
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTMAO perturbs intermolecular vibrational motions of water revealed by low-frequency modes†
Tsung-Han
Liu
 and
Masanari
Okuno
*
and
Masanari
Okuno
*
Department of Basic Science, Graduate School of Arts and Sciences, The University of Tokyo, Meguro, Tokyo 153-8902, Japan. E-mail: cmokuno@g.ecc.u-tokyo.ac.jp
First published on 8th April 2024
Abstract
Trimethylamine N-oxide (TMAO) as a representative natural osmolyte has received much attention because of its unique properties, including enhancement of hydrogen bonding networks in liquid water and stabilization of three-dimensional structures of proteins in living organisms. As a hydrogen bond maker and/or a protein stabilizer, its hydrated structures and orientation dynamics in aqueous solutions have been investigated by various spectroscopic methods. Particularly, distinct from other natural osmolytes, it has been found that TMAO molecules form complexes with water molecules even at low concentrations, showing extraordinarily long lifetimes and much larger effective dipole moments. In this study, we demonstrated that collective motions of water molecules are closely correlated to TMAO molecules, as revealed by the changes of the librational modes observed in hyper-Raman (HR) spectra in the low-frequency region (<1000 cm−1) for the first time. Based on HR spectra of the TMAO solutions at submolar concentrations, we observed that the librational bands originating from water apparently upshift (∼15 cm−1) upon the addition of TMAO molecules. Compared to the OH stretching band of water showing a negligible downshift (<5 cm−1), the librational bands of water are more sensitive to reflect changes in the hydrogen bonding networks in the TMAO solutions, suggesting formation of transient TMAO–water complexes plays an essential role toward surrounding water molecules in perturbing their librational motions. We expect to provide a supplementary approach to understand that water molecules in TMAO aqueous solutions are strongly affected by TMAO molecules, different from other osmolytes.
Introduction
Natural osmolytes have received much attention, especially in the biological research fields.1,2 In general, natural osmolytes generated during metabolism in organisms are small organic molecules with neutral charge at physiological pH.1,2 They effectively protect cells from water pressure and help organisms survive against external extreme osmotic stress.1,3 In particular, for those with hydrophilic functional groups such as amine, hydroxyl, and carboxyl groups, the osmolytes, including amino acids and derivatives, polyols and sugars, methylamines, methylsulfonium compounds, and urea, can easily form hydrogen bonds to surrounding water molecules and then locally change the hydrogen bonding network of water.1 For example, trimethylamine N-oxide (TMAO) is a well-known hydrogen bond maker to enhance the hydrogen bonding networks.4,5 Meanwhile, urea is recognized as a hydrogen bond breaker to weaken the hydrogen bonds among water molecules selectively.6 In addition, osmolytes are also found to associate with