
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Attractive acceptor–acceptor interactions in self-complementary quadruple hydrogen bonds for molecular self-assembly†
Usman
Ahmed
a,
Christopher D.
Daub
 a,
Dage
Sundholm
*a and
Mikael P.
Johansson
*b
a,
Dage
Sundholm
*a and
Mikael P.
Johansson
*b
aDepartment of Chemistry, Faculty of Science, University of Helsinki, P. O. Box 55 (A. I. Virtasen aukio 1), FI-00014, Helsinki, Finland. E-mail: Usman.Ahmed@helsinki.fi; Christopher.Daub@helsinki.fi; Dage.Sundholm@helsinki.fi
bCSC-IT Center for Science Ltd, P.O. Box 405, FI-02101 Espoo, Finland. E-mail: Mikael.Johansson@csc.fi
First published on 6th September 2024
Abstract
Molecular self-assembly provides the means for creating large supramolecular structures, extending beyond the capability of standard chemical synthesis. To harness the power of self-assembly, it is necessary to understand its driving forces. A potent method is to exploit self-complementary hydrogen bonding, where a molecule interacts with its own copy by suitable positions of hydrogen-bond donor (D) and acceptor (A) groups. With four hydrogen bonds, there are two possible self complementary patterns: the DDAA/AADD and the DADA/ADAD motifs. Of these, the DDAA pattern is usually more stable. The traditional explanation assumes that the secondary interactions between equal groups, that is, between donors (D⋯D) or acceptors (A⋯A), are repulsive. DDAA arrays would then have two, and DADA arrays six repulsive interactions. Here, using high-end quantum chemical analysis, we show that contrary to the traditional explanation, the secondary A⋯A interactions are, in fact, attractive. We revise the model of secondary interactions accordingly.
1 Introduction
In intermolecular self-assembly, a collection of initially disordered molecules spontaneously adopt a specific arrangement, without the need for an outside influence. Self-assembly provides a powerful means of creating large supramolecular structures.1,2 This order-out-of-chaos process is guided by non-covalent interactions between the individual molecules. Understanding the underlying mechanisms that guide the self-organising process is crucial, as this makes it possible to design rules for guiding rational design of supramolecular species. The rules should be as simple as possible to facilitate their use, but also as complicated as necessary to provide usefully accurate a priori predictions.Of the non-covalent interactions, hydrogen bonding is one of the most important ones, utilized both in living systems and in the laboratory for self-assembly due to its directionality, selectivity, and strength.3,4 One of the most famous examples of the importance of hydrogen bonding is found in the structure of DNA, where hydrogen bonds help to maintain the three-dimensional structure of this biological information carrier. As can be expected, the strength of the total hydrogen bonding increases with increasing number of hydrogen bonds. Here, we study the co-operativity between individual hydrogen bonds in quadruple hydrogen-bond systems, where the chemical species have four sites amenable to hydrogen bonding.5
Quadruple hydrogen bonding motifs come in six distinct configurations, with different acceptor (A) and donor (D) arrangements on the two hydrogen-bonding entities: AAAA–DDDD, AAAD–DDDA, AADA–DDAD, ADDA–DAAD, DDAA–AADD, and DADA–ADAD.6 Of these six possible arrays, DDAA–AADD and DADA–ADAD are self complementary, that is, the two molecules binding with each other can be identical. This is a highly advantageous property for self-assembly, where large supramolecular structures can be constructed from simpler similar, or even identical, building blocks.
The preparation and study of self-complementary four-fold hydrogen-bonded motifs have been intense during the past few decades.7–10 Four-fold hydrogen-bonded systems have a wide range of potential applications, ranging from light-capturing devices11,12 to supramolecular polymers.13–19 Quadruple hydrogen bonding has been used for molecular recognition with high fidelity and affinity.20–22
The strength and stability of hydrogen-bonded motifs is generally determined by several factors, including intramolecular hydrogen bonding, tautomerization, electronic substituent effects, secondary interactions, and pre-organization.9 Among all these factors, secondary interactions have a significant impact on the stability of hydrogen bonding associations, as set out in the secondary interaction hypothesis (SIH).23,24 Of the two self-complementary groups, the DDAA and DADA molecules, the DDAA pairs usually form stronger interactions. In the SIH model, this is traditionally explained by repulsive interactions between A⋯A and D⋯D pairs, as shown in the two upper pictures of Fig. 1. Then, in the DDAA motif, there would be four attractive and two repulsive secondary interactions, while all six secondary interactions in the DADA arrays would be repulsive.
