
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Continuous flow oxidation of HMF using a supported AuPd-alloy†
Dominik
Neukum
 a,
Ajai R.
Lakshmi Nilayam
cd,
Maya E.
Ludwig
a,
Athanasios A.
Vadarlis
a,
Jan-Dierk
Grunwaldt
ab and
Erisa
Saraçi
*ab
a,
Ajai R.
Lakshmi Nilayam
cd,
Maya E.
Ludwig
a,
Athanasios A.
Vadarlis
a,
Jan-Dierk
Grunwaldt
ab and
Erisa
Saraçi
*ab
aInstitute of Catalysis Research and Technology, Karlsruhe Institute of Technology, Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany. E-mail: erisa.saraci@kit.edu
bInstitute for Chemical Technology and Polymer Chemistry, Karlsruhe Institute of Technology, Engesserstraße 20, 76131 Karlsruhe, Germany
cInstitute of Nanotechnology, Karlsruhe Institute of Technology, Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
dKarlsruhe Nano Micro Facility (KNMFi), Karlsruhe Institute of Technology, Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
First published on 28th February 2024
Abstract
The oxidation of 5-(hydroxymethyl)furfural (HMF) to 2,5-furandicarboxylic acid (FDCA) holds significant promise for replacing fossil-based monomers. Continuous flow operation enhances the process in terms of green chemistry by improving heat and mass transfer, enabling easier scalability of the reaction and ensuring higher safety with a smaller reactor volume. In this study, we investigated the use of heterogeneous catalysts in a fixed-bed reactor for the continuous oxidation of HMF. Air served as a green oxidant, water as a non-toxic solvent, and Na2CO3 as a mild base. An AuPd-alloy-based catalyst supported on activated carbon demonstrated remarkable performance, yielding 81% FDCA at a liquid hourly space velocity of 31.4 h−1. This corresponds to a productivity of 68 molFDCA molAuPd−1 h−1, which is, to our knowledge, one order of magnitude higher than typically reported for the heterogeneously catalyzed continuous oxidation of HMF. In addition, the catalyst showed a good stability over 90 h time on stream without any detectable deactivation. The formation of humins led to a progressive catalyst deactivation. The developed catalytic system and continuous process offer a more sustainable and efficient approach to future production of the renewable monomer FDCA.
Introduction
The production of renewable monomers, which are key products in the chemical industry, is of great interest to transition towards a bio-based chemical industry.1–3 The use of biomass brings a substantial opportunity, leveraging inherent functional groups that only necessitate simple transformation, instead of introducing such groups via oxidation of fossil-based hydrocarbons.3,4 The effective H/C-ratio of bio-derived base chemicals is closer to that of the desired monomer products, in comparison to their fossil-derived counterparts, reducing the amount of transformation steps and fostering more sustainable processes.3,5 The oxidation of 5-(hydroxymethyl)furfural (HMF), ideally sourced from lignocellulose or other biomass sources, to yield 2,5-furandicarboxylic acid (FDCA) emerges as a promising avenue for the production of renewable monomers.6–10 FDCA can be used to produce various polyesters, particularly as a substitute for fossil-derived terephthalic acid.11,12 The oxidation of HMF has been explored through various approaches, encompassing stoichiometric reagents, homogeneous catalysts, heterogeneous catalysts, and bio-catalysts.1,13–15 In the context of sustainability, heterogeneous catalysts emerge as the preferred choice due to their facile separation from the reaction medium, effective reusability, and high stability.16 Extensive investigations employing both non-noble and noble metal-based catalysts have been reported.1,13,17,18 Despite the higher cost associated with noble metal-based catalysts, their superior activity and stability, coupled with the utilization of air as a green and safe oxidant make them compelling candidates.6,19–21 A list of noble metals, including Au,6,7,19,22,23 Pt,20,24–26 Pd,27,28 Ru,29–31 and Ag,32,33 and various bimetallic combinations8,21,34–38 have been reported for the oxidation of HMF. Their successful deployment not only attests to their catalytic efficiency but also aligns with a closed-cycle approach, wherein the catalysts exhibit reusability and end-of-life noble metals can be recycled for subsequent catalyst preparation.39The oxidation of HMF can proceed via two different reaction routes depending on the alkalinity of the reaction medium and the active metal species involved.6,37,40,41 The initial oxidation step targets either the alcohol or the aldehyde functional group (Scheme 1), resulting in the formation of 5-(hydroxymethyl)furan-2-carboxylic acid (HFCA) or 2,5-diformylfuran (DFF). Subsequent oxidation leads to 5-formyl-2-furancarboxylic acid (FFCA) and finally FDCA.
 | ||
| Scheme 1 Simplified reaction mechanism and reactor scheme for HMF oxidation.1 | ||
Despite the vast knowledge on HMF oxidation with heterogeneous catalysts, most studies have been confined to batch processes, although continuous processes offer various advantages, especially in the context of green chemistry. The adoption of continuous flow synthesis presents several advantages, including easier scalability, improved heat and mass transfer favouring energy efficiency, and a safety enhancement due to reduced reactor volume, mitigating the risk of runaway reactions.42–44 Recognizing these merits, continuous flow synthesis has been acknowledged by the International Union of Pure and Applied Chemistry (IUPAC) as one of ten chemical innovations contributing to a sustainable future.45,46 Despite the obvious advantages, reports on the continuous flow oxidation of HMF utilizing heterogeneous catalysts are very scarce. Lilga et al. studied the continuous oxidation of HMF with Pt-based catalysts in a tubular reactor, noting the highest reaction rates for the formation of FDCA with an alkaline feed compared to neutral and acidic solutions.47 They observed a high initial selectivity to FDCA, which decreased slightly over the following hours of time on stream (ToS). The FDCA yield increased with an increase of pressure or gas hourly space velocity (GHSV), or a decrease of the liquid hourly space velocity (LHSV). Similar observations were made for the continuous oxidation of HMF with a Ru/Al2O3 catalyst, however, only a moderate FDCA yield of 63 ± 2% was obtained.48 An enhancement of the FDCA yield with higher reaction temperature up to 140 °C was found. Motagamwala et al. used a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of H2O and γ-valerolactone as solvent for the continuous oxidation, resulting in an improved solubility of FDCA, which reduces the risk of blocking active sites.26 A good stability of the Pt/C catalyst with an average FDCA yield of about 94% over 60 h ToS was achieved. Finally, base-free conditions with Pt supported on a resin were explored, yielding 99% FDCA over 44 h ToS.49 However, limited HMF-concentration (0.025 M) was used to avoid precipitation of FDCA, lowering the FDCA productivity considerably.