stabilization of native proteins, as they are classified into protein stabilizers and denaturants.1,2,6,7 TMAO and urea showing opposite characters in hydrogen bonding are a classical pair of protein stabilizers and denaturants, commonly counteracting with each other in certain organisms in the oceans.1,7,8 However, scientific issues including how these osmolytes interact with biomacromolecules at the molecular level in detail and whether surrounding water molecules are mediated or not are still under investigation.9,10TMAO and tert-butyl alcohol (TBA) are isosteric compounds with the same hydrophobic part but different hydrophilic heads, and their physical properties, hydration behaviors, and dynamics have been frequently compared and examined.11–18 In aqueous solutions of TMAO/TBA, two or three hydrogen bonds per TMAO/TBA molecule are formed between their hydrophilic sites and water molecules.12 Compared to water molecules in bulk, some water molecules show slowdowns of orientation and hydrogen bond dynamics even at low TMAO concentrations.14,18,19 In contrast, TMAO counteracts the osmotic stress due to urea giving opposite effects on hydrogen bonding.1,8,13,20,21 In urea aqueous solutions, one urea molecule can form six hydrogen bonds at most with adjacent water molecules because its carbonyl group and two primary amine groups provide two and four bonding sites, respectively.6 Previous spectroscopic and simulation results indicate that a small fraction of water molecules (approximately one water molecule per urea molecule) can simultaneously form two hydrogen bonds with one urea molecule, while most water molecules do not show remarkable differences in orientation dynamics from the bulk water.6,22
The intermolecular interactions in aqueous solutions containing osmolytes have been experimentally investigated through neutron scattering,3,13,23 light scattering,15,17 X-ray scattering and absorption spectroscopy,23–27 NMR,8,20 Raman spectroscopy,4,21,28 steady-state IR absorption spectroscopy,14,29 dielectric spectroscopy and femtosecond infrared pump–probe spectroscopy,5,6,14,20,22 and OKE spectroscopy and THz spectroscopy.28,30–34 In IR and Raman spectroscopies, frequency shifts of the OH stretching and HOH bending bands have been frequently discussed to judge whether hydrogen bonds are enhanced or weakened.14,21,29,35 On the other hand, vibrational modes in the ultralow-frequency region (<300 cm−1) have been studied by OKE and THz spectroscopies, showing the addition of osmolytes changes orientation dynamics of water.28,30–34 However, to the best of our knowledge, the low-frequency region (<1000 cm−1), in which the librational modes of water locate,36,37 between the ultralow-frequency and the fingerprint regions, has not been explored yet. Hyper-Raman (HR) spectroscopy has been recently applied to water and small organic molecules in solutions as a vibrational spectroscopic technique complementary to conventional IR and Raman spectroscopies.38–43 In particular, HR spectroscopy has the advantage of observing signals in the low-frequency region in addition to the bending and stretching vibrational