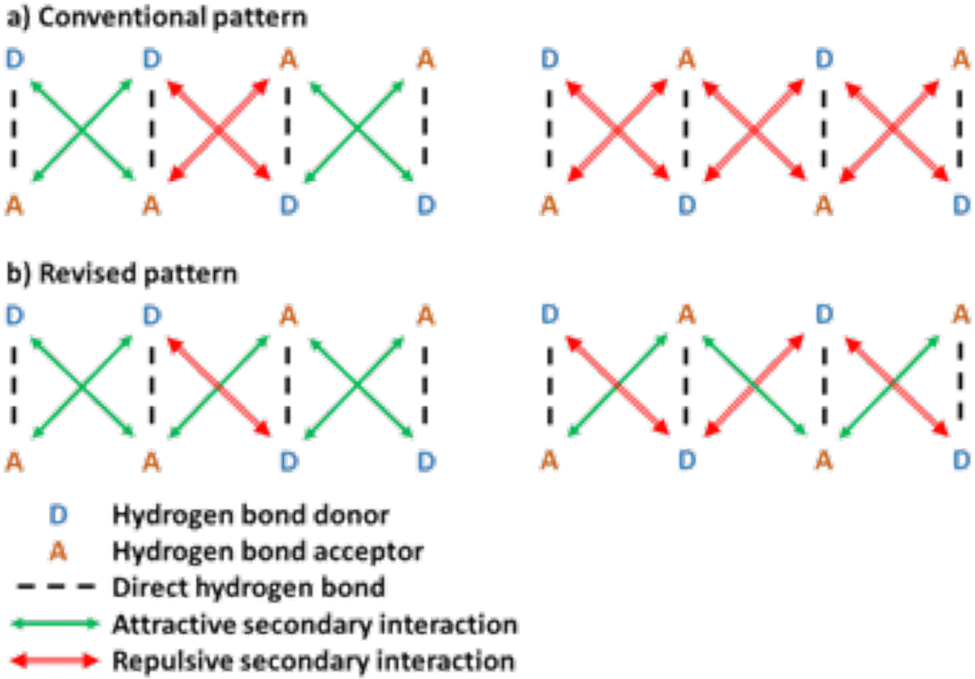 | ||
| Fig. 1 Attractive and repulsive secondary interactions in quadruple hydrogen bonded systems: (a) the conventional pattern and (b) the revised pattern, presented in this work. | ||
The secondary interactions hypothesis provides an appealing rationale due to its simplicity. The quantitative predictive power of the model has been questioned, however, by a few studies on the details of the simultaneous interactions within hydrogen-bonded complexes. For example, based on quantum chemical studies of DNA base pairs, Popelier and Joubert concluded that the SIH model is too simplistic.25 In general, the emerging consensus is, that while the SIH provides a good base to build upon, quantification of the interaction strength requires considering more than the individual, acceptor and donor atoms.7,26–34
2 Methodology
We have studied the interactions of a selection of quadruply hydrogen-bonded dimers with DDAA and DADA motifs using high-end quantum chemical methodologies, that is, symmetry-adapted perturbation theory (SAPT) and coupled cluster theory (CCSD(T)). Our results suggest that, contrary to the traditional view, the secondary A⋯A interactions are, in fact, usually attractive. The SIH model would thus not only be too simplistic, but qualitatively misleading.We have studied a representative set of sixteen dimers having quadruple hydrogen bonding: eight dimers with the DDAA motif and eight with the DADA motif, see Fig. 2. and 3 To elucidate the complex interactions within the complementary hydrogen-bonded dimers, we dissected the individual interaction components using the symmetry-adapted perturbation theory (SAPT) approach.35,36 SAPT provides a means of directly computing non-covalent interactions between molecules and allows a decomposition of the interaction energy into physically meaningful components: electrostatic, exchange, induction, and dispersion terms. With the recent extension of the SAPT functionality to further decompose the origins of the different interactions into individual functional groups within the interacting molecules,36 the method provides the means required for a detailed investigation of the secondary interactions in hydrogen-bonded arrays. Fig. 4 shows how the intramolecular grouping is performed in the SAPT analysis.
 | ||
| Fig. 2 The DDAA species considered in this study. Atoms in red act as hydrogen bond acceptors, and hydrogen atoms in blue as donors during complementary self-assembly. | ||
 | ||
| Fig. 3 The DADA species considered in this study. Atoms in red act as hydrogen bond acceptors, and hydrogen atoms in blue as donors during complementary self-assembly. | ||
 | ||
| Fig. 4 Two quadruply hydrogen-bonded dimers exemplifying the bonding patterns of the less favourable DADA (left) and the more favourable DDAA (right). The groupings used in the SAPT interaction analysis are circled and labelled; D for hydrogen-bond donor and A for acceptor. The division into acceptor and donator domains of the studied molecules is shown in the ESI.† | ||
In the following, we present the results of the interaction energy decomposition of the DDAA and DADA motifs, we estimate the effect of solvation on the interaction energies, and we corroborate the results using state-of-the-art quantum chemical wave function theory.
