
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
CO2 activation by copper oxide clusters: size, composition, and charge state dependence†
Pavol
Mikolaj
 a,
Barbara
Zamora Yusti
b,
László
Nyulászi
bc,
Joost M.
Bakker
d,
Tibor
Höltzl
*ce and
Sandra M.
Lang
*a
a,
Barbara
Zamora Yusti
b,
László
Nyulászi
bc,
Joost M.
Bakker
d,
Tibor
Höltzl
*ce and
Sandra M.
Lang
*a
aInstitute of Surface Chemistry and Catalysis, University of Ulm, Ulm 89069, Germany. E-mail: sandra.lang@uni-ulm.de
bDepartment of Inorganic and Analytical Chemistry, Budapest University of Technology and Economics, Műegytem rkp. 3, Budapest-1111, Hungary
cHUN-REN-BME Computation Driven Chemistry research group, Műegytem rkp. 3, Budapest-1111, Hungary
dRadboud University, Institute of Molecules and Materials, FELIX Laboratory, 6525 ED, Nijmegen, The Netherlands
eFurukawa Electric Institute of Technology, Nanomaterials Science Group, Késmárk utca 28/A, Budapest 1158, Hungary. E-mail: tibor.holtzl@furukawaelectric.com
First published on 2nd September 2024
Abstract
The interaction of CO2 with copper oxide clusters of different size, composition, and charge is investigated via infrared multiple-photon dissociation (IR-MPD) spectroscopy and density functional theory (DFT) calculations. Laser ablation of a copper target in the presence of an O2/He mixture leads to the preferred formation of oxygen-rich copper oxide cluster cations, CuxOy+ (y > x; x ≤ 8), while the anionic cluster distribution is dominated by stoichiometric (x = y) and oxygen-deficient (y < x; x ≤ 8) species. Subsequent reaction of the clusters with CO2 in a flow tube reactor results in the preferred formation of near-stoichiometric CuxOy(CO2)+/− complexes. IR-MPD spectroscopy of the formed complexes reveals the non-activated binding of CO2 to all cations while CO2 is activated by all anions. The great resemblance of spectra for all sizes investigated demonstrates that CO2 activation is largely independent of cluster size and Cu/O ratio but mainly determined by the cluster charge state. Comparison of the IR-MPD spectra with DFT calculations of the model systems Cu2O4(CO2)− and Cu3O4(CO2)− shows that CO2 activation exclusively results in the formation of a CO3 unit. Subsequent CO2 dissociation to CO appears to be unfavorable due to the instability of CO on the copper oxide clusters indicating that potential hydrogenation reactions will most likely proceed via formate or bicarbonate intermediates.
1 Introduction
Based on the possibility of harnessing renewable energy to form chemical products and to reduce the environmental impact of the current petrochemical-based industry, extensive research has accelerated the development of better catalysts for the efficient CO2 conversion into valuable products.1 Copper based catalysts are well known for their high activity.2–4 Besides copper, oxygen plays an important role in these catalysts, e.g., at the copper metal-oxide interface in thermal catalysis5,6 or in electrocatalysis using oxide derived copper,7–9 where the subsurface oxygen helps to stabilize the activated (chemisorbed) bent CO2 on the catalyst surface10 and can enhance the formation of C2+ products. It is worth noting that copper-oxide itself is also catalytically active, e.g., in oxidative dehydrogenation of alcohols,11 propylene partial oxidation,12 or in electrocatalysis for oxygen evolution reaction.13 Copper–oxygen intermediates often play an important role in different oxygenation and oxidation reactions, including biologically important cases.14Nanoparticles or clusters are promising catalyst candidates on their own, as well as model compounds whose investigation provides invaluable information to understand the influence of the active site composition and charge in bulk catalysts.15 Accordingly, (sub-)nanometer copper oxide clusters have become popular in heterogeneous catalysis because of their catalytic activities in selective oxidation reactions, e.g., in the reduction of NO and CO2,16 or the methane-to-methanol conversion.17 It has also been shown that copper oxide nanoparticles effectively catalyze the formation of environmentally hazardous molecules, such as polychlorinated dibenzo-p-dioxins and dibenzofurans.18 Amorphous Cu(II) oxide nanoclusters are reported to be highly active for producing CO both in photo- and electrocatalysis.19 Alumina-supported Cu4 clusters were found to catalyze the CO2 hydrogenation to methanol with an exceptionally high turn-over frequency.20
Rooted in the central role that metal nanoparticles have in heterogeneous catalysis, fundamental research in physical chemistry has provided a set of tools to study simplified systems such as metal nanoparticles and an important subclass constituted by metal clusters. These model systems are characterized by their exact elementary composition and charge state.21 Isolated copper oxide clusters were investigated experimentally by negative ion photoelectron spectroscopy,22 ion-mobility23 and thermal desorption mass spectrometry.24 Quantum chemical calculations were carried out for certain charged and neutral copper oxide clusters.23,25–29
Gas-phase vibrational spectroscopy in conjunction with theoretical first-principles calculations is a useful tool to reveal the structure of metal clusters and their reaction products and thus provides insight into the binding and activation of small molecules.15,30 In the present work, we have employed these techniques to study the potential activation of CO2 by free copper oxide clusters as a function of charge state (+1, −1) as well as cluster size and composition (CuxOy+ with x = 1–6 and CuxOy− with x = 1–3).
2 Methods
2.1 Experimental methods
Copper oxide clusters were produced via laser ablation of an isotopically enriched 65Cu foil in the presence of a 1% O2/He mixture and subsequently reacted with 1.7% CO2/He mixture in an adjacent flow tube reactor, which was held at room temperature throughout the experiments. The reaction mixture was then expanded into vacuum forming a molecular beam before entering the intracavity region where it was irradiated by the IR laser beam of the Free-Electron Laser for Intra Cavity Experiments (FELICE, 240–1800 cm−1; 10 μs pulse duration, spectral bandwidth 0.5% FWHM of the central wavenumber) crossing it at an angle of 35°. A few μs after the interaction with FELICE, all clusters were extracted into a reflectron time-of-flight mass spectrometer by a set of pulsed high voltage plates and detected with a microchannel plate detector.31,32To correct for long-term source fluctuations, the experiment was operated at twice the FELICE repetition rate, allowing for the recording of reference mass spectra in between successive FELICE pulses. Whenever FELICE was in resonance with an IR active vibrational mode of a given cluster, multiple IR photons were absorbed sequentially, leading to heating of the complex and finally to its fragmentation. The IR-MPD spectra shown in this contribution represent the depletion yield Y(![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) ) at wavenumber , calculated as Y() = −ln[I()/I0], where I() and I0 are the mass peak intensities with and without laser light, respectively. To reduce the IR fluence with which the complexes are irradiated, the whole instrument can be translated up to 300 mm from the focus position leading to a 30-fold reduction in intensity but increased overlap between the laser and molecular beam and thus an increased signal to noise ratio. All spectra presented in this work were recorded between 300 and 290 mm from the focus. To further reduce the IR fluence and increase the spectral resolution, the overlap between laser and molecular beams can be purposely misaligned such that the molecular beam only observes the lower intensity part of the laser beam. Information on this position is given in the caption of Fig. 3 below.
) at wavenumber , calculated as Y() = −ln[I()/I0], where I() and I0 are the mass peak intensities with and without laser light, respectively. To reduce the IR fluence with which the complexes are irradiated, the whole instrument can be translated up to 300 mm from the focus position leading to a 30-fold reduction in intensity but increased overlap between the laser and molecular beam and thus an increased signal to noise ratio. All spectra presented in this work were recorded between 300 and 290 mm from the focus. To further reduce the IR fluence and increase the spectral resolution, the overlap between laser and molecular beams can be purposely misaligned such that the molecular beam only observes the lower intensity part of the laser beam. Information on this position is given in the caption of Fig. 3 below.
