
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Controlling nanocluster growth through nanoconfinement: the effect of the number and nature of metal–organic framework functionalities†
James
King
a,
Zhipeng
Lin
 a,
Federica
Zanca
b,
Hui
Luo
c,
Linda
Zhang
de,
Patrick
Cullen
a,
Mohsen
Danaie
f,
Michael
Hirscher
de,
Simone
Meloni
g,
Alin M.
Elena
b and
Petra Á.
Szilágyi
*h
a,
Federica
Zanca
b,
Hui
Luo
c,
Linda
Zhang
de,
Patrick
Cullen
a,
Mohsen
Danaie
f,
Michael
Hirscher
de,
Simone
Meloni
g,
Alin M.
Elena
b and
Petra Á.
Szilágyi
*h
aSchool of Engineering and Materials Science, Queen Mary University of London, Mile End Campus, E1 4NS, London, UK
bScientific Computing Department, Science and Technologies Facilities Council, Daresbury Laboratory, Keckwick Lane, Daresbury, WA4 4AD, UK
cDepartment of Chemical Engineering, Imperial College London, South Kensington Campus, London, SW7 2AZ, UK
dAdvanced Institute for Materials Research (WPI-AIMR), Tohoku University, Sendai 980-8577, Japan
eHydrogen Storage Group, Max Planck Institute for Intelligent Systems, Heisenbergstrasse. 3, Stuttgart 70569, Germany
felectron Physical Science Imaging Centre (ePSIC), Diamond Light Source, Didcot, OX11 0DE, UK
gDepartment of Chemical, Pharmaceutical and Agricultural Sciences, University of Ferrara, via Luigi Borsari 46, Ferrara 44121, Italy
hCentre for Materials Science and Nanotechnology (SMN), Department of Chemistry, University of Oslo, P.O. Box 1033, Blindern, Oslo N-0315, Norway. E-mail: p.a.szilagyi@kjemi.uio.no
First published on 10th September 2024
Abstract
Controlled nanocluster growth via nanoconfinement is an attractive approach as it allows for geometry control and potential surface-chemistry modification simultaneously. However, it is still not a straight-forward method and much of its success depends on the nature and possibly concentration of functionalities on the cavity walls that surround the clusters. To independently probe the effect of the nature and number of functional groups on the controlled Pd nanocluster growth within the pores of the metal–organic frameworks, Pd-laden UiO-66 analogues with mono- and bi-functionalised linkers of amino and methyl groups were successfully prepared and studied in a combined experimental–computational approach. The nature of the functional groups determines the strength of host–guest interactions, while the number of functional groups affects the extent of Pd loading. The interplay of these two effects means that for a successful Pd embedding, mono-functionalised host matrices are more favourable. Interestingly, in the context of the present and previous research, we find that host frameworks with functional groups displaying higher Lewis basicity are more successful at controlled Pd NC growth via nanoconfinement in MOFs.
Introduction
Noble metal nanoparticles (NPs) such as Pd have significantly different physical and chemical properties than their bulk counterparts,1–3 resulting in high potential for applications in a number of different fields such as imaging,4,5 cancer therapy6 and catalysis.7–9 Nanoclusters (NCs) have even smaller dimensions, typically ≤1 nm for transition metals, i.e. the size regime wherein ‘every atom matters’, resulting in even higher surface-to-volume ratios than those observed for nanoparticles, with the vast majority of atoms being on the particle surface.10–12 Thus, the difference in physical and chemical properties observed between bulk materials and NCs is even greater than those for NPs.13–16 In the nanocluster regime particle size significantly controls physical properties for certain applications it is important for NCs to be of uniform size as a non-uniform size distribution may lead to divergent behaviours. NCs have a high surface free energy due to their high surface-to-volume ratio, compromising their thermodynamic stability.17,18 As a consequence of this instability, NCs tend to sinter, or agglomerate to form larger particles, thereby increasing their stability by decreasing surface energy19 and, in this process, lose the unique properties that arise from their scale. As such, strongly binding, i.e. ‘anchoring’, supports are required to immobilise and stabilise NCs. Typically, 2D surfaces with functionalities are used which can interact with a side of the nanocluster.20,21 However, with only one side of the NCs held in place by the support, this anchoring effect is limited and particle aggregation can still occur, as has been observed for such materials used as catalysts in recurrent reaction cycles.22 On the other hand, 3D porous supports which can encapsulate NCs, thus anchoring them from multiple sides, overcome this issue while still allowing liquids and gases to reach the particles through open channels.23,24 Metal–organic frameworks (MOFs)25–28 are a particular class of porous materials that can act as 3D supports for NCs29–31 given their high surface area and crystallinity, with numerous other potential applications in fields such as gas storage32,33 and energy applications.34–36 These porous crystal lattices consist of inorganic nodes interconnected by organic linkers, which form extended frameworks on self-assembly, with a wide range of possible pore topologies and sizes, depending on the coordinative nature and geometry of the node and linker building units. Precursors for NCs can be introduced into the pores and subsequently reduced to form the desired metallic particles.18 Of particular interest with using MOFs to support NCs is the ability to engineer and fine-tune the dimensions of the particles. By virtue of the crystalline nature of MOFs, the pores are part of their crystal structure. Therefore, the size of clusters within their pores are geometrically limited by these dimensions allowing for their accurate size control, as long as the conditions for growing them are mild enough to maintain the bonds between building units within the framework. In addition, the ability to functionalise the MOF linkers coupled with the facile nature of interchanging linkers with different functional groups for MOFs of the same topology can allow for strong host–guest interactions between these groups and the guest. This approach enables the fixation of NCs within the MOF pores,37–39 thus inhibiting particle agglomeration, as well as to tailor the properties of the guest NCs by controlling their geometry and surface chemistry.40–42As previously stated, our focus of interest is in embedding Pd NCs in the pores of MOFs for accurate size control and reduced agglomeration, i.e. nanoparticle formation, a process that, in the case of catalysts would otherwise lead to deactivation.19 In addition, the proximity of these active sites to Lewis acid sites on the Zr6O8 nodes of UiO-66 may be utilised for phenomena such as tandem catalysis.43–46 Despite the benefits of MOF-supported NCs, i.e. controlled geometry and surface chemistry as well as increased morphological stability, the steering of the growth of guest particles within the pores remains a challenge47–49 and one that we have been exploring. Even with the use of computational modelling, only general tendencies could be previously identified.50,51 Given the set of analogues studied thus far, i.e. Br–, NH2–, (OH)2–, H–, (Cl)2–, the only obvious common denominator for a successful embedding was that there was one single functional group on the terephthalate linker (i.e. Br– and NH2–). However, this set of specimens has not enabled the independent study of the nature and number of functional groups on the linker, i.e. we have been unable to unequivocally assign this phenomenon to electronic effects and exclude potential symmetry and spatial contributions, and their effect on the successful embedment of Pd in the MOFs’ pores.51