3 Computational methods
The molecular geometries were optimized at the dispersion-corrected density functional theory level (DFT-D3)37,38 using the TPSSh39,40 functional and the def2-TZVPP41,42 basis set. The secondary interaction energies were computed using symmetry-adapted perturbation theory (SAPT)35,36 recently extended for functional group analysis, in conjunction with the jun-cc-pVDZ43–45 basis set. Point charges for simulating a water environment in the SAPT calculations were computed at the GFN2-xTB level.46 Domain-based local pair natural orbital coupled cluster calculations with single, double, and perturbative triple excitations, DLPNO-CCSD(T),47 were performed with the correlation-consistent triple and quadruple-ζ basis sets augmented with diffuse basis functions (aug-cc-pVTZ and aug-cc-pVQZ).43,44 The electron correlation energies were extrapolated towards the complete basis set (CBS) limit using the robust two-point formula by Halkier, Helgaker and Jørgensen.48 The coupled cluster energies were further extrapolated toward the full configuration-interaction (FCI) limit, using Goodson's continued-fraction methodology.49 Thus, obtained interaction energies are as near to the full solution of the Schrödinger equation as currently feasible. To obtain the configurations of the solvated DADA and DDAA dimers that are needed for the molecular dynamics (MD) simulations, we first prepared a configuration of 1000 SPC/E water molecules.50 The SPC/E model is thoroughly studied and found to agree better with experiments on bulk water than other three-site models for water.51 It has been used to successfully model aqueous solutions of monosodium glutamate,52 and is expected to reliably model water solutions containing the studied molecules. However, we do not expect that our results differ much from those obtained using other water models. The water molecules were initially placed on a lattice in a large cubic simulation box. We use a cubic simulation box with standard periodic boundaries. Then, a simulation of 100 ps duration was done in the NVT ensemble at 300 K with a Nosé–Hoover thermostat,53 combined with a gradual compression of the simulation box to obtain a configuration of liquid SPC/E water at 1 g cm−3 density. This configuration was used to initialize a new simulation for 300 ps in the NVT ensemble at 300 K to assure equilibration. The integration timestep was 0.5 fs for all of our simulations. A standard PPPM Ewald summation was used to compute electrostatic interactions. After the preparation of the liquid water configurations, the dimer was introduced into the centre of the simulation box. All water molecules with their oxygen atom within 3 Å of any solute atom were removed. The resulting configurations had 969 water molecules remaining, in both the DADA and DDAA cases. The partial charges of each dimer were determined with the restricted electrostatic potential (RESP) method and the geometries of the dimers were obtained by geometry optimizations. Both partial charges and the solute geometry were held fixed during the MD simulations. The Lennard-Jones interaction terms for the solute were taken from the OPLS parameter set.54 A simulation in the NPT ensemble with T = 300 K and P = 0 was done over 125 ps to re-equilibrate the system density. This resulted in systems with a box length L = 30.82 Å for the DDAA dimer, and L = 30.90 Å for the DADA dimer. A final simulation run was done for 500 ps to generate configurations for subsequent analysis. For simplicity, these production runs were done in the NVT ensemble. A total of 21 snapshots were collected for each case, each separated by 25 ps. The DFT calculations were performed with Turbomole,55 the SAPT calculations with PSI4,56 the DLPNO-CCSD(T) calculations with Orca,57,58 and the MD calculations with LAMMPS.59,604 Results and discussion
4.1 The DDAA motif