For comparison between the experimental and theoretical spectra, the intensity of the latter were divided by the wavenumber, which we find leads to a better match of relative intensities. We rationalize this as due to the inverse proportionality of the laser's spectral brightness to wavenumber, mirroring the proportionality of the spectral bandwidth to wavenumber (cf. Section S1 of the ESI†).
2.2 Theoretical methods
The search for the global minimum bare cluster geometries of Cu2O4− and Cu3O4− (which were chosen as representatives for the experimentally investigated clusters) was performed using the CALYPSO program,33 a swarm-intelligence based structure prediction method, interfaced with the Gaussian 1634 package for local structure optimization in different spin states. For the structure relaxation, the geometry optimizations were carried out using the LANL2DZ basis set and BP86 functional without any symmetry constraint. Further optimization and analytical second derivatives of the molecular energy with respect to the nuclear coordinates were computed on the optimized structures obtained from CALYPSO to confirm that each located structure had no imaginary vibrational frequency, i.e. true minima are located. For the post-optimization of the CALYPSO found bare clusters, the def2-TZVPD basis set and the TPSSh functional were used, implemented in the Q-Chem35 software. While the polarized triple-ζ quality def2-TZVP basis set has been found to yield accurate CO2 binding energies for different (doped) copper clusters,36,37 here we also included diffuse functions to ensure the adequate description of the anion electronic structures.To systematically investigate carbon dioxide adsorption in different binding modes to low-energy bare copper-oxide clusters, we used an in-house code as e.g. in ref. 36. In the binding modes considered, CO2 was either non-activated (in η1(O) binding mode), activated (in η2(C,O) binding mode; activation is evidenced from a bending of the CO2 moiety and an increased CO distance), or dissociated – decomposing into CO and O. The ESI,† specifically section S6, provides comprehensive data on the energies, spin multiplicities and XYZ coordinates.
3 Results and discussion
3.1 Formation and reactivity of copper oxide clusters
Fig. 1 displays typical copper oxide cluster distributions for cationic (Fig. 1a) and anionic (Fig. 1b) species as color map representations. Under these cluster production conditions, CuxOy+ cations with up to eight copper and nine oxygen atoms are formed and the distribution is dominated by highly oxidized (y > x) clusters. In contrast, the size distribution of anionic CuxOy− clusters is narrower, and the mass spectrum is dominated by stoichiometric (x = y) and slightly oxygen-rich clusters for x ≤ 4 as well as oxygen-deficient (y < x) clusters for x > 4. In addition, while Cu+ and CuOy+ (y = 2,4,6) are formed in considerable amounts, the anionic counterparts are hardly produced.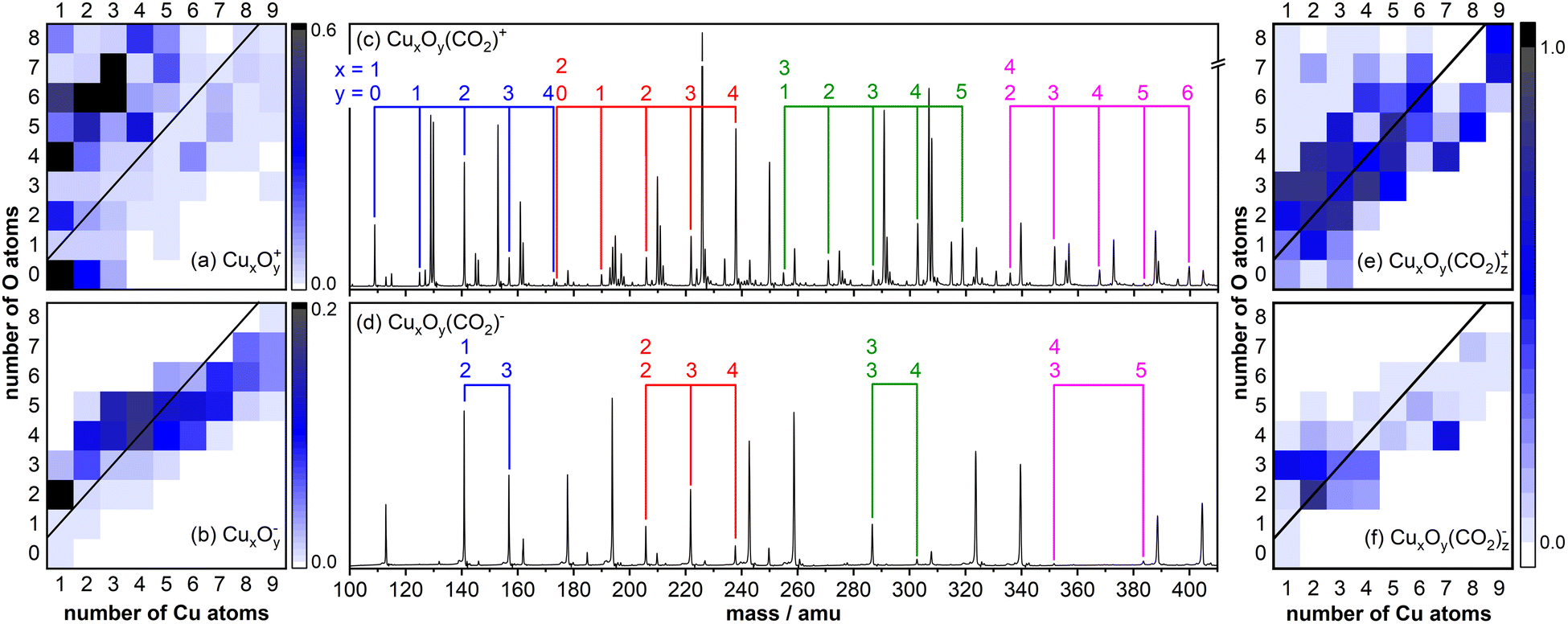 | ||
| Fig. 1 (a) and (b) Mass spectral intensities of the (a) cationic and (b) anionic copper oxide cluster distributions produced via laser ablation (intensities are given in arbitrary units). The black lines in (a) and (b) indicate stoichiometric clusters CuxOy+/− (x = y). (c) and (d) Mass spectra obtained after reaction of (c) cationic and (d) anionic clusters with CO2 in a flow tube reactor. The colored indicators identify the mass peaks of CuxOy(CO2)+/−. (e) and (f) Adsorption efficiency for (e) cationic and (f) anionic clusters calculated with eqn (1). | ||
Subsequent reaction of the clusters with CO2 in a flow tube reactor typically leads to the adsorption of one or two CO2 molecules. Fig. 1c and d display mass spectra obtained for (c) cationic and (d) anionic clusters showing that CuxOy(CO2)+, products are mainly formed for oxygen deficient and near stoichiometric cationic clusters, while products of highly oxidized clusters (y > x + 2) are not observed in notable quantities. This can be either caused by the inherent inactivity of these clusters towards CO2 or by reaction via O2/CO2 exchange (CuxOy+ + CO2 → CuxOy−2(CO2)+ + O2). In any case, this shows that CuxOy(CO2)+ complexes with excess oxygen content are rather unstable. In case of anionic clusters only few CuxOy(CO2)− products with sufficient intensity are produced. This is on the one hand caused by the narrower size distribution of the bare CuxOy− clusters (Fig. 1b) but on the other hand the mass spectra also show that CuxOy(CO2)− products are mainly formed on a much smaller number of clusters, limited to near-stoichiometric clusters (with x ≤ 3 and y ≤ x + 1), while larger clusters (with x > 3) appear to be rather unreactive. This is also illustrated in a color map representation (Fig. 1e and f) showing the adsorption efficiency (AE) calculated as
 | (1) |
3.2 IR-MPD spectra of cationic and anionic CuxOy(CO2) complexes
Although mass spectrometry can provide information on the overall reactivity, it does not give insight into the potential activation of CO2 or bonding motifs. Therefore, we have additionally probed the formed CuxOy(CO2) complexes via IR-MPD spectroscopy.Free CO2 is linear and has four normal modes: the symmetric (ν1 = 1333 cm−1) and asymmetric (ν3 = 2349 cm−1) stretching vibration and the doubly degenerate bending vibration (ν2 = 667 cm−1).38ν1 and the first overtone of ν2 couple into a Fermi resonance and instead of a single peak at 1333 cm−1 typically a pair of peaks is observed at 1388 cm−1 and 1285 cm−1 (ν+ and ν−).39–42 Although IR-inactive for gas-phase CO2, the Fermi dyad can become IR-active due to symmetry breaking when CO2 is weakly adsorbed on extended metal oxide surfaces43 or clusters.44,45 In contrast, if CO2 is activated by partial electron transfer from the (cluster) surface, the C–O bonds weaken and the molecule adopts a bent geometry.46,47 This change of geometry results in the breakdown of the Fermi dyad and a red-shift of the asymmetric stretching mode – depending on the binding geometry – by several hundreds of wavenumbers.30,48–50 Thus, weak adsorption of CO2 on a (cluster) surface can be clearly distinguished from activated adsorption via infrared spectroscopy.