:1 mixture of H2O and γ-valerolactone as solvent for the continuous oxidation, resulting in an improved solubility of FDCA, which reduces the risk of blocking active sites.26 A good stability of the Pt/C catalyst with an average FDCA yield of about 94% over 60 h ToS was achieved. Finally, base-free conditions with Pt supported on a resin were explored, yielding 99% FDCA over 44 h ToS.49 However, limited HMF-concentration (0.025 M) was used to avoid precipitation of FDCA, lowering the FDCA productivity considerably.
In our study, we systematically explore the oxidation of HMF in a continuous flow reactor utilizing AuPd-based catalysts renowned for their efficacy in achieving high FDCA yields in batch reactors.21,34,37,38,50 We implemented air as a sustainable and economical oxidant, water as a safe and green solvent, Na2CO3 as a mild and non-toxic base, and heterogeneous catalysts in a fixed-bed reactor to avoid separation of the catalytic system from the reaction mixture. These measures collectively enable a more sustainable approach to the production of the important renewable monomer FDCA. Our investigation delves into the influence of support materials, reaction temperature, pressure, and base-equivalents on the oxidation process, providing comprehensive insights into the key factors that influence the efficiency of the oxidation reaction, thereby informing strategies for optimizing FDCA production. A critical facet of our study involves the assessment of the long-term stability of the catalytic system, which is of utmost importance for ensuring the sustainability of the entire process.
Experimental
Catalyst synthesis
AuPd-alloyed particles with a targeted ratio of 52:48 (Au:Pd) were prepared according to a method previously published by our group.21 In brief, PdCl2 (m = 0.0222 g) and HAuCl4·3H2O (m = 0.0533 mg) were dissolved in diluted HCl (3 mL conc. HCl in 77 mL H2O) for about 3 h. Subsequently, 620 mL H2O and poly(vinylalcohol) (PVA; m = 37.8 mg) were added. The solution was stirred for 10 min and then a diluted NaBH4 (m = 39.4 mg) solution was added. After an additional 30 min of stirring, the pH of the solution was adjusted by addition of either 50 vol% H2SO4 or KOH (activated carbon (AC-ROX): pH = 13, ZrO2: pH = 1, TiO2: pH = 1). Following this, the support material (m = 1.96 g) was added to the suspension corresponding to a AuPd-loading of 2 wt%. The catalyst was subjected to overnight stirring before being separated from the solution via filtration. The catalyst was thoroughly washed and dried overnight.
Characterization methods
Continuous flow oxidation of HMF
The catalyst was loaded into a tubular reactor with a 3/8-inch diameter, preheated to the specified reaction temperature before introducing the reactant solution. The aqueous HMF-solution with a concentration of 0.2 M and the aqueous Na2CO3-solution were pumped separately into the reactor at identical flow rates, resulting in a final HMF concentration of 0.1 M within the reactor. Upon mixing of the two aqueous streams, air was introduced at the given flow rate and reaction pressure. The product solution was collected after the backpressure regulator, and samples for HPLC analysis were collected. In certain instances, solid impurities in the HMF-solution caused fluctuations in the flow rate inducing variations in observed product yields. Hence, averaged product yields and productivities after reaching a steady state were compared to account for fluctuations in the HMF-concentration in the feedstock. The measurement of the ToS started when the reactants reached the reactor. Additional information on the experimental setup can be found in the ESI.†Results and discussion
Characterization
The metal loading for each of the tested catalysts was determined by ICP-OES (Table 1). The total metal loading varied between 1.4 wt% (AuPd/ZrO2 and AuPd/AC-ROX) and 1.7 wt% (AuPd/TiO2). The deviation from the targeted loading of 2 wt% might be attributed to the supporting process of the particles. Apart from AuPd/ZrO2, which had a Au:Pd ratio of 49:51, the nanoparticles generally displayed a slightly higher Au content compared to the intended ratio of 52:48. Nevertheless, the discrepancy was marginal for all the catalysts, and no significant differences in catalytic activity were anticipated due to these variations.21
| Catalyst | Au-loading/wt% | Pd-loading/wt% |
n
Au:nPd |
![[d with combining macron]](https://www.rsc.org/images/entities/i_char_0064_0304.gif) particle/nm
particle/nm |
|---|---|---|---|---|
| AuPd/ZrO2 | 0.9 ± 0.1 | 0.5 ± 0.1 | 49:51 |
3.3 ± 1 |
| AuPd/TiO2 | 1.2 ± 0.1 | 0.5 ± 0.1 | 56:44 |
4.2 ± 0.9 |
| AuPd/AC-ROX | 1.0 ± 0.1 | 0.4 ± 0.1 | 57:43 |
3.5 ± 0.9 |
All the catalysts showed comparable and narrow particle size distributions (Fig. 1) with mean particle diameters ranging from 3.3 nm to 4.2 nm (Table 1). Particle agglomeration was observed on the activated carbon support (AC-ROX). In the case of TiO2 and ZrO2, predominantly isolated particles were found in the STEM images. The particles showed a homogeneous distribution of Au and Pd in the EDX-mapping (Fig. S3†).
 | ||
| Fig. 1 STEM images and particle size distributions (inset: particle size distribution, n: particle count, d: average particle diameter) of (a) AuPd/AC-ROX (n = 126), (b) AuPd/TiO2 (n = 212), and (c) AuPd/ZrO2 (n = 203). | ||
X-ray diffraction analysis was employed to identify reflections indicative of metallic AuPd-alloys, confirming the successful alloy formation (Fig. S4†). For AuPd/AC-ROX, the presence of alloyed AuPd-particles with a broad reflection at 39.04° was found, aligning with the intended 1:1 ratio in the AuPd-alloys. In the case of AuPd/ZrO2 and AuPd/TiO2, the alloy reflections were overlaid by the reflections from the respective support materials. Specifically, the TiO2-based catalyst showed a combination of anatase and rutile phases, while the reflections of Au/ZrO2 were assigned to a monoclinic ZrO2-phase.