Table 1 shows the direct hydrogen bond energies and secondary interaction energies between the donor (D) and acceptor (A) groups, as the averages of all the studied DDAA dimers. Complete information on all the individual dimers is found in the ESI.† The total interaction energies have been decomposed into electrostatic, exchange, induction, and dispersion components. Here, we concentrate on the secondary interactions. According to the traditional secondary interactions hypothesis (SIH) model, the DDAA–AADD motif has two repulsive secondary interactions, namely A⋯A and D⋯D. The SAPT analysis shows, however, that the secondary A⋯A interactions in quadruply H-bonded DDAA systems are actually attractive. The average attraction energy between A1 and A4 is −26.8 kJ mol−1, which is of the same size as the direct hydrogen bonding contribution between the A and D moieties. Thus, there is only one repulsive and five attractive interactions in the DDAA motif. The individual interaction energies of the DDAA dimers are reported in Table S3.1 of the ESI.† Analyzing the individual terms of the interactions, we see that the exchange interaction almost vanishes due to the long distance between the two moieties. The induction and dispersion interactions are smaller than for the direct terms. Electrostatic interactions dominate the binding energy of the secondary interactions. The main component of the attractive interaction in the A⋯A cross-terms is actually near-field electrostatic, because it has previously been shown that the long-ranged dipole-interaction energy is significantly smaller than near-field electrostatic interactions.34 The short-ranged electrostatic attraction has a similar quantum mechanical origin as electron affinities implying that the attraction cannot be properly modeled at classical levels of theory.| E stat | E exch | E ind | E disp | E tot | |
|---|---|---|---|---|---|
| H bonds | |||||
| D1–A3/A2–D4 | −53.7 | +66.7 | −22.6 | −13.2 | −22.8 (×2) |
| D2–A4/A1–D3 | −68.0 | +57.6 | −24.7 | −12.1 | −47.2 (×2) |
| Secondary | |||||
| D1–A4/A1–D4 | +0.2 | +1.2 | −2.8 | −2.6 | −4.1 (×2) |
| D2–A3/A2–D3 | −2.4 | +2.2 | −4.7 | −3.4 | −8.3 (× 2) |
| D2–D3 | +8.4 | +1.8 | −2.0 | −3.0 | +5.3 |
| A1–A4 | −17.4 | +2.0 | −8.3 | −3.1 | −26.8 |
| Rest | −23.0 | +0.2 | −4.2 | −8.5 | −33.4 |
| Total | −279.6 | +259.3 | −128.2 | −71.0 | −219.5 |
4.2 The DADA motif
The DADA–ADAD hydrogen-bonding motif is less stable than the DDAA–AADD motif. According to the SIH model, this is due to the presence of six repulsive secondary interactions in the DADA motif, compared to only two in the DDAA motif.A detailed SAPT analysis again shows this explanation to be too simplistic and even qualitatively misleading. Table 2 shows the direct hydrogen bonding terms and the cross terms for the secondary interactions. The three D⋯D secondary interactions are indeed repulsive because depletion of electron charges usually leads to higher energies corresponding to repulsion in this case. On average, the three A⋯A secondary interactions are, however, again attractive, as in the case of the DDAA dimers discussed above. The average A1–A3 and A2–A4 attraction energies are −24.3 kJ mol−1, whereas the average A1–A4 attraction energy is −10.7 kJ mol−1 yielding a total stabilization energy of −59.3 kJ mol−1 from the secondary interaction between the acceptor moieties. The only exception is DADA04, which has a repulsive A1–A4 interaction of 21.7 kJ mol−1. And again, electrostatic interactions dominated by short-ranged contributions make up for the majority of the attraction. Dispersion effects also notably contribute to the total attractive interaction between the A⋯A pairs in the DADA motif.
| E stat | E exch | E ind | E disp | E tot | |
|---|---|---|---|---|---|
| H bonds | |||||
| D1–A3/A2–D4 | −56.6 | +66.3 | −23.9 | −15.4 | −29.7 (×2) |
| D2–A4/A1–D3 | −40.5 | +39.0 | −13.7 | −10.4 | −25.7 (×2) |
| Secondary | |||||
| D1–D3/D2–D4 | +7.4 | +1.0 | +0.6 | −2.4 | +6.6 (×2) |
| A1–A3/A2–A4 | −19.1 | +2.6 | −4.9 | −2.9 | −24.3 (×2) |
| D2–D3 | +6.7 | +2.0 | +0.6 | −2.3 | +7.0 |
| A1–A4 | −8.9 | +1.6 | −0.5 | −3.0 | −10.7 |
| Rest | −0.5 | +0.1 | −0.1 | −1.4 | −1.9 |
| Total | −220.4 | +221.4 | −83.9 | −68.9 | −151.8 |
The distance between the hydrogen bonds is about twice longer in DADA07 than in DADA08. The binding energy of DADA07 is −209 kJ mol−1 and −138 kJ mol−1 for DADA08. They differ because the direct A–D interaction of DADA08 is only −57 kJ mol−1, the A⋯A interaction is −93 kJ mol−1, and the D⋯D interaction of 4 kJ mol−1 is weakly repulsive. For DADA07, the corresponding energies are −204 kJ mol−1, −35 kJ mol−1 and 31 kJ mol−1. The individual interaction energies of the DADA dimers are reported in Table S3.2 of the ESI.†
Still, even with attractive A⋯A secondary interactions, the DADA motif is less stable than the DDAA motif, as the two motifs have three and one repulsive secondary interaction pairs, respectively, as shown in Fig. 1.