Fig. 2 displays the IR-MPD spectra of cationic and anionic CuxOy(CO2) complexes in the 1050–1800 cm−1 spectral region which is most diagnostic for the activation of the attached CO2. The left column shows that for all cationic complexes two peaks centered at 1225–1278 cm−1 and 1372–1393 cm−1 (the exact position slightly depends on the cluster size) are observed (indicated by blue dashed lines). The spectral position of these bands is close to the frequency of the characteristic Fermi dyad of the unperturbed CO2, which indicates a linear CO2 adsorption geometry and only a small perturbation of the CO2 molecule upon interaction with the clusters.44 Since metal–oxygen vibrations51 are typically found below 1100 cm−1 the remaining strong bands shown in the IR-MPD spectra most likely arise from motions of non-activated52–54 (1400–1550 cm−1) or activated52–57 (<1200 cm−1) O2. Due to the large amount of bare clusters as well as CO2 complexes in the mass spectrum (with potentially overlapping fragmentation channels) it is not easy to trace fragmentation pathways for all of the clusters. However, for some complexes such as (Cu2O3(CO2)+, Cu2O4(CO2)+, Cu3O4(CO2)+) we find fragmentation via loss of O2 units indicating the rather weak binding of O2 in agreement with the observation of the bands around 1500 cm−1 that are indicative for a non-activated O2 unit. In addition, a low intensity band at around 1190–1200 cm−1 is present. A similar band was previously also observed for Cux(CO2)+ and assigned to the antisymmetric CO2 stretching mode (ν3 = 2349 cm−1 for free CO2).38 This band can potentially become visible at around 1200 cm−1 due to the likely presence of second harmonic radiation of the free electron laser.45
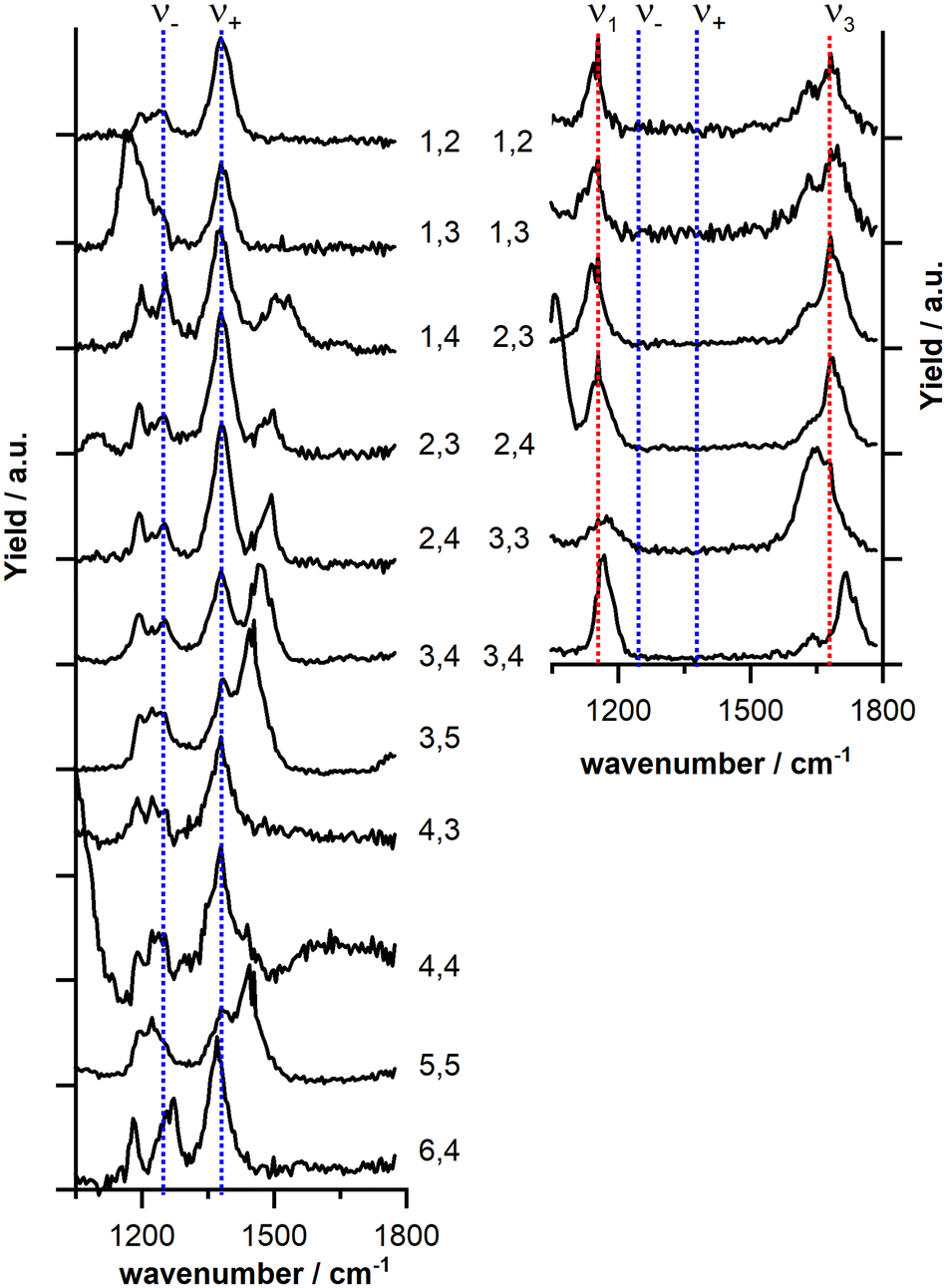 | ||
| Fig. 2 IR-MPD spectra of (left column) cationic CuxOy(CO2)+ and (right column) anionic CuxOy(CO2)− complexes (labeled x,y). The blue dotted lines indicate the Fermi dyad and the red dotted lines modes of activated CO2. The black tick marks on the y-axis indicate the zero depletion yield for each spectrum. The spectra of the anionic complexes were recorded at reduced IR laser fluence (cf. Section 2.1). | ||
In marked contrast, the IR-MPD spectra of the anionic complexes (Fig. 2, right column) do not show the Fermi dyad (indicated by blue dashed lines) but instead two bands are observed, centered around 1150 cm−1 and 1680 cm−1, respectively. These two bands fall in the range where CO2 stretching vibrations were observed earlier for bent CO2 geometries and thus indicate the activation of CO2via electron transfer.30,48–50 It should be noted that in case of the anionic complexes, the assignment of fragmentation channels is more difficult, but e.g. for Cu2O4(CO2)− we have indications that fragmentation also occurs via loss of O2. Based on these observations we conclude that in the investigated cluster size (number of Cu atoms) and composition (Cu/O ratio) range, cluster charge is the decisive parameter for CO2 activation. Although cluster size and composition to some extend affect the reactivity towards CO2 (cf.Fig. 1) they seem to play only a minor role in the activation of CO2.
To gain more insight into the CO2 activation and bonding to anionic copper oxide clusters we will in the following discuss the IR-MPD spectra of Cu2O4(CO2)− and Cu3O4(CO2)− in more detail, serving as representatives for all CuxOy(CO2)− species.