motions of water molecules,38 while IR spectroscopy has difficulties measuring the whole spectral region, including the low-frequency region. Low-frequency signals of water and aqueous solutions directly reflect information on the intramolecular hydrogen bond formation and collective motions of water,38 closely correlated with the initiation of rearrangement dynamics of the hydrogen bonding network.44–47 Frequencies of the librational modes are proportional to the stiffness of water's rotational potential around intact hydrogen bonds, enabling us to discuss tiny local changes in conformations of hydrogen bonds, water structures, and the dynamics in detail.46 Recent Raman studies on water molecules confined inside biological cells suggest the librational modes of water are more sensitive to reflect different molecular environments due to various types of biomolecules than the bending mode.48,49
Here, we demonstrate HR spectroscopy of aqueous solutions of osmolytes at submolar concentrations comparable to the estimated concentrations in organisms to investigate changes in collective motions of water induced by osmolytes.1,8 We focus on the librational and OH stretching modes of water molecules in aqueous solutions of TMAO, TBA, and urea. From the HR spectra of TMAO aqueous solutions, we clarified that the librational motions of water molecules are changed upon the addition of TMAO molecules, as revealed by the shifted signals in the low-frequency region. Compared to the OH stretching band of water, the librational bands of water are more sensitive to reflect changes in the hydrogen bonding networks in the submolar TMAO solutions. In contrast, it turns out that the additions of TBA or urea did not induce any change in the librational motion or OH stretching regions of water. We expect to provide a supplementary point of view to understand how water molecules are affected by osmolytes.
Experimental section
Samples
Anhydrous TMAO (>95.0%, Tokyo Chemical Industry), TBA (guaranteed reagent, Nacalai Tesque), and urea (guaranteed reagent, Nacalai Tesque) were used without further purification. TMAO and urea are hygroscopic, and all of them are highly water-soluble. To prepare their aqueous solutions of various concentrations ranging from 0.1 to 1 or 2 M (mol L−1), ultrapure water (resistivity; 18.2 MΩ cm) obtained from a water purification system (Purelab flex 3, ELGA) and deuterated water (99.9 D atom%, Eurisotop) were used as solvents to dissolve them completely at room temperature.Hyper-Raman spectroscopic system
Our HR spectroscopic system has been described in detail elsewhere.41,50,51 The excitation light source is an Nd:YVO4 laser (Cepheus, Photon Energy; 1064 nm, 100 kHz, ∼15 ps). We used the second harmonic light (532 nm) of the output of the laser source. The average laser power was 150–180 mW. The vertically-polarized laser beam was focused by a lens onto a 1-cm square quartz cuvette inserted in a temperature-controlled holder (Luma 40, Quantum Northwest). The HR scattered signals at 90° were collected by a lens and passed through an analyzer and a short-pass filter. The scattered light