4.3 Solvent effects
Hydrogen bonding is different in the gas phase and the solution phase. In the gas phase, the hydrogen bond donors and acceptors from both the monomers have only each other to interact with. In a solution, there are solvent molecules present around the actual donors and acceptors, which change the environment. The solvent environment may interfere with the donors and acceptors, especially when the solvent is polar. In the case of water where a single water molecule possesses two hydrogen-bond donors and can accept two hydrogen bonds for a total of four hydrogen bonds per water, the solvent molecules can form competing hydrogen bonds with the original monomers. It is therefore of interest to see how the pattern of hydrogen bonding as well as the secondary interactions for the studied molecules change in a water environment. To see the effect of the solvent, the two tautomeric dimers DDAA06 and DADA08 were studied in the solution phase, using molecular dynamics to sample the configuration space of water molecules around the hydrogen-bonded dimers. Being tautomeric, The DDAA06/DADA08 pair allows for a direct comparison of the two donor–acceptor motifs, with minimal influence of other contributions.From the simulated time series, snapshots were selected at regular intervals, and subjected to a SAPT analysis, as in the case of the gas phase species discussed in the previous sections.
Table 3 shows the differences in the interaction energies of the dimers in the gas phase and in the simulated solution phase. There are some interesting changes imposed by the solution phase. The electrostatic and the induction interactions become even more attractive, while the exchange repulsion is enhanced. Overall, the total interaction energy of the dimers becomes more attractive in the solution phase. The attraction is strengthened by 14 and 7 kJ mol−1 for the DDAA and DADA tautomer, respectively. The strengthening of the interaction energy in the solution phase might at first sight seem to be counter intuitive. Here, we should note that the interaction energy is not the same as the complexation energy: in a water solution phase, where competing hydrogen bonds between monomers and water molecules is possible, the complexation energy will be lower than in gas phase, where the monomers by definition have no interaction with other molecules before dimerization. In solution, the dipole moments already of individual water molecules increase, which enhances the hydrogen bond strength. Going from the individual water dimer towards more bulk-like environments has been found to notably increase the H-bonding strengths.61 Thus, cooperative effects from surrounding polar molecules would be expected to increase the strength of the intermolecular forces such as hydrogen bonds as compared to their strength in isolation. Importantly, the analysis of the solution phase interactions shows that the general findings of the gas phase still hold, with the same pattern of attractive A⋯A secondary interaction. In fact, for the DDAA tautomer, all of the secondary interactions are attractive, both in the solution and gas phase. For the DADA tautomer, the solution phase decreases the repulsion also between the D⋯D secondary interactions, so that the SAPT analysis classifies two of them as minutely attractive as well.
| E stat | E exch | E ind | E disp | E tot | |
|---|---|---|---|---|---|
| DDAA06 | |||||
| H bonds | |||||
| D1–A3/A2–D4 | +2.6 | +3.7 | +0.1 | −0.3 | +6.1 (× 2) |
| D2–A4/A1–D3 | −3.3 | −0.2 | −0.7 | 0.0 | −4.1 (× 2) |
| Total H bonds | −1.5 | +7.1 | −1.2 | −0.5 | +4.0 |
| Secondary | |||||
| D1–A4/A1–D4 | −1.2 | +0.3 | 0.0 | −0.1 | −1.4 (× 2) |
| D2–A3/A2–D3 | −1.1 | +0.1 | 0.0 | 0.0 | −1.0 (× 2) |
| D2–D3 | −5.1 | 0.0 | −1.1 | 0.0 | −6.3 |
| A1–A4 | −4.0 | −0.1 | −0.5 | 0.0 | −4.5 |
| Rest | −2.0 | 0.0 | −0.6 | 0.0 | −2.6 |
| Total | −17.3 | +7.8 | −4.0 | −0.6 | −14.2 |
| DADA08 | |||||
| H bonds | |||||
| D1–A3/A2–D4 | +2.3 | +2.0 | −0.4 | −0.1 | +3.7 (× 2) |
| D2–A4/A1–D3 | −6.9 | +0.7 | −2.0 | −0.1 | −8.3 (× 2) |
| Total H bonds | −9.3 | +5.4 | −4.8 | −0.4 | −9.2 |
| Secondary | |||||
| D1–D3/D2–D4 | −2.0 | +0.1 | −1.5 | −0.1 | −3.4 (× 2) |
| A1–A3/A2–A4 | +1.3 | +0.1 | +0.0 | 0.1 | +1.5 (× 2) |
| D2–D3 | +2.7 | 0.0 | +1.0 | 0.0 | +3.8 |
| A1–A4 | +6.9 | +0.1 | +2.4 | −0.1 | +9.3 |
| Rest | −7.7 | +0.0 | +1.0 | +0. | −6.7 |
| Total | −8.9 | +5.9 | −2.1 | −0.5 | −6.8 |
4.4 Assessing the accuracy of the SAPT approach
Now, one might, and should naturally question the reliability of the analysis method, when the results go against established expectations. Therefore, we have also computed the interaction energies using the DLPNO-CCSD(T) coupled cluster level of theory. CCSD(T) is colloquially considered to be the gold standard of quantum chemical accuracy. This does require the use of large basis sets, however. Therefore, we have used both triple-zeta and quadruple-zeta basis sets, augmented by diffuse functions that is important for describing weak interactions. In addition, we have extrapolated the results towards the complete basis set (CBS) limit. The CCSD(T) results agree very well with the SAPT model, corroborating the findings here. The interaction energies of DDAA01 and DADA07 could not be calculated at the coupled cluster levels, due to their size. For the other dimers, the average total hydrogen bonding energy for the DDAA dimers is 220 kJ mol−1 at the SAPT level and 197 kJ mol−1 at the CCSD(T) level; for the DADA dimers, the corresponding average interaction energies are 152 kJ mol−1 (SAPT) and 127 kJ mol−1 (CCSD(T)). The full CCSD(T) data is found in the ESI.†5 Summary and conclusions