3.3 CO2 activation by Cu2O4− and Cu3O4− – computations
Fig. 3 displays the IR-MPD spectrum of Cu2O4(CO2)− (gray shaded spectrum) exhibiting six bands (labeled I–VI). As discussed above, bands I (centered at 1681 cm−1) and II (1154 cm−1) are likely due to vibrations of a bent CO2 and band III is likely due to the O–O stretch of a superoxide-like O2 unit (1057 cm−1, cf. 1090 cm−1 for free O2−).58 An intense structural search, from which we present the detailed product structures below, revealed that CO2 is activated by Cu2O4−via exclusive formation of a CO3 unit (a formal adduct of an oxide ion and CO2), which can bind in different ways to the cluster. It should be noted, that in the structural search also isomers with activated bent CO2 bound to one of the Cu atoms were considered. However, such a structure was found to be considerably higher in energy (2.36 eV; cf. Fig. S2, ESI†). In the structural search CO2 dissociation into carbon monoxide was also considered, but this appears to be unfavorable: the lowest energy isomer containing a copper bound CO was found 3.55 eV higher in energy than the located lowest energy isomer with an intact CO2. Furthermore, for all investigated structures the doublet spin state is lower in energy than the quartet. Accordingly, unless otherwise stated, only the doublet state isomers are discussed below.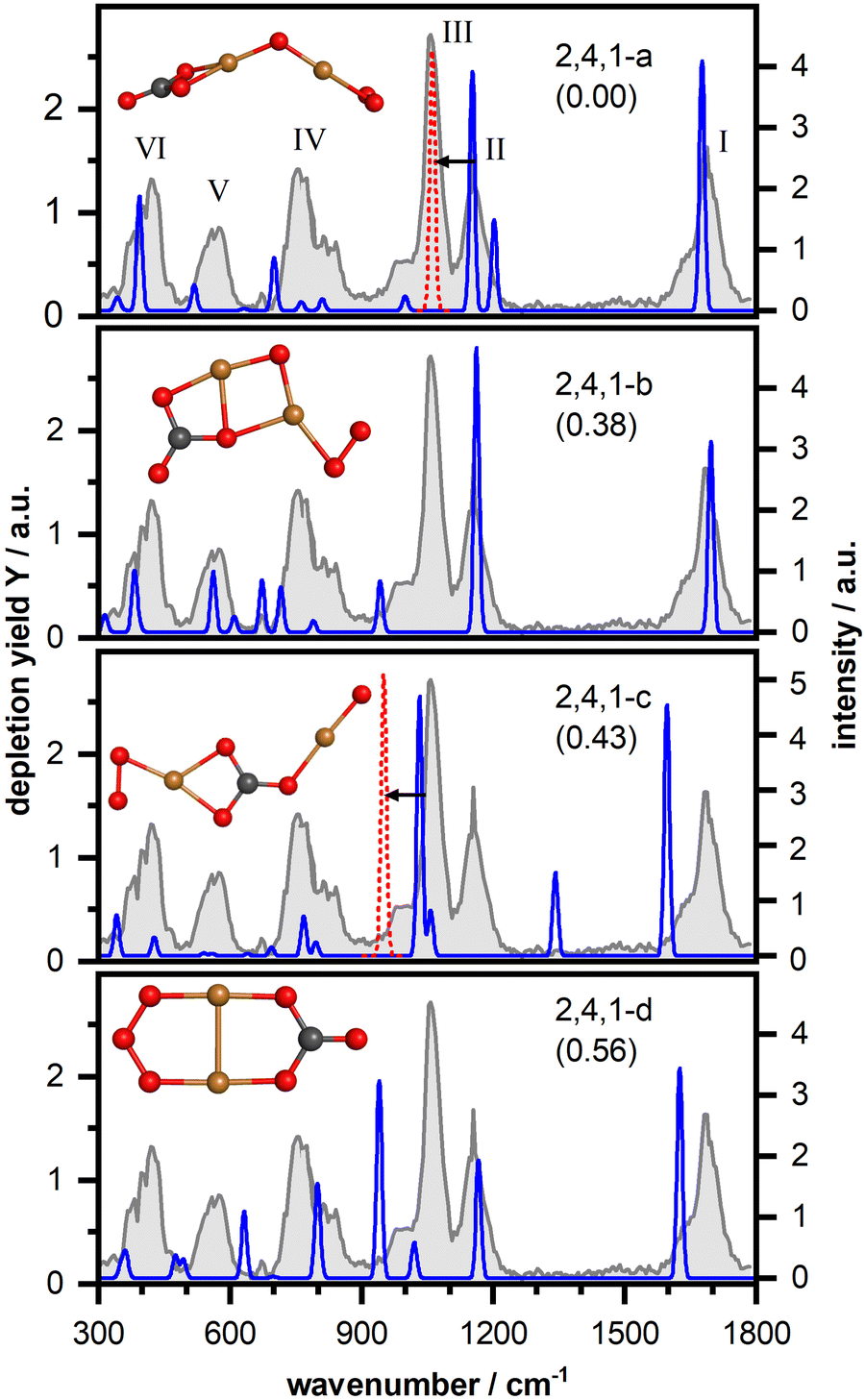 | ||
| Fig. 3 IR-MPD spectrum (in gray) of Cu2O4(CO2)− together with calculated vibrational spectra (in blue) of several isomeric structures (computed relative energies in eV are given between parentheses). The experimental spectrum was measured in two independent runs (1800–660 cm−1 and 800–240 cm−1, with the former range recorded at reduced FELICE fluence). To obtain an intensity match of the peaks in the overlapping spectral region the depletion yield of the 800–240 cm−1 spectrum was multiplied by a factor 0.6. The intensities of the calculated spectrum were scaled the wavelength (cf. methods section). The red dashed peak corresponds to the O–O stretching mode resulting from the scaling with a factor of 0.921, the shift is indicated by a black arrow. Cu, C, and O atoms are depicted as yellow, black, and red spheres, respectively. | ||
The lowest energy isomer for Cu2O4(CO2)− (isomer 2,4,1-a in Fig. 3) is based on the bent chain-like O2–Cu–O–Cu–O cluster that we identified as the lowest energy isomer of the bare cluster (Fig. S5 (ESI†); a similar but more linear structure has previously been reported59) with the CO2 molecule forming a CO3 unit with the terminal oxygen atom. The resulting CO3 group is η2(O,O) bound to one of the Cu atoms leading to a stabilization of 1.54 eV relative to the reactants (Cu2O4− + CO2). However, the properties of this metal-bound CO3 deviate from those of the unperturbed, highly symmetric (D3h) carbonate ion. Here, the trigonal planar geometry is distorted with an internal O–C–O angle of 109° as well as a charge of −0.86 e (cf. Fig. S5, ESI†), both significantly affecting the vibrational frequencies. Morgan and Staats, in analyzing the IR spectra of copper carbonate dilute solid solution in potassium-halides,60 have noted the significant difference between the observed 1785 and 1265 cm−1 frequencies and the 1415 cm−1 frequency of the doubly degenerate asymmetric stretching vibration of the naked carbonate dianion, attributing the splitting to the lowered symmetry. They tentatively attributed their 1785 cm−1 vibration to a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch in an OCOO resonance structure (the two oxygens bound to C by a single bond interacting with Cu). In agreement, our calculations show that the degenerate E' pair of CO32− (1292 cm−1) is split in two components for 2,4,1-a (1202 cm−1 and 1676 cm−1; see also Fig. S3, ESI†). Considering the different charges in the systems (carbonate dianion and 2,4,1-a monoanion) the numerical frequency values are not directly comparable, but the qualitative similarity is clear. Likewise, the experimental observations of the neutral copper-carbonate in the potassium-halide matrix are not directly comparable with the frequencies of the gas phase anion Cu2O4(CO2)−. Nevertheless, qualitatively, it is clear that the observed spectrum is in accordance with the activation of CO2, resulting in a distorted CO3 structure and the observed frequency near 1700 cm−1 is a clear indication for a CO double bond.