Results and discussion
To address this, we synthesised frameworks to differentiate between the effect of the nature of the functional group and the number of them present. Four analogues of UiO-66 were studied, two of which had one functional group per linker and their bi-functional counterparts; NH2-UiO-66, (NH2)2-UiO-66, CH3-UiO-66 and (CH3)2-UiO-66. It should be pointed out that the selected functional groups have different acid–base characters and therefore the insights herein generated may have significant implications for a range of applications, particularly in catalysis. First, we shall evaluate the MOFs’ ability to control the Pd NC growth within their pores, then we rationalise the observed behaviours in terms of the strength of the host–guest interactions between Pd and the MOFs.We would like to note that we have already disclosed some of our early findings (TEM, PXRD and XPS, which data will not be shown here) on the amino-functionalised NH2-UiO-66(Zr), where it was established that the MOF successfully controls the Pd NC growth in its pores and the resultant composite features strong host–guest interactions through charge transfer between the amino group of the framework and the Pd.50 The three new samples were synthesised using a solvothermal process following the work of Farha et al.52 The samples were characterised using Fourier-transform infrared (FTIR) spectroscopy (Fig. 3) and powder X-ray diffraction (PXRD) to determine the integrity of the frameworks, the latter of which enabled us to establish that none of the MOF hosts have undergone loss of crystallinity on the in situ Pd NC growth through nanoconfinement (Fig. S1, ESI†). Brunauer–Emmett–Teller (BET) specific surface area measurements have also allowed us to ascertain that the frameworks’ porosity has been maintained (Table S5, ESI†). High-angle annular dark field scanning transmission electron microscopy (STEM) was used to image the Pd-laden samples to investigate the success of controlling nanocluster growth via nanoconfinement within the MOF pores (Fig. 1). This was carried out alongside transmission electron microscopy (TEM), which offers an alternative contrast for identifying Pd within the sample (Fig. S2, ESI†). As was observed in previous work, Pd was seen to be successfully embedded in the pores of NH2-UiO-66 with no large Pd particles on the surface, yet a clear presence of Pd observed using X-ray photoelectron spectroscopy (XPS), which is assigned to Pd nanoconfined within the MOF pores near the particle surface.50 In the case for CH3-UiO-66, we also found evidence of Pd NCs embedded in the pores from the microscopic as well as spectroscopic evidence from energy-dispersive X-ray (EDX) spectroscopy and XPS, similarly to our previous observations, in which other mono-functionalised linkers (–NH2 and –Br) facilitated the embedment of Pd NCs in the MOF pores.51 However, some Pd NPs, larger than the UiO-66 pore diameters, were also observed for CH3-UiO-66, suggesting that some degree of agglomeration had occurred (Fig. 1c). It is worth noting that, although not the case for all MOFs of the UiO topology, they are in general beam sensitive samples that rapidly amorphise under the high-energy electron beam, with our measurements suggesting that more functional groups on the linkers can decrease their beam stability. In addition to a lowering of the Z contrast with Pd for analogues of higher-Z functionalities, as seen with Br-UiO-66 in our previous work,51 this makes it difficult to acquire high resolution images without significant loss of detail.
 | ||
| Fig. 1 HAADF-STEM micrographs of (a) Pd in (NH2)2-UiO-66, (b) and (c) Pd in CH3-UiO-66 and (d) Pd in (CH3)2-UiO-66 demonstrating the extent of embedment of Pd (bright spots) in the frameworks. Some examples of the Pd NCs have been circled, with NPs apparent as large bright areas in (c) and (d). | ||
We therefore conclude that for CH3-UiO-66, an obvious degree of agglomeration of Pd particles occurs. In fact, Pd NPs up to 20 nm in diameter have been observed on CH3-UiO-66 (Fig. 1c). On a closer inspection of the EDX maps (Fig. S3b, ESI†), it is apparent that the areas of MOF around these larger particles have little to no Pd, which is in line with Ostwald ripening whereby Pd would favourably aggregate to form larger particles on the surface of the MOF. This reveals high Pd mobility in the CH3-UiO-66 framework, indicative of a lack of strong host–guest interactions. For (CH3)2-UiO-66, hardly any small Pd NPs could be observed in the HAADF-STEM micrographs, which display large particles up to 50 nm in diameter (Fig. 1d). This is also clearly evident from the EDX map (Fig. S3c, ESI†). When we compare this observation with those made for the CH3-UiO-66 framework, it appears that the presence of a single functional group is favourable for the controlled growth of Pd NC via nanoconfinement in the MOF pores. As seen in the case of the mono-functionalised amino framework, the MOF is able to strongly anchor the Pd NCs inside the pores through charge transfer.50 As the HAADF-STEM micrographs display no large Pd NPs, which would be indicative of agglomeration if present, one possible explanation would be the effective anchoring of the Pd NCs. In fact, EDX spectroscopy reveals an even distribution of Pd throughout the (NH2)2-UiO-66 framework, highlighting the abundance of Pd in the host matrix and the lack of nanoparticles (Fig. S3a, ESI†). We note that this observation is not trivial, as the BET surface area (Table S5, ESI†) analysis suggests that (NH2)2-UiO-66 does not appear to be porous, indeed its crystallinity also appears to be lower (Fig. S1, ESI†). We note that such lower degree of porosity would limit the compound's applicability in catalysis, sensing, etc., however such apparent lowering of the surface area (and crystallinity) may originate in other effects, such as an extended, less regular H-bonding network within the pores. Nevertheless, our Pd nanoconfinement results clearly show that a significant amount of Pd NCs (Table S4, ESI†) may grow within the MOF crystallites, effectively in its pores. Therefore, we attribute the apparently lower specific surface area of (NH2)2-UiO-66 to a significant hydrogen-bonding network inside the pores and the pore apertures, between the amino groups, which hinders N2 diffusion. Indeed, the comparison of the FTIR spectra of the two NH2– functionalised UiO-66 analogues (Fig. 3e and g) suggests the presence of H-bonding in the bi-functionalised framework. In fact, the reduction of the Pd-precursor in the (NH2)2-UiO-66 could only be achieved at harsher conditions, i.e. at 200 °C compared with that in the NH2-UiO-66 at 150 °C. This can be rationalised with the strongly stabilised Pd precursor in this framework, with some evidence from our density functional theory (DFT) calculations reporting a higher bond strength for the precursor with the bi-functionalised amino-MOF compared with the monofunctionalised one (Table S7, ESI†). However, it may also be possible that such requirement for harsher precursor reduction conditions is of a kinetic nature and could be linked with the hindered pore access (as also seen in the case of N2 adsorption). Importantly, even the harsher reduction conditions did not affect the degree of Pd agglomeration as no Pd particles larger than the pore diameter of (NH2)2-UiO-66 or could be observed in the HAADF-STEM micrographs (Fig. 1a).