was then introduced into a polychromator (MS3504i, SOL; 1800 grooves per mm, 270-nm blaze grating) and was detected by a CCD camera (iDus DU420A-OE, Andor). The polarization directions of the incident laser light and the scattered light were set to be vertical, referred as VV. In the measurements of TBA solutions, we also measured horizontally polarized HR scattered signals, referred as VH. The slit width was set to 100 μm. Exposure time was 600 s, and each HR spectrum is an average of 12 exposures. Wavenumber calibration was performed by using the emission lines of a handheld mercury lamp (GL-4, Panasonic).Results and discussion
HR spectra of TMAO aqueous solutions
Fig. 1 shows HR spectra of water and TMAO aqueous solutions at various concentrations. Each set of spectra is normalized in the low-frequency and OH stretching regions, respectively. In HR spectra of water, the OH stretching band around 3400 cm−1 and the HOH bending band around 1650 cm−1 appear as discussed in the reported IR and Raman spectroscopic results.4,29,35 In addition, two broad bands ranging from 400–900 cm−1 ascribed to librational motions of water molecules are observed as in the previous HR spectra of water and aqueous solutions.38,50 For convenience, we name the librational bands at the lower and higher vibrational frequencies the L1 and L2 bands, respectively. There is no remarkable difference between the spectra of water and the TMAO solution at 0.1 M. In contrast, at higher concentrations of TMAO, in addition to intense signals originating from water molecules, weak intramolecular vibrational bands of TMAO molecules appear. Based on the reported Raman spectra of TMAO aqueous solutions, the vibrational bands around 490, 940, and 1140–1500 and 2920–3030 cm−1 are assigned to the NO rocking mode, the CN and NO stretching modes, and the CH3 rocking/deformation/stretching modes, respectively,4 consistent with our HR spectra of the TMAO aqueous solutions.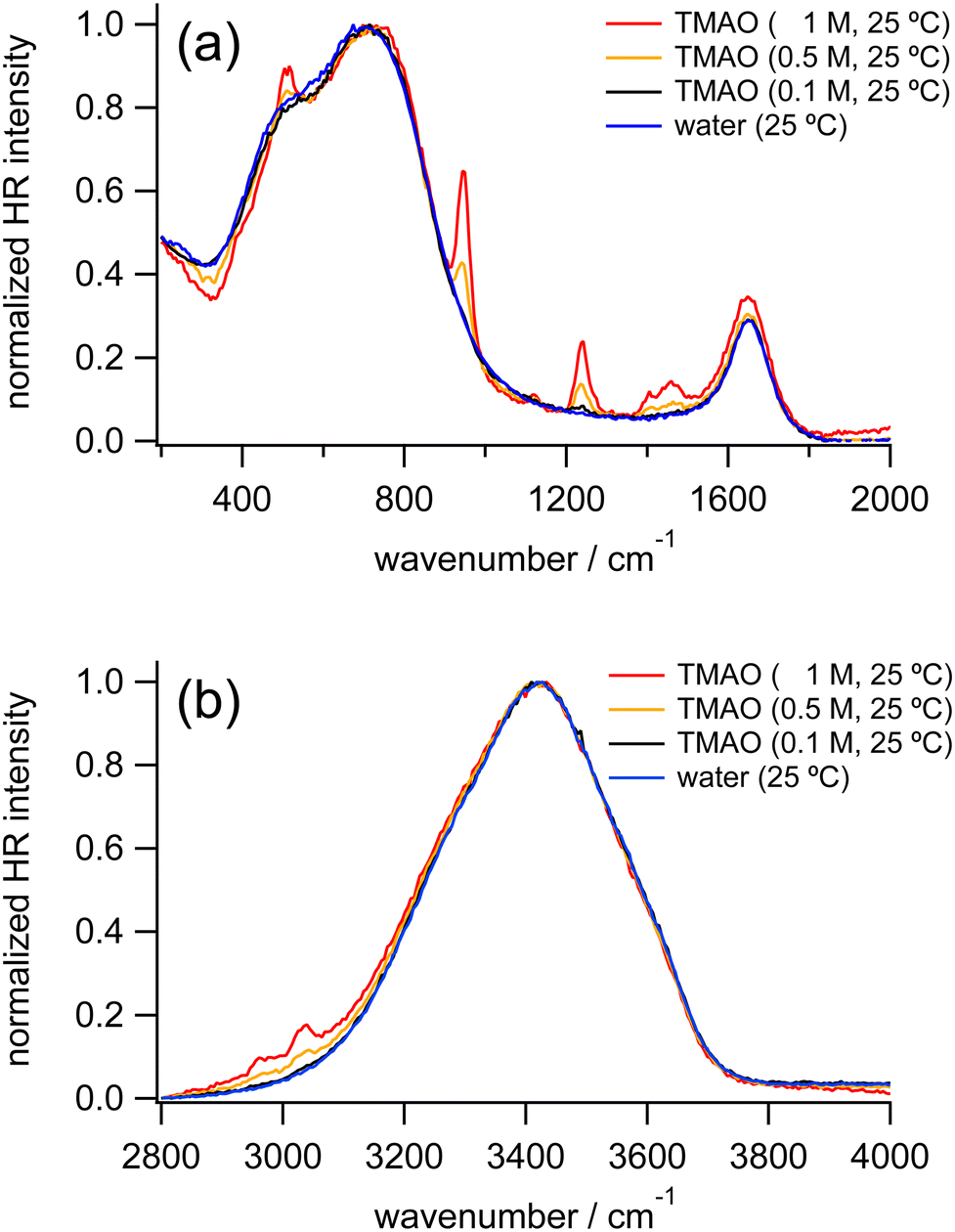 | ||
| Fig. 1 HR spectra of water (blue) and TMAO aqueous solutions at 0.1 M (black), 0.5 M (orange), and 1 M (red) measured at 25 °C, normalized in the low-frequency region (a) and the OH stretching region (b), respectively. | ||
To discuss changes in hydrogen bonding induced by the osmolytes, we focus on HR signals originating from water molecules, especially the librational and OH stretching bands. Fig. 2 shows the enlarged HR spectra of water and TMAO aqueous solutions normalized in the low-frequency region. We fitted the spectra with two Gaussian functions for the librational bands and two Lorentzian functions for the intramolecular bands of TMAO in a range of 200 and 1200 cm−1 after baseline subtraction. The fitted band positions of the librational bands are summarized in Table 1. As the TMAO concentration increases from 0 to 1 M, the OH stretching band around 3420 cm−1 downshifts but to a small extent (∼5 cm−1), suggesting the hydrogen bonding network among water molecules is slightly enhanced, as reported in the IR spectra of TMAO in a mixture of H2O/HDO and D2O.14,29 Similar to the OH stretching band of water, the HOH bending