O stretch in an OCOO resonance structure (the two oxygens bound to C by a single bond interacting with Cu). In agreement, our calculations show that the degenerate E' pair of CO32− (1292 cm−1) is split in two components for 2,4,1-a (1202 cm−1 and 1676 cm−1; see also Fig. S3, ESI†). Considering the different charges in the systems (carbonate dianion and 2,4,1-a monoanion) the numerical frequency values are not directly comparable, but the qualitative similarity is clear. Likewise, the experimental observations of the neutral copper-carbonate in the potassium-halide matrix are not directly comparable with the frequencies of the gas phase anion Cu2O4(CO2)−. Nevertheless, qualitatively, it is clear that the observed spectrum is in accordance with the activation of CO2, resulting in a distorted CO3 structure and the observed frequency near 1700 cm−1 is a clear indication for a CO double bond.
The calculated vibrational spectrum itself is shown in light and dark blue lines in Fig. 3, where the light blue part of the spectrum is vertically scaled by a factor 0.2 to improve visibility. This calculated spectrum provides a very reasonable match for all experimentally observed bands, except for band III, although the frequencies of bands IV–VI seem somewhat underestimated by the calculations. Analysis of the distortion vectors reveals that the modes calculated at 1676 cm−1, 1202 cm−1, and 998 cm−1 correspond to the C–O stretching vibration (matching band I), the asymmetric (band II) and the symmetric (low-frequency shoulder of band III) OCO stretching vibrations of the CO3 unit, in accordance with the qualitative discussion above, while the mode at 1152 cm−1 arises from the O–O stretching vibration of the η2-bound O2 unit. All other modes mainly arise from vibrations of the cluster core. Two linear but non planar structures with the same cluster and CO3 binding motif were found to be only 0.04 eV and 0.24 eV, respectively, higher in energy (isomers 2,4,1-e and 2,4,1-f – multiplicity 4 – in Fig. S2, ESI†). These isomers have similar vibrational spectra but fail to explain bands V and III.
A second more compact structure (isomer 2,4,1-b, +0.39 eV) consists of a rhombic Cu2O2 cluster with O2 η2-bound to one of the Cu atoms and the CO2 bridge bound to the Cu–O, forming a CO3 unit. This isomer has a similarly good match for bands I (C–O stretching vibration predicted at 1696 cm−1) and II (two modes predicted at 1162 cm−1 and 1168 cm−1 both arising from coupled asymmetric OCO and O–O stretching motions), but fails to reproduce the low-frequency part of the spectrum (mainly vibrations of the cluster core), including band III. We found two more structures with a similar CO3 binding motif (isomers 2,4,1-g,h in Fig. S2, ESI†). Both structures are even higher in energy (0.51 eV and 0.83 eV) and the computed harmonic vibrational frequencies and intensities are not in agreement with the IR-MPD spectrum either. Insertion of the CO2 molecule into a chain-like Cu2O4-core results in isomer 2,4,1-c. Such a structure can, however, be excluded based on a mismatch with bands I and II and a rather intense band predicted at 1343 cm−1 (asymmetric OCO stretching vibration) which is not observed in the IR-MPD spectrum. Finally, we found an isomer (2,4,1-d; +0.56 eV) containing an ozone-like O3 and a CO3 unit, for which the vibrational spectrum is largely in disagreement with the experimental spectrum.
Thus, we conclude that the IR-MPD spectrum is best described by isomer 2,4,1-a, although some of the calculated frequencies are shifted compared to the experimental ones. The largest question mark is for the mismatch for band III. To address this, we take a closer look at the O–O stretch vibration, calculated at 1152 cm−1. In previous studies it was shown that DFT calculations tend to overestimate the O–O bond strength of η1 and η2-bound O2 units.52,53,55,61 For manganese oxide clusters with an experimental O–O stretching vibration around 1200 cm−1 a moderate frequency scaling factor of 0.9806 was required to match the experimental data.55 For gold oxides experimental O–O stretching vibrations were observed around 1060 cm−1 (close to frequency of band III) and significantly stronger scaling (0.907–0.936) was required.52,53,61 If we scale the frequency of the O–O stretching vibration of isomer 2,4,1-a by a factor of 0.921 (the average of 0.907 and 0.936) it matches band III very well, as indicated by the red dashed band and black arrow in Fig. 2. In case of isomer 2,4,1-b such a scaling is difficult since both modes predicted at 1162 cm−1 and 1168 cm−1 are not pure O–O stretching vibrations but arise from coupled asymmetric OCO and O–O stretching motions. For isomer 2,4,1-c scaling clearly leads to an even poorer match. To conclude, we assign the spectrum recorded for the Cu2O4(CO2)− cluster to structure 2,4,1-a, under the assumption that the frequency of the O–O stretching vibration is significantly overestimated by the calculations.
Next, we have investigated the complex Cu3O4(CO2)− in more detail. Fig. 4 shows the IR-MPD spectrum (in gray) and the calculated vibrational spectra of selected isomers (more structures and spectra can be found in Fig. S4, ESI†). The IR-MPD spectrum exhibits eight bands (labeled I–VIII). Similar to Cu2O4(CO2)− bands I and II are characteristic CO2 bands, but band I is somewhat blue shifted (to 1717 cm−1) with respect to the corresponding Cu2O4(CO2)− band. Based on our analysis for Cu2O4(CO2)−, band III can be tentatively assigned to the O–O stretching vibration of a super- or even peroxo-like O2 unit.
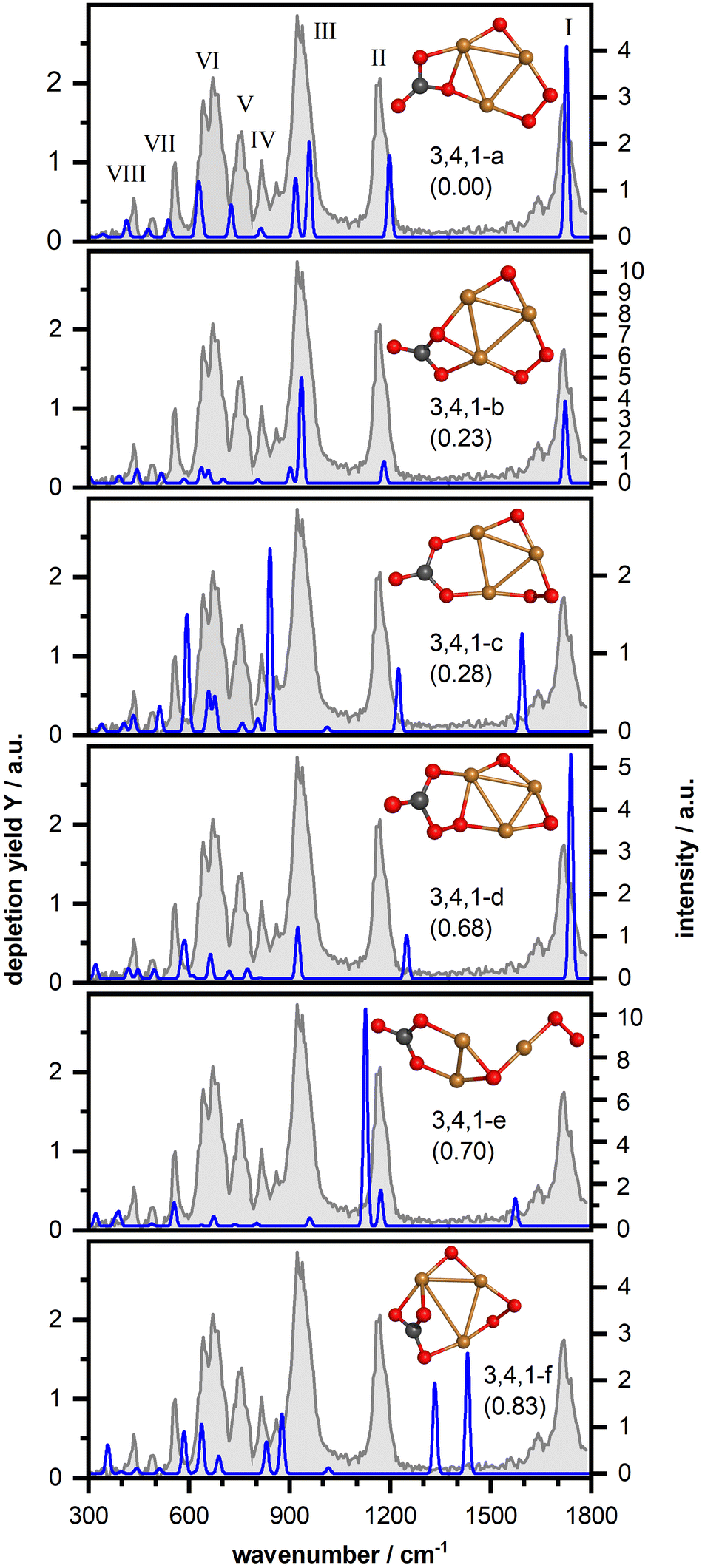 | ||
| Fig. 4 IR-MPD spectrum (in gray) of Cu3O4(CO2)− together with calculated harmonic vibrational spectra (in blue) of several isomeric structures. For more details, see caption of Fig. 3. | ||
Our structural search revealed thirteen isomeric structures, all differing by an energy of up to about 1 eV and, similar to Cu2O4(CO2)−, all containing a CO3 unit. Once more, as with Cu2O4(CO2)−, an isomer with metal-bound activated CO2 is, at 2.34 eV, considerably higher in energy than the lowest energy isomer (cf. Fig. S4, ESI†). Again, like for Cu2O4(CO2)−, CO2 dissociation into CO is energetically unfavorable and the lowest energy isomer containing CO is 3.19 eV higher in energy than the lowest energy isomer 3,4,1-a. Therefore, we will in the following only discuss selected structures with different structural motifs and reference to the ESI† for further similar structures or structures that are considerably higher in energy. Since band III suggests the presence of a (su)peroxo O2 we will not discuss any structures here that do not contain such a unit. All structures are found with the triplet spin state lower in energy than the singlet one.