At a first glance therefore, we can see that the presence of NH2-groups facilitates the controlled Pd NC growth by nanoconfinement in MOFs pores. This is not the case for the CH3-groups, wherein the formation of large nanoparticles outside of the MOF pores was observed. Since the Pd precursor infusion is carried out in all cases from an acetonitrile solution of the precursor, one may consider that the different outcome is a result of the different pore hydrophobicity of the pore walls, a property conferred to the frameworks by the amino (hydrophilic) and methyl (hydrophobic) functional groups. However, earlier results on (OH)2-UiO-66 MOF show that controlled NC growth in the pores of even highly hydrophilic materials may be hindered.51 Furthermore, our DFT calculations revealed that the precursor Pd2+ state is unstable in the methyl-functionalised, CH3-UiO-66 and (CH3)2-UiO-66 frameworks and the distance between the Pd and the NO3 increases compared to that of the precursor alone, indicating that what is been optimised is already a reduced version of the Pd (Table S6, ESI†). Table S6 (ESI†) reports the bonding energies of Pd(NO3)2 on the MOFs. Each MOF is optimised using different starting points, and here are reported 2 local minima (configurations 1 and 2 for each MOF). For the CH3 structures we observe that Pd(NO3)2 in configuration 1 maintains its original geometry (Pd–N distance is 2.4 Å, which is the same in Pd(NO3)2 optimised outside of the MOF), while in configuration 2 the Pd and the NO3 are more distant, indicating that the geometry optimisation tends to go toward the direction in which the Pd alone attaches to the framework i.e. it is reduced. The second structure has a more thermodynamically favourable bonding energy than the oxidised one. In the NH2-structures there is no such significant difference in the Pd–N distances between the two optimisations, indicating that the precursor manages to bond with the framework. However, for the mono-functionalised NH2 the bonding energy is much smaller while the binding of the precursor on the –(NH2)2 MOF is more favourable. In addition, the charge-neutral reduced Pd atoms are much more mobile, particularly at higher temperatures and under gas flow, the methyl-MOFs’ inability to strongly anchor nascent NCs in their pores therefore is laid bare. This is in extremely good agreement with our experimental observations.
In order to experimentally probe the existence or lack of strong host–guest interactions in the mono-methylated MOF, we have carried out XPS (Fig. 2), and indeed the same binding energy was determined as that of the bulk metal, confirming the lack of strong interactions. Similarly, in the case of the bi-functionalised methyl framework, there was no sign of any host–guest interactions, which is clearly in line with the frameworks evident inability to control the Pd NC growth inside its pores. On the other hand, –NH2 groups have been shown to engage in strong interactions with the Pd guest,50,53 as demonstrated by a shift in the binding energies of both Pd and the nitrogen atom in the amino functional group52 revealed by XPS (Tables S2 and S3, ESI†). Atomistic simulations at the DFT level confirmed this trend, with the bonding energies of Pd nanoclusters with –NH2 being more than double compared with those with –CH3 (see Table S7, ESI†).
 | ||
| Fig. 2 XPS spectra of (a) the Pd 3d region and (b) the N 1s region of Pd in (NH2)2-UiO-66, (c) the Pd 3d region, (d) the C 1s region of Pd in CH3-UiO-66, (e) the Pd 3d region and (f) the C 1s region of Pd in (CH3)2-UiO-66. On the top of each spectrum is the raw data, while on the bottom the fitted data is displayed. For comparisons of the regions associated with the functional groups, spectra for the empty MOFs have been included in black, while the spectra for the Pd-laden samples are in green (Pd in (NH2)2-UiO-66), grey (Pd in CH3-UiO-66) and magenta (Pd in (CH3)2-UiO-66). | ||
Interactions between the frameworks and the nascent Pd particles has been probed using FTIR (Fig. 3). In accordance with the PXRD data, our spectra reveal no significant changes on Pd growth, specifically for all samples bands have been observed in the 800–600 cm−1 region, which can be assigned to the Zr–O bonds,54 highlighting that MOF integrity has been maintained. We note however a degree of dampening of the bands assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretches, which may be related to pore loading. For the empty and Pd-laden (NH2)2-UiO-66, there are some distinct peaks in the 3500–3400 cm−1 range that correspond to N–H stretching modes for primary amines, as would be expected given the presence of the –NH2 functional group. These peaks are less obvious in the empty and Pd-laden NH2-UiO-66 and are more convoluted with the broad O–H peak from water, with lower intensity of the peaks as there are stoichiometrically half the number of –NH2 groups as in the bi-functional analogue. Intriguingly, these N–H stretches show no significant shift on Pd addition in the case of the bi-functional framework, which could be expected based on the charge transfer observed in the XPS, and which is what is observed for the mono-functionalised MOF. However, it must be recalled that the Pd precursor's reduction temperature is higher for the diamino MOF, furthermore previous EXAFS data shows that the Pd–N bond is unstable at and above 200 °C,‡ where the preparation of the Pd in (NH2)2-UiO-66 takes place. This suggests that the Pd NCs formed in the (NH2)2-UiO-66 pores are not anchored by the NH2-functionalities, which is in line with the XPS showing that the Pd is in the metallic state.