To further confirm the alloy formation at the level of an individual nanoparticle, a SAED pattern of a single AuPd nanoparticle on AuPd/AC-ROX was recorded (Fig. 2). The diffraction pattern of the particle could be assigned to the crystal planes in the fcc-structure of a AuPd (1:1) reference (ICSD collection code 58571). The determined lattice constant of 0.396 nm closely approximated the reference data with 0.398 nm (ICSD collection code 58571). The detailed analysis was only feasible for AuPd/AC-ROX, as the other two catalysts predominantly showed reflections of the metal oxide supports.
 | ||
| Fig. 2 SAED diffraction pattern of a AuPd nanoparticle supported on activated carbon ROX (AuPd/AC-ROX). | ||
Catalytic testing in batch mode
Before subjecting the catalysts to continuous flow testing, a comprehensive investigation of all the catalysts was conducted in the batch oxidation of HMF (0.1 M HMF, 100 °C, 15 bar air, 2 eq. Na2CO3, 5 h, M:HMF 1:92). All the catalysts were used in powder form in these preliminary assessments (Table 2). Each catalyst demonstrated a quantitative conversion of HMF under the tested reaction conditions. AuPd/AC-ROX showed the highest FDCA yield, achieving a quantitative conversion of HMF to FDCA, thus representing the most proficient catalyst in these batch tests. AuPd/TiO2 and AuPd/ZrO2 followed with FDCA yields of 97% and 94%, respectively. In their powder form, all three catalysts delivered high yields of FDCA in the batch process. However, the catalytic activity exhibited variations in continuous flow testing, highlighting the importance of understanding how these catalysts function under dynamic, continuous conditions.
:HMF 1:92)
| Catalyst | HMF conversion/% | FDCA yield/% |
|---|---|---|
| AuPd/ZrO2 | >99 | 94 |
| AuPd/TiO2 | >99 | 97 |
| AuPd/AC-ROX | >99 | >99 |
Comparison of catalysts in continuous flow
In the assessment of catalysts for continuous flow oxidation of HMF, TiO2, ZrO2, and AC-ROX were employed as support materials to identify the most suitable catalyst. Fig. 3 displays the FDCA yield as a function of ToS for the three catalysts, revealing a striking distinction with AuPd/AC-ROX achieving the highest FDCA yield with a maximum over 90%. The other two catalysts followed in the order of AuPd/TiO2 and AuPd/ZrO2, presenting similar FDCA yields. | ||
| Fig. 3 Support influence on continuous flow HMF oxidation with AuPd/ZrO2 (LHSV: 7.7 min−1), AuPd/TiO2 (LHSV: 5 min−1), AuPd/AC-ROX (LHSV: 1.9 min−1; 0.1 M HMF, 4 eq. Na2CO3, 100 °C, 51 ± 3 bar, liquid-flow: 0.1 mL min−1, air-flow: 20 mL min−1). | ||
Previous work by Lilga et al.47 observed the highest FDCA-productivity for inorganic metal oxides with a low surface area. The use of carbon-based supports led to product inhibition due to the strong adsorption of HMF and the oxidized products. Nevertheless, AC-ROX was shown in the literature to play an active role in the base-free HMF-oxidation, possessing good adsorption properties for HMF, which potentially increases the catalytic activity in continuous flow oxidation under alkaline conditions.51
AuPd/AC-ROX emerged as the superior catalyst, giving the highest average FDCA yield at 87%, in contrast to 74% for AuPd/TiO2 and 61% for AuPd/ZrO2 (Fig. 3). Furthermore, AuPd/TiO2 and AuPd/ZrO2 demonstrated the tendency to form humins at higher flow rates, resulting in lower FDCA yields, rendering them less suitable for increasing the overall productivity. The different density of the support materials resulted in a variation of the LHSV for all tested catalysts. AuPd/AC-ROX achieved higher FDCA yields even at higher LHSV compared to the other two catalysts. Over a range of 5.3 h−1, which is close to the LHSV of AuPd/TiO2, up to 16.3 h−1 an average FDCA yield >89% was achieved (Fig. S5†). Only for the highest tested LHSV of 31.4 h−1 a drop in the average FDCA yield to 63.2% was visible. This proves the superior intrinsic catalytic activity of AuPd/AC-ROX over a broad LHSV range. The higher catalytic activity might originate from the superior adsorption properties of AC-ROX,51 particularly for HMF, which was not adsorbed on the surfaces of TiO2 and ZrO2 (Table S2†). AC-ROX thus proved to be the most suitable support material for further testing, offering the potential to enhance the productivity of FDCA further.