The first class of isomers is based on a planar Cu3O4− consisting of a triangular Cu3 frame with two bridging oxygen atoms and a bridging O2 molecule bound to the third Cu–Cu side (cf. Fig. S5 (ESI†) for the structure of the bare cluster, which is largely in agreement with the structure previously determined via photoelectron spectroscopy62) to which a CO2 molecule is η2(C,O) coordinated forming a μ2–η2(O,O)-bound CO3 unit with one of the bridging oxygen atoms. In the lowest energy isomer (3,4,1-a in Fig. 4) one of the CO3 oxygen atoms bridges two Cu atoms and a second O atom is bound to one of the Cu atoms. The calculated vibrational spectrum of this isomer shows a reasonable agreement with the experimental spectrum. However, besides some spectral shifts in the low frequency region as well as for band II, the right part of the double-band VI is missing in the calculated spectrum and the intensity of band III is underestimated. These three bands are better reproduced by isomer 3,4,1-b (with a different position of the CO3 group), but now the spectrum in the region of bands V and VII is less favorable. With isomer 3,4,1-c we found a similar structure where the CO2 has incorporated the oxygen atom, and the resulting CO3 group symmetrically bridges two Cu atoms. This leads to considerable shifts of three high-frequency modes (the terminal C–O stretch, the OCO asymmetric stretch of CO3, and the O–O stretch respectively), which disagrees with the experimental spectrum. In particular isomers 3,4,1-a and 3,4,1-b describe band I very well and thus also the blue shift observed with respect to the corresponding Cu2O4(CO2)− band. Since this frequency corresponds to the carbon–oxygen stretching (see Fig. S3, ESI†) the blue shift indicates a strengthening of this bond in Cu3O4(CO2)−. Indeed, in the calculated structure of 2,4,1-a, the carbon–oxygen distance is 1.221 Å, against 1.215 Å in 3,4,1-a, with both values well in the CO double bond range (ca. 1.20 Å). Thus, the size of the copper-oxide cluster seems to affect the extent of the CO2 activation, which might potentially also affect any subsequent reactions of this unit, for example in a hydrogen reduction reaction. Finally, it should be noted that for all structures in this class, the O–O stretch vibration of the bridging μ2-bound O2 appears to be considerably better described than in the case of the μ1-bound O2 of Cu2O4(CO2)−.
Other classes of isomers were also found, but their spectral properties do not provide a much better comparison. In the first of these, the CO2 binds to the O2 unit of the reactant cluster, now forming CO3 that coordinates to Cu and O atoms forming a CuOOC(O)O ring (isomer 3,4,1-d; for more see Fig. S4, ESI†). Such a binding motif has a much weaker O–O stretch intensity (very weak mode at 812 cm−1) and a blue-shift of the C–O (1739 cm−1) and asymmetric OCO stretching vibrations (1248 cm−1) and thus an inferior match with the IR-MPD spectrum. In a third class of isomers (isomer 3,4,1-e; for more see Fig. S4, ESI†) the triangular Cu3 core is strongly distorted with the CO3 unit bridging two Cu atoms and the O2 unit η2-bound. This leads to a weakening and red-shift of the C–O stretching vibration (1574 cm−1) and a considerable blue-shift of the O–O stretching vibration (1126 cm−1). An attempt to rationalize the mismatch of the O–O stretch on the same grounds as in the case of Cu2O4(CO2)− (i.e. applying a scaling factor of 0.921) results in an even larger mismatch. Finally, a fourth class of isomers found, contains a η3-bound CO3 group (isomer 3,4,1-f, for more see Fig. S4, ESI†). The additional coordination of the terminal C–O results in a red-shift of its stretching mode by about 300 cm−1 and a blue-shift of the OCO asymmetric stretching mode of more than 100 cm−1. Both end up in a spectral region where we do not observe any bands in the IR-MPD spectrum, disqualifying this class.
Overall, we conclude that the IR-MPD spectrum is best described by the structures containing a formal CO3 group species with a terminal C–O and one oxygen atom bridging two Cu atoms, such as isomers 3,4,1-a or 3,4,1-b. Since none of these isomers provide a perfect match, we hesitate to make a firm assignment but assume that multiple, quite similar species are present in the molecular beam. This assumption is supported by the small energy difference between isomers 3,4,1-a, -b, and -c raising the possibility of a frustrated rotation of the formal CO3 unit linking the different structures. We finally would like to note that there is no ready explanation for the low frequency satellite of band I (which also appears to be present for other anionic clusters shown in Fig. 2). One possibility would be an overtone of the O–O stretch (found at 930 cm−1), but the required anharmonicity exceeding 20% seems too much. Another possibility would be a second isomer with a slightly differently bound CO2. However, as discussed above for Cu3O4(CO2)− no such isomer was located. Therefore, it can be speculated that the weak satellite bands could be hot bands, originating from vibrationally excited states insufficiently cooled during the expansion of the molecular beam into the vacuum. It is known that vibrational cooling is poor in any molecular beam expansion and especially for those from a flow-tube, where pressures are already lower than stagnation pressures of conventional molecular beams.
4 Summary and conclusions
We have produced cationic and anionic copper oxide clusters via laser ablation of a 65Cu target in the presence of an O2/He gas pulse. Under the given experimental conditions, the cluster distribution of cationic clusters is dominated by highly oxidized clusters, while mainly stoichiometric and oxygen-deficient anionic clusters were formed. Subsequent reaction of the formed clusters with CO2 in a flow tube reactor showed that primarily near-stoichiometric CuxOy(CO2)+/− complexes are formed. Furthermore, IR-MPD spectroscopy of the formed products in conjunction with DFT calculations on two selected systems (Cu2O4(CO2)− and Cu3O4(CO2)−) revealed that (1) CO2 binds to cationic clusters as a linear molecule without noteworthy activation, while activation occurs on all anionic clusters. Thus, in the investigated size range, cluster charge is the decisive parameter for CO2 activation and size or composition (Cu/O ratio) only play a minor role. (2) activation of CO2 upon binding to anionic copper oxide clusters leads to formation of a CO3 unit. The activation of CO2 is fine-tuned by the size of the copper oxide cluster influencing the strength of the CO bond in the CO3 group. (3) CO2 dissociation is highly unlikely due to the relatively weak binding of CO to the copper oxide clusters.