O stretches, which may be related to pore loading. For the empty and Pd-laden (NH2)2-UiO-66, there are some distinct peaks in the 3500–3400 cm−1 range that correspond to N–H stretching modes for primary amines, as would be expected given the presence of the –NH2 functional group. These peaks are less obvious in the empty and Pd-laden NH2-UiO-66 and are more convoluted with the broad O–H peak from water, with lower intensity of the peaks as there are stoichiometrically half the number of –NH2 groups as in the bi-functional analogue. Intriguingly, these N–H stretches show no significant shift on Pd addition in the case of the bi-functional framework, which could be expected based on the charge transfer observed in the XPS, and which is what is observed for the mono-functionalised MOF. However, it must be recalled that the Pd precursor's reduction temperature is higher for the diamino MOF, furthermore previous EXAFS data shows that the Pd–N bond is unstable at and above 200 °C,‡ where the preparation of the Pd in (NH2)2-UiO-66 takes place. This suggests that the Pd NCs formed in the (NH2)2-UiO-66 pores are not anchored by the NH2-functionalities, which is in line with the XPS showing that the Pd is in the metallic state.
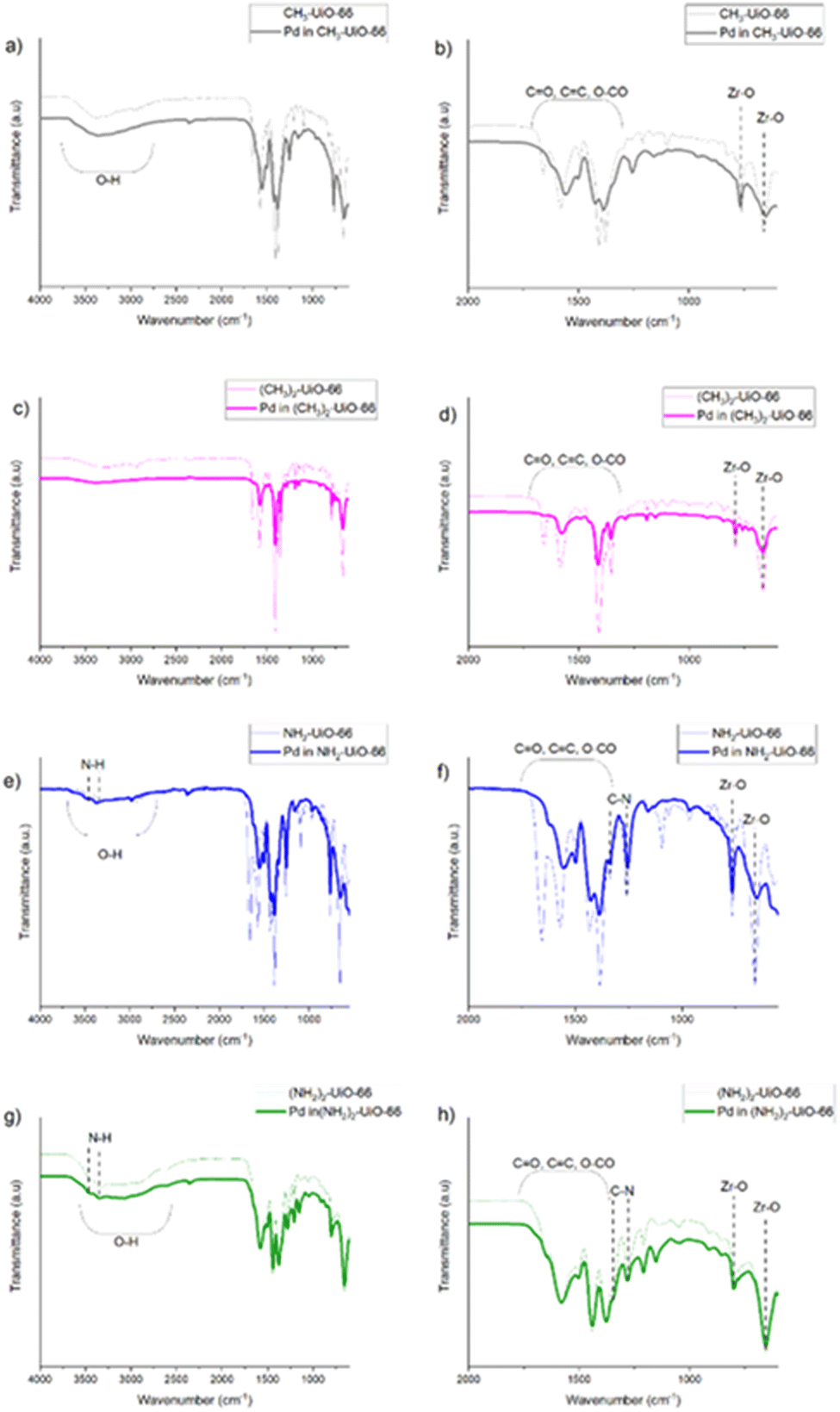 | ||
| Fig. 3 FTIR spectra of Pd-laden MOFs compared with the empty parent framework: (a) and (b) CH3-UiO-66, (c) and (d) (CH3)2-UiO-66, (e) and (f) NH2-UiO-66, and (g) and (h) (NH2)2-UiO-66. | ||
We note that our data analysis reveals that the number of functional groups on the linkers has a negligible effect on the charge transfer between the ligands and the Pd guest, i.e. on the host–guest interaction strength (Tables S2 and S3, ESI†). This observation was supported by the computational results. The number of functional groups strengthens the binding by 3.3 eV in the NH2-functionalised MOF and 2 eV in the CH3-functionalised MOF (Table S6, ESI†), and the charge transfer takes place between the linker and the Pd cluster (Fig. S6 and Table S7, ESI†). In the NH2-MOFs, the clusters appear to be distorted after geometry optimisation (Fig. S15 and S16, ESI†) compared with the ‘ideal’ structure of the cluster outside the framework (Fig. S12, ESI†), which is more symmetrical. The clusters seem to bind to the NH2 group, which is ∼2.2 Å far from the closest Pd (Table S7, ESI†). The clusters in the CH3-MOFs (Fig. S13 and S14, ESI†), on the other hand, seem to form a stronger bond with the benzene ring rather than the functional group (Table S7, ESI†). Specifically, the Pd17 cluster in the mono-functionalised CH3-UiO-66 shows very little distortion of the Pd cluster, suggesting that the interaction between the cluster and the framework is more of a steric origin, as the degree of distortion increases in the bi-functionalised MOF (Fig. S9, S10, S13 and S14, ESI†). To re-iterate, the EDX and TEM measurements have revealed a lack of agglomerated NPs in the amino-functionalised MOFs, and an even distribution of Pd throughout samples of these analogues, strongly suggesting that the controlled growth of Pd NCs via nanoconfinement within the frameworks’ pores was successful.