We have studied the hydrogen bonding of quadruple hydrogen-bonded dimers using symmetry-adapted perturbation theory (SAPT) and by performing state-of-the-art coupled-cluster calculations. Dimers bound through four hydrogen bonds can form two complementary bonding patterns, namely DDAA–AADD or DADA–ADAD, where D is the donor and A is the acceptor. The established notion in the secondary interactions hypothesis (SIH) is that the DDAA motif is more stable due to the presence of four attractive and two repulsive secondary interactions, compared to the purely repulsive secondary interactions in the DADA motif. The quantum-chemical analysis presented here shows, however, that this model is qualitatively misleading. The secondary A⋯A interactions are, in fact, attractive. Furthermore, the main component of the attractive interaction comes from the near-field electrostatic attraction between the two acceptor groups. The near-field electrostatic interaction dominates. The long-ranged dipole interaction energy has previously been found to be about one order of magnitude smaller than the energy of the secondary interactions.34 The A⋯A attraction cannot be simulated at the classical level because negative point charges are always repulsive, whereas the interaction becomes attractive at the quantum mechanical level due to a similar mechanism as the one lowering the energy of anions with respect to the corresponding neutral species.The study highlights the importance of considering molecular interactions with sufficient sophistication. While it is tempting, and often useful, to reduce for example electrostatic interactions to a point charge model, reality is more complex. In the case presented here, the intricacies of electronic structure lead to what at first glance might seem counter intuitive. We note that the striking failure of the point charge model for reproducing Coulomb interactions in transition metal compounds, notorious for their complex electronic structure, has been highlighted before by Frenking.62 As demonstrated here, it is necessary to consider the interactions of charge concentrations as the interaction of diffuse, malleable electron clouds even in seemingly simple interactions between hydrogen-bonded organic species. Our findings do not change the stability order of the DDAA versus DADA motifs. In the revised model presented here, on average, the DDAA motif has only one repulsive secondary interaction, while the DADA motif still has three. Thus, the simplicity of the original model is retained, but with a more correct description of the nature of the secondary A⋯A interactions. The fundamental difference between the secondary A⋯A and D⋯D interactions, the former being attractive and the latter usually repulsive, has direct implications for the design of self-complementary species and self-assembly in general. The realization that the interactions are opposite supports the design process and provides a tool for assessing the effect of secondary interactions on the total interaction energy through qualitative predictions based on the true physical and chemical nature of intramolecular interactions.
Author contributions
The project was initiated by MPJ. UA performed most of the electronic structure calculations under the supervision of MPJ and DS. CD performed the MD simulations. The results have been analyzed by MPJ, UA and DS. All authors have contributed to writing the article.Data availability
The data supporting this article are included as part of the ESI.† The MD data for this article are available at the NOMAD repository webpage https://nomad-lab.eu/prod/v1/gui/user/uploads/upload/id/jn9QfzEjR_OYPBv-Ry32WA. The DFT calculations were performed with Turbomole version 7.7.1, the SAPT calculations with PSI4, the DLPNO-CCSD(T) calculations with Orca version 5.0, and the MD calculations with LAMMPS version 7 Aug 2019. The Turbomole webpage is https://www.turbomole.org/. The PSI4 webpage is https://psicode.org/. The code for PSI4 can be found at https://github.com/psi4/. The ORCA webpage is https://orcaforum.kofo.mpg.de/app.php/portal. The LAMMPS webpage is https://www.lammps.org. The code for LAMMPS can be found at https://github.com/lammps/lammps.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work has been supported by the Academy of Finland through project numbers 289179, 314821, 319453, and 340583, by the Magnus Ehrnrooth Foundation, the Oskar Öflund Foundation, the Swedish Cultural Foundation in Finland, and Waldemar von Frenckells stiftelse. We acknowledge computational resources from CSC – IT Center for Science, Finland. We are grateful to Dr Joanna Johansson for critical comments on the manuscript and Dr Chapin E. Cavender for assistance with the PSI4 software.References
- G. M. Whitesides, J. P. Mathias and C. T. Seto, Science, 1991, 254, 1312–1319 CrossRef CAS PubMed.