These findings are important for the potential rational design of copper-based CO2 hydrogenation catalysts. Our study shows that anionic copper oxide clusters are able to activate CO2, but that the most dominant factor is the presence of excess negative charge, something which was already found by Weber and co-workers for single-atomic ions,47,50 and later confirmed by others for few-atom metal clusters.48,49,63,64 Furthermore, we observe the exclusive formation of a CO3 unit while dissociation to CO is energetically unfavorable. This indicates that the presence of oxygen atoms stabilizes CO3 and prevents CO2 dissociation. Similar CO3 intermediates have previously been observed for some anionic mono65–67- and few-metal oxide clusters64 as well as (basic) metal-oxide surfaces68–71 and even a few cationic species forming a CO3 intermediate have been reported.72,73 The presence of CO3 and the lack of CO2 dissociation suggest that any further hydrogenation reaction proceeds via hydrogenation of the activated CO2 (CO3), potentially leading to formate or bicarbonate intermediates. Consequently, the formation of carbon monoxide is less likely. As carbon monoxide is a key intermediate in the C2+ product formation route,74,75 we speculate that C1 products are more likely to form (via the formate route) which can lead to methane or methanol.76–78
Data availability
The data for this article are shown in the Figure. Further data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie Grant Agreement No. 955650. We also thank the Nederlandse Organisatie voor Wetenschappelijk Onderzoek (NWO) for the support of the FELIX Laboratory. The research leading to these results has received funding from LASERLAB-EUROPE (grant agreement no. 654148, European Union's Horizon 2020 research and innovation program). T. H. is grateful for the János Bolyai Research Scholarship of the Hungarian Academy of Sciences (grant number BO/00642/21/7).References
- J. Klankermayer, S. Wesselbaum, K. Beydoun and W. Leitner, Angew. Chem., Int. Ed., 2016, 55, 7296–7343 CrossRef.
- K. M. K. Yu, I. Curcic, J. Gabriel and S. C. E. Tsang, ChemSusChem, 2008, 1, 893–899 CrossRef PubMed.
- K. C. Waugh, Catal. Today, 1992, 15, 51–75 CrossRef.
- F. Studt, M. Behrens, E. L. Kunkes, N. Thomas, S. Zander, A. Tarasov, J. Schumann, E. Frei, J. B. Varley, F. Abild-Pedersen, J. K. Nørskov and R. Schlögl, ChemCatChem, 2015, 7, 1105–1111 CrossRef.
- A. Beck, M. Zabilskiy, M. A. Newton, O. Safonova, M. G. Willinger and J. A. van Bokhoven, Nat. Catal., 2021, 4, 488–497 CrossRef.
- A. R. Woldu, Z. Huang, P. Zhao, L. Hu and D. Astruc, Coord. Chem. Rev., 2022, 454, 214340 CrossRef.
- S. Wang, T. Kou, S. E. Baker, E. B. Duoss and Y. Li, Mater. Today Nano, 2020, 12, 100096 CrossRef.
- A. Eilert, F. Cavalca, F. S. Roberts, J. Osterwalder, C. Liu, M. Favaro, E. J. Crumlin, H. Ogasawara, D. Friebel, L. G. M. Pettersson and A. Nilsson, J. Phys. Chem. Lett., 2017, 8, 285–290 CrossRef PubMed.
- F. Yang, S. Jiang, S. Liu, P. Beyer, S. Mebs, M. Haumann, C. Roth and H. Dau, Commun. Chem., 2024, 7, 1–13 CrossRef.
- M. Favaro, H. Xiao, T. Cheng, W. A. Goddard, J. Yano and E. J. Crumlin, Proc. Natl. Acad. Sci., 2017, 114, 6706–6711 CrossRef.
- R. Poreddy, C. Engelbrekt and A. Riisager, Catal. Sci. Technol., 2015, 5, 2467–2477 RSC.
- J. B. Reitz and E. I. Solomon, J. Am. Chem. Soc., 1998, 120, 11467–11478 CrossRef.
- X. Liu, S. Cui, M. Qian, Z. Sun and P. Du, Chem. Commun., 2016, 52, 5546–5549 RSC.
- C. E. Elwell, N. L. Gagnon, B. D. Neisen, D. Dhar, A. D. Spaeth, G. M. Yee and W. B. Tolman, Chem. Rev., 2017, 117, 2059–2107 CrossRef PubMed.
- S. M. Lang and T. M. Bernhardt, Phys. Chem. Chem. Phys., 2012, 14, 9255–9269 RSC.
- G. Sun, A. N. Alexandrova and P. Sautet, ACS Catal., 2020, 10, 5309–5317 CrossRef.
- D. Palagin, A. J. Knorpp, A. B. Pinar, M. Ranocchiari and J. A. van Bokhoven, Nanoscale, 2017, 9, 1144–1153 RSC.
- S. Nganai, B. Dellinger and S. Lomnicki, Environ. Sci. Technol., 2014, 48, 13864–13870 CrossRef PubMed.
- G. Yin, M. Nishikawa, Y. Nosaka, N. Srinivasan, D. Atarashi, E. Sakai and M. Miyauchi, ACS Nano, 2015, 9, 2111–2119 CrossRef.
- C. Liu, B. Yang, E. Tyo, S. Seifert, J. DeBartolo, B. von Issendorff, P. Zapol, S. Vajda and L. A. Curtiss, J. Am. Chem. Soc., 2015, 137, 8676–8679 CrossRef CAS PubMed.
- T. Tsukuda and H. Häkkinen, J. Phys. Chem. C, 2021, 125, 4927–4929 CrossRef CAS.
- L.-S. Wang, H. Wu, S. R. Desai and L. Lou, Phys. Rev. B, 1996, 53, 8028–8031 CrossRef CAS.
- M. Abdul Latif, J. W. J. Wu, R. Moriyama, M. Nakano, K. Ohshimo and F. Misaizu, ACS Omega, 2018, 3, 18705–18713 CrossRef CAS PubMed.
- K. Morita, K. Sakuma, K. Miyajima and F. Mafuné, J. Phys. Chem. A, 2013, 117, 10145–10150 CrossRef.
- G.-T. Bae, B. Dellinger and R. W. Hall, J. Phys. Chem. A, 2011, 115, 2087–2095 CrossRef PubMed.
- G.-T. Bae, Bull. Korean Chem. Soc., 2016, 37, 638–642 CrossRef.
- M. Jadraque and M. Martín, Chem. Phys. Lett., 2008, 456, 51–54 CrossRef.
- S. Das, S. Nigam, P. Sharma and C. Majumder, Phys. Chem. Chem. Phys., 2024, 26, 11506–11515 RSC.
- G.-T. Bae, Comput. Theor. Chem., 2021, 1204, 113377 CrossRef CAS.
- A. Fielicke, Chem. Soc. Rev., 2023, 52, 3778–3841 RSC.
- J. M. Bakker, V. J. F. Lapoutre, B. Redlich, J. Oomens, B. G. Sartakov, A. Fielicke, G. von Helden, G. Meijer and A. F. G. van der Meer, J. Chem. Phys., 2010, 132, 074305 CrossRef.
- M. Haertelt, V. J. F. Lapoutre, J. M. Bakker, B. Redlich, D. J. Harding, A. Fielicke and G. Meijer, J. Phys. Chem. Lett., 2011, 2, 1720–1724 CrossRef CAS.
- Y. Wang, J. Lv, L. Zhu and Y. Ma, Comput. Phys. Commun., 2012, 183, 2063–2070 CrossRef CAS.