To confirm this with an independent method, we looked at the hydrogenation and dehydrogenation properties of Pd embedded in MOFs. When exposed to hydrogen pressure, Pd undergoes hydride formation. Firstly, a non-stoichiometric solid solution (α-phase) is formed and when the plateau pressure is reached at the give temperature, the interstitial PdH0.67, β-hydride, is produced.55 As the size of the Pd particles strongly affects the concentration and indeed the very existence of interstices, it is regarded that interstitial β-hydride cannot form on Pd NCs, i.e. from ca. 1 nm downwards in size.56,57 It has been demonstrated that thermal desorption spectroscopy (TDS) can be employed to distinguish between the different hydride phases58 and we have therefore carried out TDS on the Pd in (NH2)2-UiO-66 sample to probe the size of Pd NCs within (Fig. S5, ESI†).
It has been shown that when Pd NPs are present, the TDS spectrum features two characteristic desorption peaks; for pure Pd NPs, there is a relatively intense characteristic peak at ca. 260 K superimposed on a broad, lower-intensity, desorption peak with onset temperature of ca. 170 K.‡ From our previous studies, we knew that when Br-UiO-66 is used for the growth of Pd NCs, nanoparticles are also formed on poorly crystalline regions,51 which are susceptible to the formation of interstitial hydrides on hydrogenation, therefore we also acquired its TDS spectrum. When observing the desorption spectrum, very similar features are displayed; a desorption peak occurs at around 250 K, superimposed on a broader peak with an onset of ca. 170 K. This gives a blueprint of what the hydrogen desorption peak of a sample containing both Pd NPs and NCs would look like. Conversely, when evaluating the TDS spectrum of Pd in (NH2)2-UiO-66, no such characteristic features are apparent, instead, one can observe two peaks coalesced into a broad signal at lower temperature, which is consistent with desorption from the solid-solution phase, associated with a lower hydrogen loading.59 When the Pd particle size is smaller than a limit (ca. 1 nm), the interstitial hydride, capable of storing larger amounts of hydrogen, cannot form.56,57 This observation also is in excellent agreement with that Pd being embedded in the pores of the bi-functionalised (NH2)2-UiO-66.
Experimental
Synthetic procedures
All materials were purchased from commercial sources and used without further purification.Structural and chemical characterisation
XPS was carried out using a Thermo Fisher Nexsa XPS System with a monochromated Al Kα X-ray source. A pass energy of 50 eV was used with a flood gun as an electron source for charge neutralisation. The data was analysed by the OriginPro software. Peaks were fitted with a Gaussian numerical convolution after baseline and background corrections. FTIR spectroscopy was carried out using an attenuated total reflectance (ATR) setup on a Bruker Tensor 27 spectrometer. A resolution of 4 cm−1 was used with a range of 400–4000 cm−1, with 32 scans. The software used to record the spectra was OPUS. PXRD data was collected on a Panalytical X’pert Pro in reflection mode using a Cu Kα anode (λ = 1.54178 Å), divergence slit, Ni-filter and a range of 5–120° 2θ. HAADF-STEM and TEM micrographs were obtained for the samples at ePSIC, Diamond Light Source. We imaged the samples using a probe-corrected ARM-200F JEOL microscope operated at 200 kV accelerating voltage using a 0.4 pA probe (10C spot size, 10 μm probe-forming aperture). The convergence semi-angle of the probe was 7.6 mrad and the collection range of the ADF detector used was 34 to 132 mrad. Some additional TEM micrographs of the Pd in (NH2)2-UiO-66 sample reduced at 200 °C were obtained using a JEOL JEM-2100F operating at 200 kV. EDX spectroscopy was carried out and element-specific EDX maps were obtained using the probe-corrected ARM-200F JEOL microscope at ePSIC. We used a probe with 171 pA beam current (4C Spot size with 30 μm probe-forming aperture). TDS spectra were collected using an in-house made apparatus. Prior to measurements, about 5 mg samples were loaded in the sample holder and activated at 400 K under vacuum for 3 h. Then, a 50 mbar equimolar D2/H2 isotope mixture was loaded on each sample at room temperature and then cooling to 20 K. The free gas was evacuated. Finally, during heating from 20 K to room temperature with a heating rate of 0.1 K s−1, the desorbing gas was continuously detected using a mass spectrometer (QMS), recognising a pressure increase in the sample chamber when gas desorbs. The area under the desorption peak was proportional to the desorbing amount of gas, which can be quantified after careful calibration of the TDS apparatus. A solid polycrystalline piece of a diluted Pd alloy Pd95Ce5 (∼0.5 g) was used for calibration. Prior to the calibration, the alloy was etched with aqua regia in order to remove the oxide layer and it was then heated up to 600 K under high vacuum to remove any hydrogen that might be absorbed during the etching procedure. Afterwards, it was weighted and inserted into the sample chamber. At 350 K, it was loaded with 40 mbar pure H2 or pure D2 for 1.5–2.5 h. As H and D were bound preferentially to the cerium atoms at low loading pressures in this diluted alloy, the alloy could be handled under ambient conditions for a short period of time. After loading, the alloy was cooled to room temperature and weighed. Afterwards, it was placed back in the sample chamber and a normal TDS measurement with 0.1 K s−1 was performed. After the desorption, the alloy was cooled back to ambient temperature and weighted again. The weight difference between unloaded state and loaded state was equal to the mass uptake of hydrogen or