Influence of reaction conditions
In depth exploration into the influence of the reaction conditions, specifically pressure, Na2CO3-equivalents, and reaction temperature, was conducted to elucidate their impact on the FDCA yield (Fig. 4). We observed that with AuPd/TiO2 an increase of the FDCA yield from 60 to 74% occurred while increasing the reaction pressure from 12 to 51 bar. This finding underscores the positive effect of heightened reaction pressure on FDCA production. The molality of oxygen at the reaction temperature of 100 °C is about 0.0008 molO2 kgH2O−1.52 The solubility increases almost linearly with escalating reaction pressure, reaching nearly 0.04 molO2 kgH2O−1 at about 50 bar pressure.52 The enhanced oxygen solubility is expected to lead to an improved availability of oxygen on the catalyst surface, thereby expediting a fast removal of hydrogen-species from the surface. | ||
| Fig. 4 Pressure influence on the continuous flow HMF oxidation with AuPd/TiO2 (0.1 M HMF, 4 eq. Na2CO3, 100 °C, liquid-flow: 0.1 mL min−1, air-flow: 20 mL min−1, LHSV: 5 min−1). | ||
Even at a pressure of 12 bar the average HFCA yield and FFCA yield (not shown in graph) were just 2% and 1%, respectively. This indicates that the lower FDCA yield cannot be solely explained by an incomplete conversion of the reaction intermediates. Consequently, the decomposition of HMF is enhanced, which might be attributed to reduced O2 availability at the active sites, or blockage of active sites by strong adsorption of FDCA and intermediate products.
We studied the influence of Na2CO3-equivalents added to the reaction solution (Fig. 5). For AuPd/TiO2, we observed a significant increase of the FDCA yield from 39% to 74%, after >4 h ToS, when transitioning from two to four equivalents of Na2CO3. A further increase to six equivalents of Na2CO3 gave a FDCA yield of 71%. The reason for this effect was attributed to the higher solubility of FDCA when a higher concentration of Na2CO3 was dosed. A similar influence was observed when AuPd/AC-ROX served as the catalyst, demonstrating a linear FDCA yield increase up to 100% at about 3.5 h ToS. Afterwards the catalyst showed a linear decrease in FDCA yield until the end of the experiment, suggesting deactivation likely due to FDCA precipitation on active sites at elevated FDCA-concentration.
 | ||
| Fig. 5 Influence of quantity of Na2CO3-equivalents on the continuous flow oxidation of HMF with (a) AuPd/TiO2 (0.1 M HMF, 100 °C, 51 ± 3 bar, liquid-flow: 0.1 mL min−1, air-flow: 20 mL min−1, LHSV: 5 min−1) and (b) AuPd/AC-ROX (0.1 M HMF, 100 °C, 65 ± 3 bar, liquid-flow: 0.18 mL min−1, air-flow: 20 mL min−1, LHSV: 3.4 min−1). | ||
In the experiment employing six equivalents of Na2CO3, no linear decrease of the FDCA yield was noticed, resulting in an impressive average FDCA yield of 97%. Notably, neither HFCA, FFCA, nor humins were detected in the product solution in either case. Hence, the deactivation of the catalyst was attributed solely to the precipitation of FDCA on the catalyst surface, with the oxidation of the intermediates proceeding sufficiently rapidly. These findings suggest that to avoid blocking of active sites and product inhibition by FDCA precipitation, it is recommended to use at least six equivalents of Na2CO3 (0.6 M) in the HMF-oxidation. Notably, the Na2CO3 concentration required for continuous flow operation is three to six times higher than that needed for the batch process.21 This divergence may be attributed to the short residence time of reactants and products in the continuous flow reactor, which, unlike in the batch process, leads to product inhibition by active site blocking. The solubility of FDCA strongly depends on the pH of the solution, with alkaline solution resulting in the formation of a Na-carboxylate, which was reported to have a higher solubility compared to the carboxylic acid.53 Thus, the higher concentration of the sodium base enhances the formation rate of the Na-salt of FDCA, which is important under the short residence time compared to the long contact time in batch, avoiding a strong adsorption of the carboxylic acid on the catalyst surface.54 This disparity underscores the importance of tailoring the reaction conditions to the specific characteristics of the continuous flow system for optimal catalytic performance.
In the investigation of temperature influence using AuPd/AC-ROX, a higher LHSV was employed, given the proximity of the FDCA yield to 100% in the initial experiments. We observed an augmentation in FDCA yield with increasing reaction temperature, reaching a maximum of 81% at 120 °C (Fig. 6). Subsequently, at an even higher temperature of 135 °C, a slightly lower average FDCA yield of 72% was obtained. However, a linear decrease in FDCA yield was observed after 3.5 h of reaction time (cf. Fig. S6†). This decline in FDCA yield might be attributed to the formation of humins resulting from the decomposition of HMF at elevated temperatures, and their deposition on the active sites of the catalyst likely contributed to the steady deactivation of the catalyst. At 80 °C the average FDCA yield was only 29%, underscoring the need for elevated temperatures in this catalytic process.
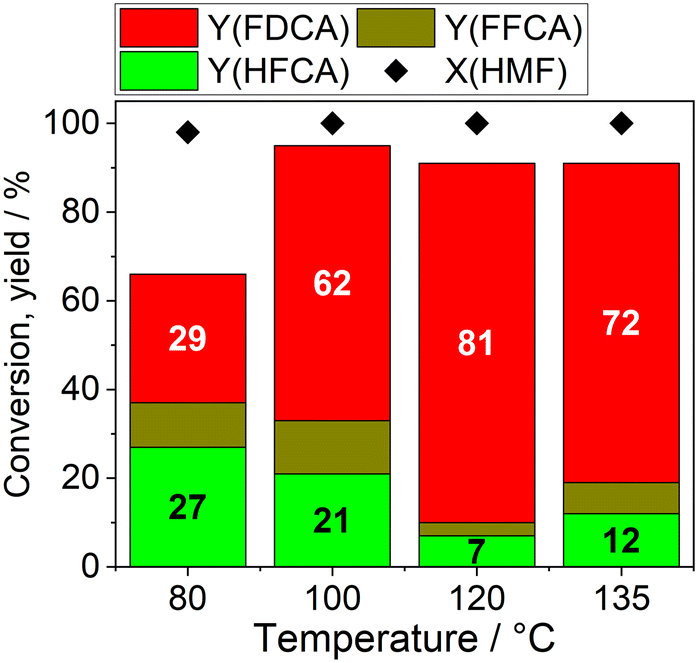 | ||
| Fig. 6 Temperature influence on the continuous flow oxidation of HMF with AuPd/AC-ROX (0.1 M HMF, 6 eq. Na2CO3, 55 ± 5 bar, liquid-flow: 0.22 mL min−1, air-flow: 20 mL min−1, LHSV: 31.4 min−1). The average yields and conversion are shown. | ||
Interestingly, the combination of 100 °C and the higher LHSV allowed for the monitoring of the yields of reaction intermediates such as HFCA and FFCA. The time-dependent yields of HFCA, FFCA, and FDCA as well as the HMF conversion are shown in Fig. S7 in the ESI.† An average HFCA yield of 20% and a FFCA yield of 12% were obtained at 100 °C. Hence, HFCA emerged as the main intermediate, and the rate-determining step was identified as the oxidation of the alcohol function.6,19 However, the FFCA yield was not significantly lower, suggesting a comparable rate of FFCA to FDCA oxidation to the oxidation of the alcohol-function in HFCA under our reaction conditions. Continuous flow operation would offer a suitable platform for more detailed kinetic and mechanistic investigations into the oxidation of HMF in future studies.