- M. J. Frisch, et al., Gaussian 16 Rev. B.01, 2016 Search PubMed.
- E. Epifanovsky, et al. , J. Chem. Phys., 2021, 155, 084801 CrossRef CAS PubMed.
- M. Szalay, D. Buzsáki, J. Barabás, E. Faragó, E. Janssens, L. Nyulászi and T. Höltzl, Phys. Chem. Chem. Phys., 2021, 23, 21738–21747 RSC.
- B. Zamora, L. Nyulászi and T. Höltzl, Chem. Phys. Chem., 2024, 25, e202300409 CrossRef CAS.
- T. Shimanouchi, U. S. N. B. of Standards, Tables of Molecular Vibrational Frequencies. Consolidated Volume I, U.S. Government, 1972.
- G. Amat and M. Pimbert, J. Mol. Spectrosc., 1965, 16, 278–290 CrossRef CAS.
- H. R. Gordon and T. K. McCubbin, J. Mol. Spectrosc., 1966, 19, 137–154 CrossRef CAS.
- H. E. Howard-Lock and B. P. Stoicheff, J. Mol. Spectrosc., 1971, 37, 321–326 CrossRef CAS.
- A. Chedin, J. Mol. Spectrosc., 1979, 76, 430–491 CrossRef CAS.
- G. Ramis, G. Busca and V. Lorenzelli, Mater. Chem. Phys., 1991, 29, 425–435 CrossRef.
- N. Zimmermann, T. M. Bernhardt, J. M. Bakker, R. N. Barnett, U. Landman and S. M. Lang, J. Phys. Chem. A, 2020, 124, 1561–1566 CrossRef PubMed.
- O. V. Lushchikova, M. Szalay, H. Tahmasbi, L. B. F. Juurlink, J. Meyer, T. Höltzl and J. M. Bakker, Phys. Chem. Chem. Phys., 2021, 23, 26661–26673 RSC.
- A. D. Walsh, J. Chem. Soc., 1953, 2260–2266 RSC.
- J. M. Weber, Int. Rev. Phys. Chem., 2014, 33, 489–519 Search PubMed.
- A. E. Green, J. Justen, W. Schöllkopf, A. S. Gentleman, A. Fielicke and S. R. Mackenzie, Angew. Chem., Int. Ed., 2018, 57, 14822–14826 CrossRef PubMed.
- O. V. Lushchikova, M. Szalay, T. Höltzl and J. M. Bakker, Faraday Discuss., 2023, 242, 252–268 RSC.
- L. G. Dodson, M. C. Thompson and J. M. Weber, Annu. Rev. Phys. Chem., 2018, 69, 231–252 CrossRef.
- K. R. Asmis, Phys. Chem. Chem. Phys., 2012, 14, 9270–9281 RSC.
- A. P. Woodham, G. Meijer and A. Fielicke, J. Am. Chem. Soc., 2013, 135, 1727–1730 CrossRef PubMed.
- A. P. Woodham and A. Fielicke, Angew. Chem., Int. Ed., 2014, 53, 6554–6557 CrossRef.
- J. Mariñoso Guiu, B.-A. Ghejan, T. M. Bernhardt, J. M. Bakker, S. M. Lang and S. T. Bromley, ACS Earth Space Chem., 2022, 6, 2465–2470 CrossRef PubMed.
- N. Zimmermann, T. M. Bernhardt, J. M. Bakker, U. Landman and S. M. Lang, Phys. Chem. Chem. Phys., 2019, 21, 23922–23930 RSC.
- F. Mafuné, K. Koyama, T. Nagata, S. Kudoh, T. Yasuike, K. Miyajima, D. M. M. Huitema, V. Chernyy and J. M. Bakker, J. Phys. Chem. C, 2019, 123, 5964–5971 CrossRef.
- C. Kerpal, D. J. Harding, A. C. Hermes, G. Meijer, S. R. Mackenzie and A. Fielicke, J. Phys. Chem. A, 2013, 117, 1233–1239 CrossRef PubMed.
- K. P. Huber and G. Herzberg, Molecular Spectra and Molecular Structure, Springer US, Boston, MA, 1979 Search PubMed.
- B. Dai, L. Tian and J. Yang, J. Chem. Phys., 2004, 120, 2746–2751 CrossRef PubMed.
- H. W. Morgan and P. A. Staats, J. Appl. Phys., 1962, 33, 364–366 CrossRef.
- A. P. Woodham, G. Meijer and A. Fielicke, Angew. Chem., Int. Ed., 2012, 51, 4444–4447 CrossRef.
- X.-L. Xu, B. Yang, Z.-Y. Wei, G.-J. Cao, H.-G. Xu and W.-J. Zheng, Phys. Chem. Chem. Phys., 2018, 20, 20622–20628 RSC.
- A. Yanagimachi, K. Koyasu, D. Y. Valdivielso, S. Gewinner, W. Schöllkopf, A. Fielicke and T. Tsukuda, J. Phys. Chem. C, 2016, 120, 14209–14215 CrossRef.
- S. Debnath, X. Song, M. R. Fagiani, M. L. Weichman, M. Gao, S. Maeda, T. Taketsugu, W. Schöllkopf, A. Lyalin, D. M. Neumark and K. R. Asmis, J. Phys. Chem. C, 2019, 123, 8439–8446 CrossRef.
- L. G. Dodson, M. C. Thompson and J. M. Weber, J. Phys. Chem. A, 2018, 122, 6909–6917 CrossRef.
- B. J. Knurr and J. M. Weber, J. Phys. Chem. A, 2015, 119, 843–850 CrossRef PubMed.
- M. Salzburger, R. T. Saragi, F. J. Wensink, E. M. Cunningham, M. K. Beyer, J. M. Bakker, M. Ončák and C. van der Linde, J. Phys. Chem. A, 2023, 127, 3402–3411 CrossRef PubMed.
- A. Álvarez, M. Borges, J. J. Corral-Pérez, J. G. Olcina, L. Hu, D. Cornu, R. Huang, D. Stoian and A. Urakawa, Chem. Phys. Chem., 2017, 18, 3135–3141 CrossRef PubMed.
- U. Burghaus, Prog. Surf. Sci., 2014, 89, 161–217 CrossRef.
- H.-J. Freund and M. W. Roberts, Surf. Sci. Rep., 1996, 25, 225–273 CrossRef.
- Y. Wang, R. Kováčik, B. Meyer, K. Kotsis, D. Stodt, V. Staemmler, H. Qiu, F. Traeger, D. Langenberg, M. Muhler and C. Wöll, Angew. Chem., Int. Ed., 2007, 46, 5624–5627 CrossRef PubMed.
- Z. Zhao, X. Kong, Q. Yuan, H. Xie, D. Yang, J. Zhao, H. Fan and L. Jiang, Phys. Chem. Chem. Phys., 2018, 20, 19314–19320 RSC.
- E. I. Brewer, A. E. Green, A. S. Gentleman, P. W. Beardsmore, P. A. J. Pearcy, G. Meizyte, J. Pickering and S. R. Mackenzie, Phys. Chem. Chem. Phys., 2022, 24, 22716–22723 RSC.
- A. J. Garza, A. T. Bell and M. Head-Gordon, ACS Catal., 2018, 8, 1490–1499 CrossRef.
- R. Kortlever, J. Shen, K. J. P. Schouten, F. Calle-Vallejo and M. T. M. Koper, J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef PubMed.
- J. Hussain, H. Jónsson and E. Skúlason, ACS Catal., 2018, 8, 5240–5249 CrossRef.
- T. Cheng, H. Xiao and W. A. I. Goddard, J. Am. Chem. Soc., 2016, 138, 13802–13805 CrossRef.
- H. Ma, E. Ibáñez-Alé, R. Ganganahalli, J. Pérez-Ramírez, N. López and B. S. Yeo, J. Am. Chem. Soc., 2023, 145, 24707–24716 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp02651a |
| This journal is © the Owner Societies 2024 |