deuterium, respectively.Atomistic modelling: all simulations were performed at DFT level with plane waves using Quantum Espresso version 6.8.60–62 The geometries and cell parameters were optimised using VDW-DF2-B86R setting a kinetic energy cut-off of 50 Ry and of 450 Ry for density, ultra-soft potentials. We used the pseudopotentials H.pbe-rrkjus_psl.1.0.0.UPF, N.pbe-n-rrkjus_psl.1.0.0.UPF, C.pbe-n-rrkjus_psl.1.0.0.UPF, O.pbe-n-rrkjus_psl.1.0.0.UPF, Zr.pbe-spn-rrkjus_psl.1.0.0.UPF and Pd.pbe-spn-rrkjus_psl.1.0.0.UPF from https://www.quantum-espresso.org. The geometry optimisation calculations were performed at gamma point. All structures had the geometry optimised with an energy convergence criteria for ionic minimisation was 1.0 × 10−6 Ry and for force was 1.0 × 10−5 Ry Bohr−1. The optimised geometries of the MOFs are shown in Fig. S7 (ESI†). The Pd cluster was optimised inside the pores of the MOF, in a single cell as shown in Fig. S8 (ESI†). Binding energies were calculated by computing the total energy of the guest inside the host from which we subtracted total energy of the guest and energy of the host. The single point calculation to obtain the energies and charges was performed setting a kinetic energy cut-off of 70 Ry and of 650 Ry for density, and a 2 2 2 k-point Monkhorst–Pack grid. Inputs and outputs were processed using atomistic simulation environment (ASE) python framework scripts.63,64 In all the simulations the size of the nano cluster used was of 17 atoms. The machine learning modelling was performed using MACE architecture65 and newly developed foundational forcefield MACE_MP large66 using ASE.
Conclusions
In conclusion, we have synthesised specimens and carried out experiments that allowed us to independently probe the effect of the nature and number of the functional groups of the linkers on the frameworks’ ability for controlled Pd nanocluster growth in its pores. We have shown that it is the ligands’ nature that determines the strength of host–guest interactions and thus controls guest growth insider the MOF pores. These effects combined mean that for design purposes of Pd NCs grown in the pores of MOFs, mono-functionalised linkers with functional groups capable of strong interactions with Pd are ideal. We find that a single functional group on the linker favours the controlled growth of Pd nanoclusters in the pores, as this was observed for all three mono-functionalised analogues (and the Br-UiO-66 published elsewhere51). We have observed that even in the case of CH3-UiO-66, where strong host–guest interactions do not exist, Pd NCs embedded in the pores are clearly discernible. When analysing these results in the context of previous literature data, we note that the following functional groups facilitate Pd embedding regardless of their number on the linker: –NH2, and Br, while –H, –Cl, –OH and –CH3 are unfavourable. Based on these results, an interesting observation can be made, namely the more the Lewis base character of the ligands, the more successful the embedding, while the higher the Lewis acidity of the functional groups, the lower the chance for Pd embedment. We therefore predict that ligands, such as –SH would also likely control guest growth inside nanometric cavities. However, care must be taken to sample quality (e.g. crystallinity) and synthetic conditions (i.e. reaction temperature), which may also affect the specimens’ behaviour. The study of the independent effects of ligand nature and number on the success of guest embedment allowed us to observe such a correlation for the first time, which will have significant impact on the field, particularly for the design and application of Pd NCs grown and anchored within the pores of MOFs in catalysis where the size control of Pd particles is of paramount importance.Author contributions
JK synthesised and carried out most materials characterisation, ZL performed data analysis, FZ and AME performed the computational analysis, LZ measured the TDS spectra under MH's supervision, HL and MD obtained TEM and STEM micrographs, while SM oversaw the computational efforts, PC contributed through discussion and analysis, and PAS initiated and designed, and supervised the research work.Data availability
The data supporting this article have been included as part of both the Main Article and the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
PAS thanks the University of Oslo for their support. JK and ZL thank the Queen Mary University of London and Chinese Scholarship Council for sponsorship. AME and FZ thank the Physical Sciences Databases Infrastructure project for funding (psdi.ac.uk) and acknowledge the usage of scarf cluster run by the Scientific Computing Department at STFC.Notes and references
- S. Liu and Z. Tang, J. Mater. Chem., 2010, 20, 24–35 RSC.
- N. Tian, Z. Y. Zhou, S. G. Sun, Y. Ding and L. W. Zhong, Science, 2007, 316, 732–735 CrossRef CAS PubMed.
- D. Astruc, F. Lu and J. R. Aranzaes, Angew. Chem., Int. Ed., 2005, 44, 7852–7872 CrossRef CAS.
- C. J. Murphy, A. M. Gole, S. E. Hunyadi, J. W. Stone, P. N. Sisco, A. Alkilany, B. E. Kinard and P. Hankins, Chem. Commun., 2008, 544–557 RSC.
- N. J. Durr, T. Larson, D. K. Smith, B. A. Korgel, K. Sokolov and A. Ben-Yakar, Nano Lett., 2007, 7, 941–945 CrossRef CAS PubMed.
- X. Huang, I. H. El-Sayed, W. Qian and M. A. El-Sayed, Nano Lett., 2007, 7, 1591–1597 CrossRef CAS PubMed.
- A. A. Herzing, C. J. Kiely, A. F. Carley, P. Landon and G. J. Hutchings, Science, 2008, 321, 1331–1335 CrossRef CAS PubMed.
- J. Lin, B. Qiao, J. Liu, Y. Huang, A. Wang, L. Li, W. Zhang, L. F. Allard, X. Wang and T. Zhang, Angew. Chem., Int. Ed., 2012, 51, 2920–2924 CrossRef CAS PubMed.
- A. Dhakshinamoorthy and H. Garcia, Chem. Soc. Rev., 2012, 41, 5262–5284 RSC.