band of water around 1650 cm−1, a direct probe for hydrogen bonding networks,35 also gives a negligible position shift (∼5 cm−1) as the TMAO concentration increases to 1 M. In contrast to the small shifts of the intramolecular vibrations, both the L1 and L2 bands upshift (∼15 cm−1). Because the librational modes originate from the hindered rotation of water molecules, the upshifts suggest the hydrogen bonding environment around TMAO molecules more strongly hinders the rotation of water molecules than bulk water. Although there is no report to directly point out that the librational bands of water upshift due to the enhancement of hydrogen bonding networks, downshifts of the librational bands of water due to temperature elevation have been suggested based on the simulated Raman spectra and the fitted IR spectra.52,53 In our HR spectra of water and the TMAO solutions, the librational signals originating from the collective motions of water molecules upshift due to the addition of TMAO molecules. Therefore, it is suggestive that in the TMAO aqueous solutions TMAO molecules strengthen the hydrogen bonds among water molecules. Compared to the OH stretching vibrational motions of water, we found that changes in the librational motions of water are more sensitive to reflect the changes in the hydration structures induced by TMAO. We will discuss the origin of the downshift of the librational mode in the later section.
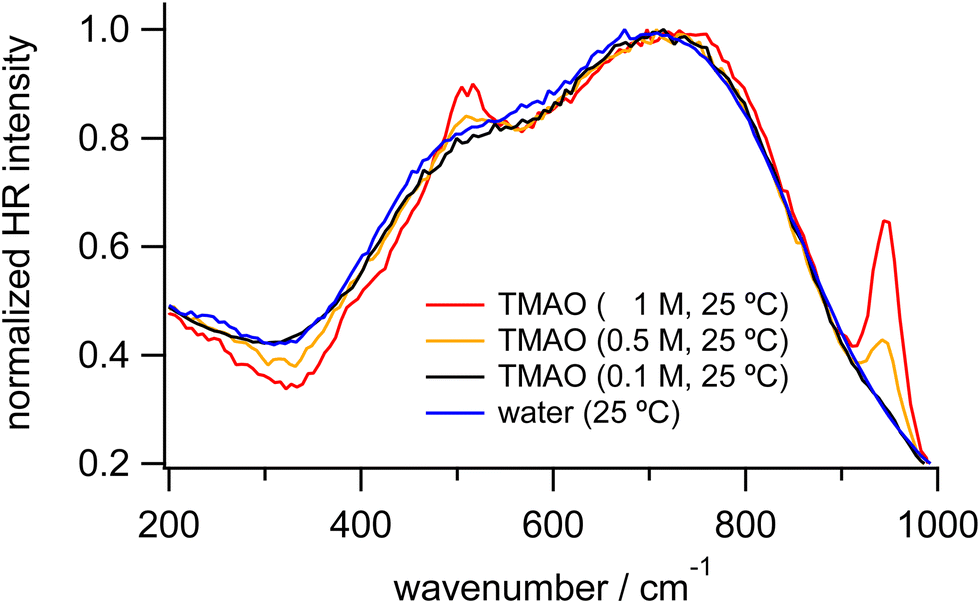 | ||
| Fig. 2 Enlarged HR spectra of water (blue) and TMAO aqueous solutions at 0.1 (black), 0.5 (orange), and 1 M (red) measured at 25 °C, normalized in the low-frequency region. | ||
| Concentration/M | L1/cm−1 | L2/cm−1 | HOH bend/cm−1 | OH stretch/cm−1 |
|---|---|---|---|---|
| 0 | 483 | 718 | 1650 | 3419 |
| 0.1 | 487 | 727 | 1650 | 3419 |
| 0.5 | 493 | 729 | 1647 | 3416 |
| 1 | 499 | 745 | 1646 | 3415 |
HR spectra of TBA aqueous solutions
Fig. 3 shows the normalized HR spectra of water and TBA aqueous solutions at various concentrations. Similar to the HR spectra of the TMAO solutions, in addition to the vibrational bands ascribed to the HOH bending mode and librational motions of water molecules, intramolecular bands of TBA molecules are observed in the HR spectra of the TBA solutions. Based on the reported Raman and IR spectra of TBA, the vibrational bands around 920 and 1020, 1210, 1240, 1370, 1455, and 2920 cm−1 are assigned to the CH3 rocking modes, the C3CO asymmetric stretching mode, the CCC stretching mode, the CH3 symmetric deformation mode, the CH3 asymmetric deformation mode, and the CH3 symmetric and asymmetric stretching modes, respectively.54 Note that in the HR spectrum of the TBA solution at 1 M the shoulder-like signals appearing around 2000 cm−1 and 3800 cm−1 are ascribed to uplifting background signals rather than signals from water molecules.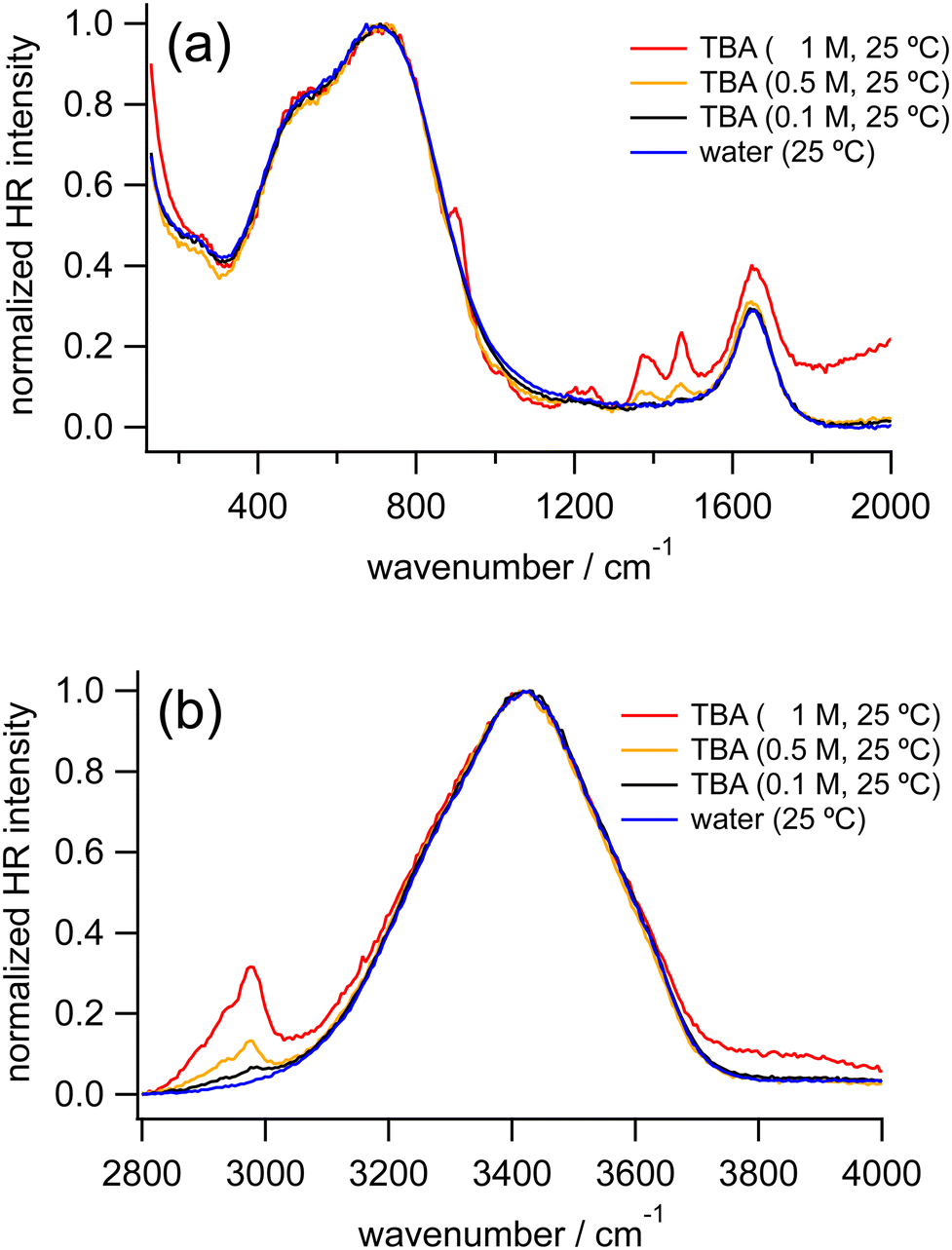 | ||
| Fig. 3 HR spectra of water (blue) and TBA aqueous solutions at 0.1 M (black), 0.5 M (orange), and 1 M (red) measured at 25 °C, normalized in the low-frequency region (a) and the OH stretching region (b), respectively. | ||
