, J. B. Maronsson, T. Maxson, T. Olsen, L. Pastewka, A. Peterson, C. Rostgaard, J. Schiøtz, O. Schütt, M. Strange, K. S. Thygesen, T. Vegge, L. Vilhelmsen, M. Walter, Z. Zeng and K. W. Jacobsen, J. Phys.: Condens. Matter, 2017, 29, 273002 CrossRef PubMed.
- B. Hammer, L. B. Hansen and J. K. Nørskov, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 7413–7421 CrossRef.
- E. Caldeweyher, S. Ehlert, A. Hansen, H. Neugebauer, S. Spicher, C. Bannwarth and S. Grimme, J. Chem. Phys., 2019, 150, 154122 CrossRef PubMed.
- J. J. Mortensen, K. Kaasbjerg, S. L. Frederiksen, J. K. Nørskov, J. P. Sethna and K. W. Jacobsen, Phys. Rev. Lett., 2005, 95, 216401 CrossRef CAS PubMed.
- J. Wellendorff, K. T. Lundgaard, A. Møgelhøj, V. Petzold, D. D. Landis, J. K. Nørskov, T. Bligaard and K. W. Jacobsen, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 235149 CrossRef.
| This journal is © The Royal Society of Chemistry 2024 |