- M. Imran, A. B. Yousaf, X. Zhou, Y.-F. Jiang, C.-Z. Yuan, A. Zeb, N. Jiang and A.-W. Xu, J. Phys. Chem. C, 2017, 121, 1162–1170 CrossRef CAS.
- J. P. Wilcoxon and B. L. Abrams, Chem. Soc. Rev., 2006, 35, 1162 RSC.
- C. Pan, K. Pelzer, K. Philippot, B. Chaudret, F. Dassenoy, P. Lecante and M.-J. Casanove, J. Am. Chem. Soc., 2001, 123, 7584–7593 CrossRef CAS PubMed.
- T. W. Hansen, A. T. Delariva, S. R. Challa and A. K. Datye, Acc. Chem. Res., 2013, 46, 1720–1730 CrossRef CAS PubMed.
- X. Zhang, H. Shi and B. Q. Xu, Angew. Chem., Int. Ed., 2005, 44, 7132–7135 CrossRef CAS PubMed.
- W. Tang, Z. Hu, M. Wang, G. D. Stucky, H. Metiu and E. W. McFarland, J. Catal., 2010, 273, 125–137 CrossRef CAS.
- S. Sun, G. Zhang, N. Gauquelin, N. Chen, J. Zhou, S. Yang, W. Chen, X. Meng, D. Geng, M. N. Banis, R. Li, S. Ye, S. Knights, G. A. Botton, T.-K. Sham and X. Sun, Sci. Rep., 2013, 3, 1775 CrossRef.
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed.
- H. R. Moon, D.-W. Lim and M. P. Suh, Chem. Soc. Rev., 2013, 42, 1807–1824 RSC.
- C. T. Campbell, Acc. Chem. Res., 2013, 46, 1712–1719 CrossRef CAS PubMed.
- X. L. Wang, X. P. Fu, W. W. Wang, C. Ma, R. Si and C. J. Jia, J. Phys. Chem. C, 2019, 123, 9001–9012 CrossRef CAS.
- X. Zhou, H. Y. Zhou, T. Y. Cheang, Z. W. Zhao, C. C. Shen, K. Liang, Y. N. Liu, Z. K. Yang, M. Imran and A. W. Xu, J. Phys. Chem. C, 2017, 121, 27528–27534 CrossRef CAS.
- K. Morgan, A. Goguet and C. Hardacre, ACS Catal., 2015, 5, 3430–3445 CrossRef CAS.
- Q.-L. Zhu and Q. Xu, Chem, 2016, 1, 220–245 CAS.
- Q. Yang, Q. Xu and H.-L. Jiang, Chem. Soc. Rev., 2017, 46, 4774–4808 RSC.
- S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed.
- S. L. James, Chem. Soc. Rev., 2003, 32, 276 RSC.
- N. Stock and S. Biswas, Chem. Rev., 2012, 112, 933–969 CrossRef CAS PubMed.
- M. O’Keeffe and O. M. Yaghi, Chem. Rev., 2012, 112, 675–702 CrossRef PubMed.
- C. Rösler and R. A. Fischer, CrystEngComm, 2015, 17, 199–217 RSC.
- M. Meilikhov, K. Yusenko, D. Esken, S. Turner, G. Van Tendeloo and R. A. Fischer, Eur. J. Inorg. Chem., 2010, 3701–3714 CrossRef CAS.
- J. Juan-Alcañiz, J. Gascon and F. Kapteijn, J. Mater. Chem., 2012, 22, 10102 RSC.
- H. Li, L. Li, R.-B. Lin, W. Zhou, Z. Zhang, S. Xiang and B. Chen, EnergyChem, 2019, 1, 100006 CrossRef PubMed.
- Y. He, W. Zhou, T. Yildirim and B. Chen, Energy Environ. Sci., 2013, 6, 2735–2744 RSC.
- K. M. Choi, H. M. Jeong, J. H. Park, Y. B. Zhang, J. K. Kang and O. M. Yaghi, ACS Nano, 2014, 8, 7451–7457 CrossRef CAS PubMed.
- H. Wang, Q.-L. Zhu, R. Zou and Q. Xu, Chem, 2017, 2, 52–80 CAS.
- M. A. Nasalevich, R. Becker, E. V. Ramos-Fernandez, S. Castellanos, S. L. Veber, M. V. Fedin, F. Kapteijn, J. N. H. Reek, J. I. Van Der Vlugt and J. Gascon, Energy Environ. Sci., 2015, 8, 364–375 RSC.
- K. Kočí, L. Obalová and Z. Lacný, Chem. Pap., 2008, 62, 1–9 Search PubMed.
- Y. B. Huang, M. Shen, X. Wang, P. Huang, R. Chen, Z. J. Lin and R. Cao, J. Catal., 2016, 333, 1–7 CrossRef CAS.
- M. Zhao, K. Yuan, Y. Wang, G. Li, J. Guo, L. Gu, W. Hu, H. Zhao and Z. Tang, Nature, 2016, 539, 76–80 CrossRef CAS PubMed.
- A. Aijaz, A. Karkamkar, Y. J. Choi, N. Tsumori, E. Rönnebro, T. Autrey, H. Shioyama and Q. Xu, J. Am. Chem. Soc., 2012, 134, 13926–13929 CrossRef CAS PubMed.
- Y. K. Hwang, D. Y. Hong, J. S. Chang, S. H. Jhung, Y. K. Seo, J. Kim, A. Vimont, M. Daturi, C. Serre and G. Férey, Angew. Chem., Int. Ed., 2008, 47, 4144–4148 CrossRef CAS PubMed.
- V. I. Isaeva, O. L. Eliseev, V. V. Chernyshev, T. N. Bondarenko, V. V. Vergun, G. I. Kapustin, A. L. Lapidus and L. M. Kustov, Polyhedron, 2019, 158, 55–64 CrossRef CAS.
- X. Li, Z. Guo, C. Xiao, T. W. Goh, D. Tesfagaber and W. Huang, ACS Catal., 2014, 4, 3490–3497 CrossRef CAS.
- Y. Z. Chen, Y. X. Zhou, H. Wang, J. Lu, T. Uchida, Q. Xu, S. H. Yu and H. L. Jiang, ACS Catal., 2015, 5, 2062–2069 CrossRef CAS.