The achievement of an 81% FDCA yield at 120 °C and a LHSV of 31.4 min−1 corresponds to an impressive productivity of 68 molFDCA molAuPd−1 h−1. These values surpassed those reported in other studies for the continuous flow oxidation of HMF with noble metal-based catalysts (cf.Table 3).47–49 In addition, the productivity exceeded previously published values for optimized batch processes for HMF oxidation, e.g. using Au/ZrO2 with a productivity of 67 molFDCA molAuPd−1 h−1.19 Considering that after a reaction in a batch process there is a dead time for draining, cleaning, and reloading the reactor, as well as reactivation of the catalyst, the time-dependent productivity is even higher for a continuous process. Table 3 provides a comparison of productivity values for various continuous and batch processes for HMF oxidation. It is essential to note that the results for the batch process are solely based on the reaction itself and do not include the dead time of the process.
| Catalyst | FDCA yield/% | Productivity/molFDCA molM−1 h−1 | LHSV/min−1 | Reference |
|---|---|---|---|---|
| a 6.5 wt% H2O2 as an oxidant. b Does not include dead time. | ||||
| a) Continuous process | ||||
| AuPd/AC-ROX | 81 | 68 | 31.4 | This work |
| AuPd/TiO2 | 74 | 3.8 | 5 | This work |
| AuPd/ZrO2 | 61 | 3.1 | 7.7 | This work |
| Ru/Al2O3 | 63 | 0.2 | 1 | 48 |
| Pt/Al2O3 | >99 | — | 4.5 | 47 |
| Pt/C | 99 | 2.2 | — | 49 |
| Ru/Ca | 47 | 1.4 | — | 55 |
| b) Batch processes | ||||
| AuPd/CNT | 99 | 8b | — | 37 |
| AuPd/ZOC | >99 | 25b | — | 56 |
| AuPd/IRA-743 | 93 | 0.4b | — | 57 |
| Au/ZrO2 | 89 | 67b | — | 19 |
| Au/HY | >99 | 18b | — | 23 |
Long-term studies on the catalyst stability
Investigations into catalyst stability were undertaken through a long-term >90 h ToS experiment with AuPd/AC-ROX (0.1 M HMF, 6 eq. Na2CO3, 100 °C, 61 ± 5 bar, liquid-flow: 0.22 mL min−1, air-flow: 20 mL min−1, LHSV: 19.6 min−1), to assess the stability of the catalyst under continuous operation over multiple days (Fig. 7). The FDCA yield increased to >90% in the initial reaction hours, but then stabilized at an average of about 75% after the first night (ToS > 16 h). This decrease at the beginning might be caused by the setting of an equilibrium of surface adsorbates on the catalyst in the first hours, leading to the formation of a steady state. In the following days, we recognized a considerable fluctuation in FDCA yields at different measurements over the day. This occurred due to pulsing in the HMF-stream caused by a pump-induced pulsation. Deposition of minor solid residues in the HMF-feed might have caused the pulsing effect of the pump, leading to the observed variations. Hence, the average FDCA yields were determined for each day and the entire 90 h experiment (cf.Fig. 7), and the daily average yields are reported in Table S1.† No decline in FDCA yield was observed after 90 h of continuous operation, affirming the high stability of AuPd/AC-ROX in the reaction medium and under the specified conditions. This persistent stability allowed for the continuous production of FDCA in high selectivity. Over the course of the experiment, the average FDCA yield over 90 h ToS was 75% (±12%), which corresponds to a high productivity of 39 molFDCA molAuPd−1 h−1. | ||
| Fig. 7 HMF oxidation with AuPd/AC-ROX (0.1 M HMF, 6 eq. Na2CO3, 58 ± 5 bar, liquid-flow: 0.22 mL min−1, air-flow: 20 mL min−1, LHSV: 19.6 min−1). The dashed line represents the average FDCA yield over 90 h ToS. The average FDCA yield for the complete experiment is given in the graph. | ||
The intermediates HFCA and FFCA were formed with an average yield of 13% and 3%, respectively, over the course of 90 h ToS experiment. The remaining 9% might be attributed to either overoxidation of HMF to CO2 or degradation of HMF. By adjusting the catalyst mass in the reactor, achieving operation at complete conversion of all intermediate oxidation products is feasible, ensuring a high selectivity to FDCA for at least 90 h without any detectable catalyst deactivation.