- D. Pochan and O. Scherman, Chem. Rev., 2021, 121, 13699–13700 CrossRef CAS PubMed.
- G. Cooke and V. M. Rotello, Chem. Soc. Rev., 2002, 31, 275–286 RSC.
- A. Buckingham, J. Del Bene and S. McDowell, Chem. Phys. Lett., 2008, 463, 1–10 CrossRef CAS.
- R. P. Sijbesma and E. W. Meijer, Chem. Commun., 2003, 5–16 RSC.
- A. J. Wilson, Nat. Chem., 2011, 3, 193–194 CrossRef CAS PubMed.
- L. J. Karas, C. Wu, R. Das and J. I. Wu, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2020, 10, e1477 CAS.
- B. Gong, Y. Yan, H. Zeng, E. Skrzypczak-Jankunn, Y. W. Kim, J. Zhu and H. Ickes, J. Am. Chem. Soc., 1999, 121, 5607–5608 CrossRef CAS.
- P. K. Baruah and S. Khan, RSC Adv., 2013, 3, 21202–21217 RSC.
- K. Liu, S. Wang, Q. Zhang, J. Jiang and L. Wang, J. Am. Chem. Soc., 2021, 143, 1162–1170 CrossRef CAS PubMed.
- S.-L. Li, T. Xiao, W. Xia, X. Ding, Y. Yu, J. Jiang and L. Wang, Chem. – Eur. J., 2011, 17, 10716–10723 CrossRef CAS PubMed.
- T. Xiao, H. Wu, G. Sun, K. Diao, X. Wei, Z.-Y. Li, X.-Q. Sun and L. Wang, Chem. Commun., 2020, 56, 12021–12024 RSC.
- R. P. Sijbesma, Y. Deng, Q. Zhang, C. Shi, R. Toyoda, D. H. Qu, H. Tian and B. L. Feringa, Science, 1997, 278, 1601–1604 CrossRef CAS PubMed.
- L. Brunsveld, B. J. B. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071–4098 CrossRef CAS PubMed.
- J. H. K. K. Hirschberg, R. A. Koevoets, R. P. Sijbesma and E. W. Meijer, Chem. – Eur. J., 2003, 9, 4222–4231 CrossRef CAS PubMed.
- G. B. W. L. Ligthart, H. Ohkawa, R. P. Sijbesma and E. W. Meijer, J. Am. Chem. Soc., 2005, 127, 810–811 CrossRef CAS PubMed.
- T. Park, S. C. Zimmerman and S. Nakashima, J. Am. Chem. Soc., 2005, 127, 6520–6521 CrossRef CAS PubMed.
- Y. Hisamatsu, N. Shirai, S. Ikeda and K. Odashima, Org. Lett., 2009, 11, 4342–4345 CrossRef CAS PubMed.
- S.-L. Li, T. Xiao, Y. Wu, J. Jiang and L. Wang, Chem. Commun., 2011, 47, 6903–6905 RSC.
- P. S. Corbin, L. J. Lawless, Z. Li, Y. Ma, M. J. Witmer and S. C. Zimmerman, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5099–5104 CrossRef CAS PubMed.
- T. Park, E. M. Todd, S. Nakashima and S. C. Zimmerman, J. Am. Chem. Soc., 2005, 127, 18133–18142 CrossRef CAS PubMed.
- M. L. Pellizzaro, K. A. Houton and A. J. Wilson, Chem. Sci., 2013, 4, 1825–1829 RSC.
- W. L. Jorgensen and J. Pranata, J. Am. Chem. Soc., 1990, 112, 2008–2010 CrossRef CAS.
- J. Pranata, S. G. Wierschke and W. L. Jorgensen, J. Am. Chem. Soc., 1991, 113, 2810–2819 CrossRef CAS.
- P. L. A. Popelier and L. Joubert, J. Am. Chem. Soc., 2002, 124, 8725–8729 CrossRef CAS PubMed.
- C. Fonseca Guerra, F. M. Bickelhaupt, J. G. Snijders and E. J. Baerends, Chem. – Eur. J., 1999, 5, 3581–3594 CrossRef.
- O. J. Backhouse, J. C. R. Thacker and P. L. A. Popelier, ChemPhysChem, 2019, 20, 555–564 CrossRef CAS PubMed.
- C.-H. Wu, Y. Zhang, K. van Rickley and J. I. Wu, Chem. Commun., 2018, 54, 3512–3515 RSC.