- Y. Pan, B. Yuan, Y. Li and D. He, Chem. Commun., 2010, 46, 2280–2282 RSC.
- C. E. Pompe and P. Á. Szilágyi, Faraday Discuss., 2021, 231, 371–383 RSC.
- S. Yoshimaru, M. Sadakiyo, A. Staykov, K. Kato and M. Yamauchi, Chem. Commun., 2017, 53, 6720–6723 RSC.
- Z. Guo, C. Xiao, R. V. Maligal-Ganesh, L. Zhou, T. W. Goh, X. Li, D. Tesfagaber, A. Thiel and W. Huang, ACS Catal., 2014, 4, 1340–1348 CrossRef CAS.
- A. Aijaz, Q. L. Zhu, N. Tsumori, T. Akita and Q. Xu, Chem. Commun., 2015, 51, 2577–2580 RSC.
- D. E. Coupry, J. Butson, P. S. Petkov, M. Saunders, K. O’Donnell, H. Kim, C. Buckley, M. Addicoat, T. Heine and P. Szilágyi, Chem. Commun., 2016, 52, 5175–5178 RSC.
- J. King, L. Zhang, S. Doszczeczko, O. Sambalova, H. Luo, F. Rohman, O. Phillips, A. Borgschulte, M. Hirscher, M. Addicoat and P. Á. Szilágyi, J. Mater. Chem. A, 2020, 8, 4889–4897 RSC.
- M. J. Katz, Z. J. Brown, Y. J. Colón, P. W. Siu, K. A. Scheidt, R. Q. Snurr, J. T. Hupp and O. K. Farha, Chem. Commun., 2013, 49, 9449 RSC.
- P. Á. Szilágyi, D. M. Rogers, I. Zaiser, E. Callini, S. Turner, A. Borgschulte, A. Züttel, H. Geerlings, M. Hirscher and B. Dam, J. Mater. Chem. A, 2017, 5, 15559–15566 RSC.
- S. M. Yakout and H. S. Hassan, Molecules, 2014, 19, 9160–9172 CrossRef PubMed.
- Y. Fukai, The Metal-Hydrogen System, Springer Berlin Heidelberg, Berlin, Heidelberg, 2005, vol. 21 Search PubMed.
- A. Malouche, G. Blanita, D. Lupu, J. Bourgon, J. Nelayah and C. Zlotea, J. Mater. Chem. A, 2017, 5, 23043–23052 RSC.
- A. Malouche, C. Zlotea and P. Á. Szilágyi, Chem. Phys. Chem., 2019, 20, 1282–1295 CrossRef CAS PubMed.
- B. Panella, Hydrogen Storage by Physisorption on Porous Materials, University of Stuttgart, 2006 Search PubMed.
- A. Stern, A. Resnik and D. Shaltiel, J. Phys. F: Met. Phys., 1984, 14, 1625–1639 CrossRef CAS.
- P. Giannozzi, O. Andreussi, T. Brumme, O. Bunau, M. Buongiorno Nardelli, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, M. Cococcioni, N. Colonna, I. Carnimeo, A. Dal Corso, S. De Gironcoli, P. Delugas, R. A. Distasio, A. Ferretti, A. Floris, G. Fratesi, G. Fugallo, R. Gebauer, U. Gerstmann, F. Giustino, T. Gorni, J. Jia, M. Kawamura, H. Y. Ko, A. Kokalj, E. Kücükbenli, M. Lazzeri, M. Marsili, N. Marzari, F. Mauri, N. L. Nguyen, H. V. Nguyen, A. Otero-De-La-Roza, L. Paulatto, S. Poncé, D. Rocca, R. Sabatini, B. Santra, M. Schlipf, A. P. Seitsonen, A. Smogunov, I. Timrov, T. Thonhauser, P. Umari, N. Vast, X. Wu and S. Baroni, J. Phys.: Condens. Matter, 2017, 29, 465901 CrossRef CAS PubMed.
- P. Giannozzi, O. Baseggio, P. Bonfà, D. Brunato, R. Car, I. Carnimeo, C. Cavazzoni, S. De Gironcoli, P. Delugas, F. Ferrari Ruffino, A. Ferretti, N. Marzari, I. Timrov, A. Urru and S. Baroni, J. Chem. Phys., 2020, 152, 154105 CrossRef CAS PubMed.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. De Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- A. Hjorth Larsen, J. JØrgen Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Dułak, J. Friis, M. N. Groves, B. Hammer, C. Hargus, E. D. Hermes, P. C. Jennings, P. Bjerre Jensen, J. Kermode, J. R. Kitchin, E. Leonhard Kolsbjerg, J. Kubal, K. Kaasbjerg, S. Lysgaard, J. Bergmann Maronsson, T. Maxson, T. Olsen, L. Pastewka, A. Peterson, C. Rostgaard, J. SchiØtz, O. Schütt, M. Strange, K. S. Thygesen, T. Vegge, L. Vilhelmsen, M. Walter, Z. Zeng and K. W. Jacobsen, J. Phys.: Condens. Matter, 2017, 29, 273002 CrossRef PubMed.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, Clarendon Press, 1990 Search PubMed.
- I. Batatia, D. P. Kovacs, G. Simm, C. Ortner and G. Csányi, Mace: Higher order equivariant message passing neural networks for fast and accurate force fields, Adv. Neural Inf. Process. Syst., 2022, 35, 11423–11436 Search PubMed.
- I. Batatia, P. Benner, Y. Chiang, A. M. Elena, D. P. Kovács, J. Riebesell, X. R. Advincula, M. Asta, W. J. Baldwin and N. Bernstein, et al., 2023, A foundation model for atomistic materials chemistry, arXiv, 2023, preprint, arXiv:2401.00096v2 DOI:10.48550/arXiv.2401.00096.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional experimental details and data for PXRD, STEM and EDX spectroscopy, XPS, TDS, BET specific surface area analysis, an additional computational details. See DOI: https://doi.org/10.1039/d4cp02422b |
| ‡ Manuscript submitted elsewhere. |
| This journal is © the Owner Societies 2024 |