To test the stability of the catalyst properties, a spent AuPd/AC-ROX catalyst after oxidation of HMF (0.1 M HMF, 6 eq. Na2CO3, 100 °C, 67 ± 5 bar, LHSV: 31.4 min−1, 6 h time on stream) was characterized by XRD (Fig. S8†) and adsorption of HMF and FFCA (Table S3†). The AuPd alloy showed a good stability with no indications of phase segregation in XRD. Slight sintering of AuPd was observed, however, the high stability over 90 h ToS indicates that this is not a major cause of deactivation. A high capacity for the adsorption of HMF was retained, indicating a good stability of the activated carbon under reaction conditions. The results are described in detail in the ESI.†
To simulate catalyst deactivation by the formation of humins, another long-term study (0.1 M HMF, 6 eq. Na2CO3, 100 °C, 58 ± 4 bar, liquid-flow: 0.22 mL min−1, air-flow: 20 mL min−1, LHSV: 19.6 min−1) was conducted over 50 h ToS. To allow for the formation of humins in the zone prior to the catalyst bed and within the reactor, the flow was intentionally paused after 16 h for about 4 h (Fig. 8). Prior to the deactivation of the catalyst, the FDCA yield was about 87%. After the deactivation of the catalyst by humins, the FDCA yield steadily decreased over the next 35 h, stabilizing at a final yield of about 42%. At the same time, the HFCA yield gradually increased, reaching a final value of 38%. Interestingly, the FFCA yield remained stable over the 35 h period, averaging 12%. Hence, the decline in FDCA yield was attributed to a slower oxidation of the alcohol function of HFCA. The FDCA yield in this scenario was considerably lower compared to the 90 h experiment, underlining how the decomposition of HMF and the formation of humins can result in catalyst deactivation. These observations emphasize the importance of operating continuous flow oxidation of HMF under conditions that favour high selectivity towards the oxidation products while suppressing humin formation, rather than pursuing the highest possible short-term productivity. Such an approach ensures the long-term stability of the catalyst in the continuous flow process.
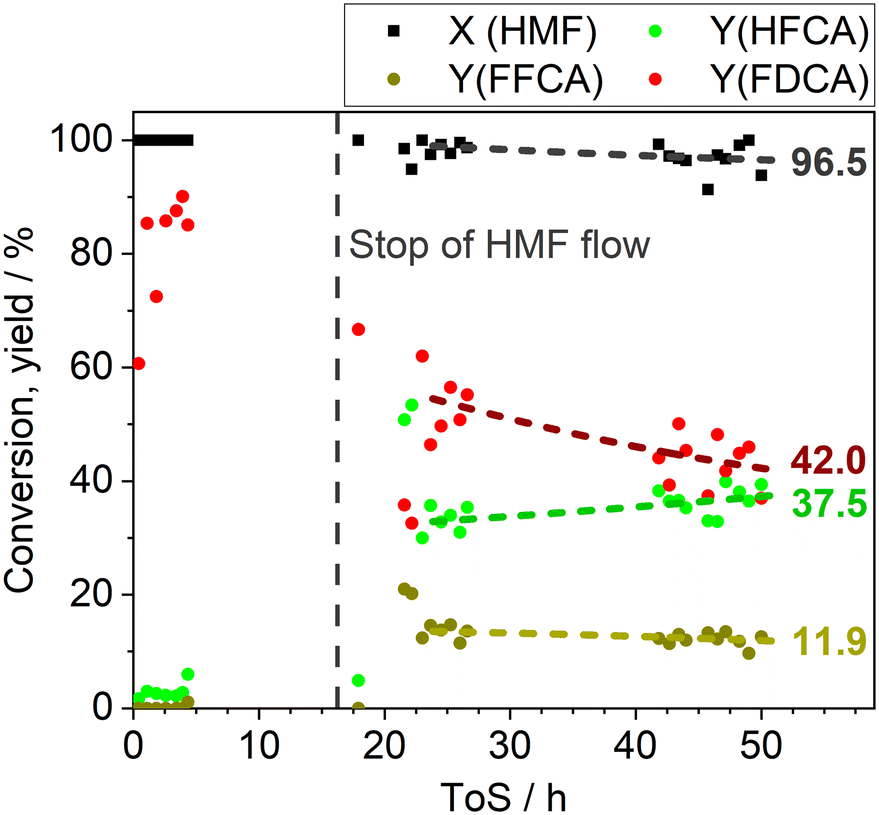 | ||
| Fig. 8 HMF oxidation with AuPd/AC-ROX (0.1 M HMF, 6 eq. Na2CO3, 58 ± 5 bar, liquid-flow: 0.22 mL min−1, air-flow: 20 mL min−1, LHSV: 19.6 min−1). The flow was stopped at the dashed line for about 4 h to induce the formation of humins in the reactor. Dashed lines represent the fitted average conversion and yields. | ||
Conclusions
The utilization of AuPd-alloys supported on activated carbon exhibited remarkable efficiency in the continuous flow oxidation of HMF, yielding the renewable monomer FDCA. Transitioning to a continuous process offers notable advantages regarding green chemistry, leveraging superior heat and mass transfer, easier scalability, and improved safety through the use of smaller reactor volumes. The achieved productivity, reaching up to 68 molFDCA molAuPd−1 h−1, surpassed existing benchmarks for continuous flow HMF oxidation by an order of magnitude. Moreover, this productivity level was comparable to optimized batch processes for HMF oxidation. The absence of significant dead time, characteristic of batch processes due to draining and filling of the reactor, positions continuous flow operation at these productivity rates as a promising alternative for HMF oxidation with heterogeneous catalysts.The AuPd/AC-ROX catalyst demonstrated robust activity and stability over 90 h ToS, establishing AuPd alloys as well-suited catalysts for continuous processes. This investigation addresses a critical gap in current knowledge, shedding light on the durability and robustness of the AuPd-based catalysts under continuous flow conditions, thereby contributing valuable insights to the broader scientific community and advancing the prospects of sustainable monomer synthesis. Future investigations should delve into a comprehensive assessment and optimization of internal and external mass transport limitations for such a continuous process to facilitate up-scaling. In addition, efforts should focus on increasing the maximum concentration of HMF in the feed. The study also revealed steady catalyst deactivation due to the formation of humins in the feed and their adsorption on the catalyst's surface. Hence, meticulous selection of reaction conditions and feed composition is imperative to suppress humin formation. In summary, the continuous flow oxidation of HMF emerges as a promising approach to up-scaling of the oxidation process, offering potential applications in decentralized plants crucial for converting agricultural by-products into important bio-based monomers.