- W. E. Vallejo Narváez, E. I. Jiménez, E. Romero-Montalvo, A. Sauza-de la Vega, B. Quiroz-García, M. Hernández-Rodríguez and T. Rocha-Rinza, Chem. Sci., 2018, 9, 4402–4413 RSC.
- W. E. Vallejo Narviáez, E. I. Jiménez, E. Romero-Montalvo, A. Sauza-de la Vega, B. Quiroz-García, M. Hernández-Rodríguez and T. Rocha-Rinza, Chem. Commun., 2019, 55, 1556–1559 RSC.
- X. Lin, W. Wu and Y. Mo, J. Org. Chem., 2019, 84, 14805–14815 CrossRef CAS PubMed.
- M. K. Tiwari and K. Vanka, Chem. Sci., 2017, 8, 1378–1390 RSC.
- L. Guillaumes, S. Simon and C. Fonseca Guerra, ChemistryOpen, 2015, 4, 318–327 CrossRef CAS PubMed.
- S. C. C. van der Lubbe, F. Zaccaria, X. Sun and C. Fonseca Guerra, J. Am. Chem. Soc., 2019, 141, 4878–4885 CrossRef CAS PubMed.
- B. Jeziorski, R. Moszynski and K. Szalewicz, Chem. Rev., 1994, 94, 1887–1930 CrossRef CAS.
- R. M. Parrish, T. M. Parker and C. D. Sherrill, J. Chem. Theory Comput., 2014, 10, 4417–4431 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- V. N. Staroverov, G. E. Scuseria, J. Tao and J. P. Perdew, J. Chem. Phys., 2003, 119, 12129–12137 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- R. A. Kendall, T. H. Dunning and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- E. Papajak, J. Zheng, X. Xu, H. R. Leverentz and D. G. Truhlar, J. Chem. Theory Comput., 2011, 7, 3027–3034 CrossRef CAS PubMed.
- C. Bannwarth, S. Ehlert and S. Grimme, J. Chem. Theory Comput., 2019, 15, 1652–1671 CrossRef CAS PubMed.
- C. Riplinger, P. Pinski, U. Becker, E. F. Valeev and F. Neese, J. Chem. Phys., 2016, 144, 024109 CrossRef PubMed.
- A. Halkier, T. Helgaker and P. Jørgensen, Chem. Phys. Lett., 1998, 286, 243–252 CrossRef CAS.
- D. Z. Goodson, J. Chem. Phys., 2002, 116, 6948–6956 CrossRef CAS.
- H. J. C. Berendsen, J. R. Grigera and T. P. Straatsma, J. Phys. Chem., 1987, 91, 6269–6271 CrossRef CAS.
- C. Vega and J. L. F. Abascal, Phys. Chem. Chem. Phys., 2011, 13, 19663–19688 RSC.
- C. D. Daub, K. Leung and A. Luzar, J. Phys. Chem. A, 2009, 113, 7687–7700 CrossRef CAS PubMed.
- S. Nosé, J. Chem. Phys., 1984, 81, 511–519 CrossRef.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS.
- S. G. Balasubramani, J. H. Jensen, H.-S. Lee, W. A. Goddard III, J. Tirado-Rives and W. L. Jorgensen, J. Chem. Phys., 2020, 152, 184107 CrossRef CAS PubMed.
- R. M. Parrish, E. G. Hohenstein, C. D. Sherrill and T. J. Martínez, J. Chem. Theory Comput., 2017, 13, 3185–3197 CrossRef CAS PubMed.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, J. Chem. Phys., 2020, 152, 224108 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- S. Plimpton, J. Comput. Phys., 1995, 117, 1–19 CrossRef CAS.
- A. P. Thompson, H. M. Aktulga, R. Berger, D. S. Bolintineanu, W. M. Brown, P. S. Crozier, P. J. in 't Veld, A. Kohlmeyer, S. G. Moore, T. D. Nguyen, R. Shan, M. J. Stevens, J. Tranchida, C. Trott and S. J. Plimpton, Comput. Phys. Commun., 2022, 271, 108171 CrossRef CAS.
- V. M. Castor-Villegas, J. M. Guevara-Vela, W. E. Vallejo Narváez, Á. Martín Pendás, T. Rocha-Rinza and A. Fernández-Alarcón, J. Comput. Chem., 2020, 41, 2266–2277 CrossRef CAS PubMed.
- G. Frenking, The Chemical Bond, Wiley-VCH Verlag GmbH & Co. KGaA, 2014, pp. 175–218 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The optimized molecular structures, total energies calculated at the CCSD(T) level, full SAPT analysis, interaction energies calculated at the SAPT and CCSD(T) levels, and pictures showing the SAPT partitioning domains. See DOI: https://doi.org/10.1039/d4cp02361g |
| This journal is © the Owner Societies 2024 |