Author contributions
The manuscript was written through contributions of all authors. D. Neukum worked on the conceptualization, investigation, visualization, and writing the original draft. M. E. Ludwig worked on the investigation. A. R. Lakshmi Nilayam helped with investigation. A. Vadarlis helped with build-up of the infrastructure. J.-D. Grunwaldt worked on the project administration, resources, and supervision. E. Saraçi took care of the project administration, resources, visualization, and supervision. All authors contributed to review & editing. All authors have given approval to the publication of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Veronika Holderied for assistance in HPLC analysis, Armin Lautenbach for ICP-OES analysis, and Shweta Sharma for assistance in conducting STEM. We thank Nikolaos Boukis for provision of a fume hood and help with the construction of the continuous flow setup. STEM imaging was carried out with the support of the Karlsruhe Nano Micro Facility (KNMFi, https://www.knmf.kit.edu), a Helmholtz Research Infrastructure at Karlsruhe Institute of Technology (KIT, https://www.kit.edu). This work was financially supported by the Federal Ministry of Food and Agriculture (BMEL) through the FNR (Fachagentur Nachwachsende Rohstoffe e. V.) based on a decision taken by the German Bundestag (funding no. 22010718).References
- M. Sajid, X. Zhao and D. Liu, Green Chem., 2018, 20, 5427–5453 RSC.
- R. A. Sheldon, ACS Sustainable Chem. Eng., 2018, 6, 4464–4480 CrossRef CAS.
- P. N. R. Vennestrøm, C. M. Osmundsen, C. H. Christensen and E. Taarning, Angew. Chem., Int. Ed., 2011, 50, 10502–10509 CrossRef PubMed.
- K. Hengst, M. Schubert, W. Kleist and J.-D. Grunwaldt, Catalytic Hydrogenation for Biomass Valorization, 2014, pp. 125–150 Search PubMed.
- C. H. Christensen, J. Rass-Hansen, C. C. Marsden, E. Taarning and K. Egeblad, ChemSusChem, 2008, 1, 283–289 CrossRef CAS PubMed.
- O. Casanova, S. Iborra and A. Corma, ChemSusChem, 2009, 2, 1138–1144 CrossRef CAS PubMed.
- E. Taarning, I. S. Nielsen, K. Egeblad, R. Madsen and C. H. Christensen, ChemSusChem, 2008, 1, 75–78 CrossRef CAS PubMed.
- S. Albonetti, T. Pasini, A. Lolli, M. Blosi, M. Piccinini, N. Dimitratos, J. A. Lopez-Sanchez, D. J. Morgan, A. F. Carley, G. J. Hutchings and F. Cavani, Catal. Today, 2012, 195, 120–126 CrossRef CAS.
- W. Partenheimer and V. V. Grushin, Adv. Synth. Catal., 2001, 343, 102–111 CrossRef CAS.
- R.-J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS PubMed.
- A. F. Sousa, C. Vilela, A. C. Fonseca, M. Matos, C. S. R. Freire, G.-J. M. Gruter, J. F. J. Coelho and A. J. D. Silvestre, Polym. Chem., 2015, 6, 5961–5983 RSC.
- E. de Jong, M. A. Dam, L. Sipos and G. J. M. Gruter, in Biobased Monomers, Polymers, and Materials, American Chemical Society, 2012, ch. 1, vol. 1105, pp. 1–13 Search PubMed.
- S. Hameed, L. Lin, A. Wang and W. Luo, Catalysts, 2020, 10, 120 CrossRef CAS.
- A. Messori, A. Fasolini and R. Mazzoni, ChemSusChem, 2022, 15, e202200228 CrossRef CAS PubMed.
- D. Troiano, V. Orsat and M.-J. Dumont, ACS Catal., 2020, 10, 9145–9169 CrossRef CAS.
- D. Astruc, F. Lu and J. R. Aranzaes, Angew. Chem., Int. Ed., 2005, 44, 7852–7872 CrossRef CAS PubMed.
- D. Zhao, T. Su, Y. Wang, R. S. Varma and C. Len, Mol. Catal., 2020, 495, 111133 CrossRef CAS.
- P. Pal and S. Saravanamurugan, ChemSusChem, 2019, 12, 145–163 CrossRef CAS PubMed.
- O. R. Schade, K. F. Kalz, D. Neukum, W. Kleist and J.-D. Grunwaldt, Green Chem., 2018, 20, 3530–3541 RSC.
- H. Ait Rass, N. Essayem and M. Besson, Green Chem., 2013, 15, 2240–2251 RSC.
- D. Neukum, L. Baumgarten, D. Wüst, B. B. Sarma, E. Saraçi, A. Kruse and J.-D. Grunwaldt, ChemSusChem, 2022, 15, e202200418 CrossRef CAS PubMed.
- N. K. Gupta, S. Nishimura, A. Takagaki and K. Ebitani, Green Chem., 2011, 13, 824–827 RSC.
- J. Cai, H. Ma, J. Zhang, Q. Song, Z. Du, Y. Huang and J. Xu, Chem. – Eur. J., 2013, 19, 14215–14223 CrossRef CAS PubMed.
- X. Han, C. Li, Y. Guo, X. Liu, Y. Zhang and Y. Wang, Appl. Catal., A, 2016, 526, 1–8 CrossRef CAS.
- S. Siankevich, G. Savoglidis, Z. Fei, G. Laurenczy, D. T. L. Alexander, N. Yan and P. J. Dyson, J. Catal., 2014, 315, 67–74 CrossRef CAS.
- A. H. Motagamwala, W. Won, C. Sener, D. M. Alonso, C. T. Maravelias and J. A. Dumesic, Sci. Adv., 2018, 4, 9722 CrossRef PubMed.
- B. Siyo, M. Schneider, M.-M. Pohl, P. Langer and N. Steinfeldt, Catal. Lett., 2014, 144, 498–506 CrossRef CAS.
- Y. Wang, K. Yu, D. Lei, W. Si, Y. Feng, L.-L. Lou and S. Liu, ACS Sustainable Chem. Eng., 2016, 4, 4752–4761 CrossRef CAS.
- J. Artz and R. Palkovits, ChemSusChem, 2015, 8, 3832–3838 CrossRef CAS PubMed.
- C. M. Pichler, M. G. Al-Shaal, D. Gu, H. Joshi, W. Ciptonugroho and F. Schüth, ChemSusChem, 2018, 11, 2083–2090 CrossRef CAS PubMed.
- Y. Y. Gorbanev, S. Kegnæs and A. Riisager, Top. Catal., 2011, 54, 1318 CrossRef CAS.
- D. Zhao, D. Rodríguez-Padrón, R. Luque and C. Len, ACS Sustainable Chem. Eng., 2020, 8(23), 8486–8495 CrossRef CAS.
- O. R. Schade, A. Gaur, A. Zimina, E. Saraçi and J.-D. Grunwaldt, Catal. Sci. Technol., 2020, 10, 5036–5047 RSC.
- A. Villa, M. Schiavoni, S. Campisi, G. M. Veith and L. Prati, ChemSusChem, 2013, 6, 609–612 CrossRef CAS PubMed.
- O. R. Schade, F. Stein, S. Reichenberger, A. Gaur, E. Saraçi, S. Barcikowski and J.-D. Grunwaldt, Adv. Synth. Catal., 2020, 362, 5681–5696 CrossRef CAS.
- G. Uzunidis, O. Schade, D. Schild, J.-D. Grunwaldt and S. Behrens, ChemNanoMat, 2021, 7, 1108–1116 CrossRef CAS.
- X. Wan, C. Zhou, J. Chen, W. Deng, Q. Zhang, Y. Yang and Y. Wang, ACS Catal., 2014, 4, 2175–2185 CrossRef CAS.
- D. Bonincontro, A. Lolli, A. Villa, L. Prati, N. Dimitratos, G. M. Veith, L. E. Chinchilla, G. A. Botton, F. Cavani and S. Albonetti, Green Chem., 2019, 21, 4090–4099 RSC.
- M. K. Jha, J.-C. Lee, M.-S. Kim, J. Jeong, B.-S. Kim and V. Kumar, Hydrometallurgy, 2013, 133, 23–32 CrossRef CAS.
- S. E. Davis, B. N. Zope and R. J. Davis, Green Chem., 2012, 14, 143–147 RSC.
- X. Han, L. Geng, Y. Guo, R. Jia, X. Liu, Y. Zhang and Y. Wang, Green Chem., 2016, 18, 1597–1604 RSC.
- L. Wan, M. Jiang, D. Cheng, M. Liu and F. Chen, React. Chem. Eng., 2022, 7, 490–550 RSC.
- C. Wiles and P. Watts, Eur. J. Org. Chem., 2008, 2008, 1655–1671 CrossRef.
- D. Dallinger and C. O. Kappe, Curr. Opin. Green Sustainable Chem., 2017, 7, 6–12 CrossRef.
- R. Gérardy, D. P. Debecker, J. Estager, P. Luis and J.-C. M. Monbaliu, Chem. Rev., 2020, 120, 7219–7347 CrossRef PubMed.
- F. Gomollón-Bel, Chemistry International, 2019, 41, 12–17 CrossRef.
- M. A. Lilga, R. T. Hallen and M. Gray, Top. Catal., 2010, 53, 1264–1269 CrossRef CAS.
- A. Danielli da Fonseca Ferreira, M. Dorneles de Mello and M. A. P. da Silva, Ind. Eng. Chem. Res., 2019, 58, 128–137 CrossRef CAS.
- F. Liguori, P. Barbaro and N. Calisi, ChemSusChem, 2019, 12, 2558–2563 CrossRef CAS PubMed.
- Z. Gao, R. Xie, G. Fan, L. Yang and F. Li, ACS Sustainable Chem. Eng., 2017, 5, 5852–5861 CrossRef CAS.
- D. Neukum, E. Saraci, B. Krause, A. R. L. Nilayam, A. Sinigalia and J.-D. Grunwaldt, ChemCatChem, 2024, e202301228 CrossRef.
- M. Geng and Z. Duan, Geochim. Cosmochim. Acta, 2010, 74, 5631–5640 CrossRef CAS.
- B. S. Rao, M. J. Hidajat, G.-N. Yun and D. W. Hwang, Catal. Sci. Technol., 2023, 13, 6921–6936 RSC.
- W. Xie, H. Liu, X. Tang, X. Zeng, Y. Sun, X. Ke, T. Li, H. Fang and L. Lin, Appl. Catal., A, 2022, 630, 118463 CrossRef CAS.
- D. Zhao, D. Rodriguez-Padron, K. S. Triantafyllidis, Y. Wang, R. Luque and C. Len, ACS Sustainable Chem. Eng., 2020, 8, 3091–3102 CrossRef CAS.
- Z. Gui, W. Cao, S. Saravanamurugan, A. Riisager, L. Chen and Z. Qi, ChemCatChem, 2016, 8, 3636–3643 CrossRef CAS.
- C. A. Antonyraj, N. T. T. Huynh, S.-K. Park, S. Shin, Y. J. Kim, S. Kim, K.-Y. Lee and J. K. Cho, Appl. Catal., A, 2017, 547, 230–236 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental information, characterization and catalytic testing. See DOI: https://doi.org/10.1039/d3cy01722b |
| This journal is © The Royal Society of Chemistry 2024 |