As the concentration of TBA increases from 0 to 1 M, the OH stretching band around 3420 cm−1 downshifts very slightly (<5 cm−1), similar to our observation in the HR spectra of the TMAO solutions. The librational bands from 400 to 900 cm−1 show no significant differences in band positions, while a new band around 250 cm−1 ascribed to the hydrogen bond (HB) stretching mode appears.
We further measured TBA solutions under a different polarization combination, VH, to confirm the independence of the peak positions of the librational modes from the TBA concentrations by suppressing the hyper-Rayleigh scattered signals, which partially overlap with the librational bands. In the VH-polarized HR spectra of water and the TBA aqueous solutions (Fig. S1 in the ESI†), the HB band and the librational bands become more prominent. As TBA concentration increases from 0 to 2 M, the HB band becomes more intense. In contrast, the librational bands do not change their intensity ratios or peak positions, as observed in the VV-polarized HR spectra. As a result, although both TMAO and TBA give a similar downshift of the OH stretching band (<5 cm−1), it turns out that the librational bands shift only in the HR spectra of the TMAO aqueous solutions. In addition, we found that the HB bands appear only in the HR spectra of the TBA aqueous solutions. These distinguishable spectral features imply that interactions between TMAO/TBA and water molecules are intrinsically different. TMAO and TBA are isosteric amphiphiles, while TMAO rather than TBA forms stable and long-lived hydrated complexes with larger dipole moments,4,5 exhibiting much more ordered hydrogen bonding networks than pure water.4,11,12,28,55 Our results suggest that the difference in the formation of the hydrated complexes gives rise to the difference in the low-frequency signals originating from the intermolecular vibrations of water molecules.
HR spectra of urea aqueous solutions
Fig. 4 shows the HR spectra of water and urea aqueous solutions at various concentrations. In the HR spectra of the urea aqueous solutions, in addition to the vibrational bands due to water molecules, intramolecular bands of urea molecules are observed. From the reported Raman and IR spectra of solid-state urea and urea aqueous solutions, the bands around 780, 1000, 1490, and 1620 cm−1 are assigned to the CO wagging mode, the CN symmetric stretching mode, the CN asymmetric stretching mode, a coupling between the CO symmetric stretching and the NH2 bending modes, respectively.56 Concerning the vibrational bands of water molecules in the HR spectra of the urea aqueous solutions, as the urea concentration increases from 0 to 1 M, the librational bands of water do not show clear shifts. Similarly, the OH stretching band in our HR spectra of the urea solutions is independent of the urea concentration, consistent with the previous IR study of the urea solutions at much higher concentrations.29 The results suggest that urea molecules are perfectly embedded inside an extended hydrogen bonding network of water molecules without disturbance because urea has the approximate size of a water dimer.6,31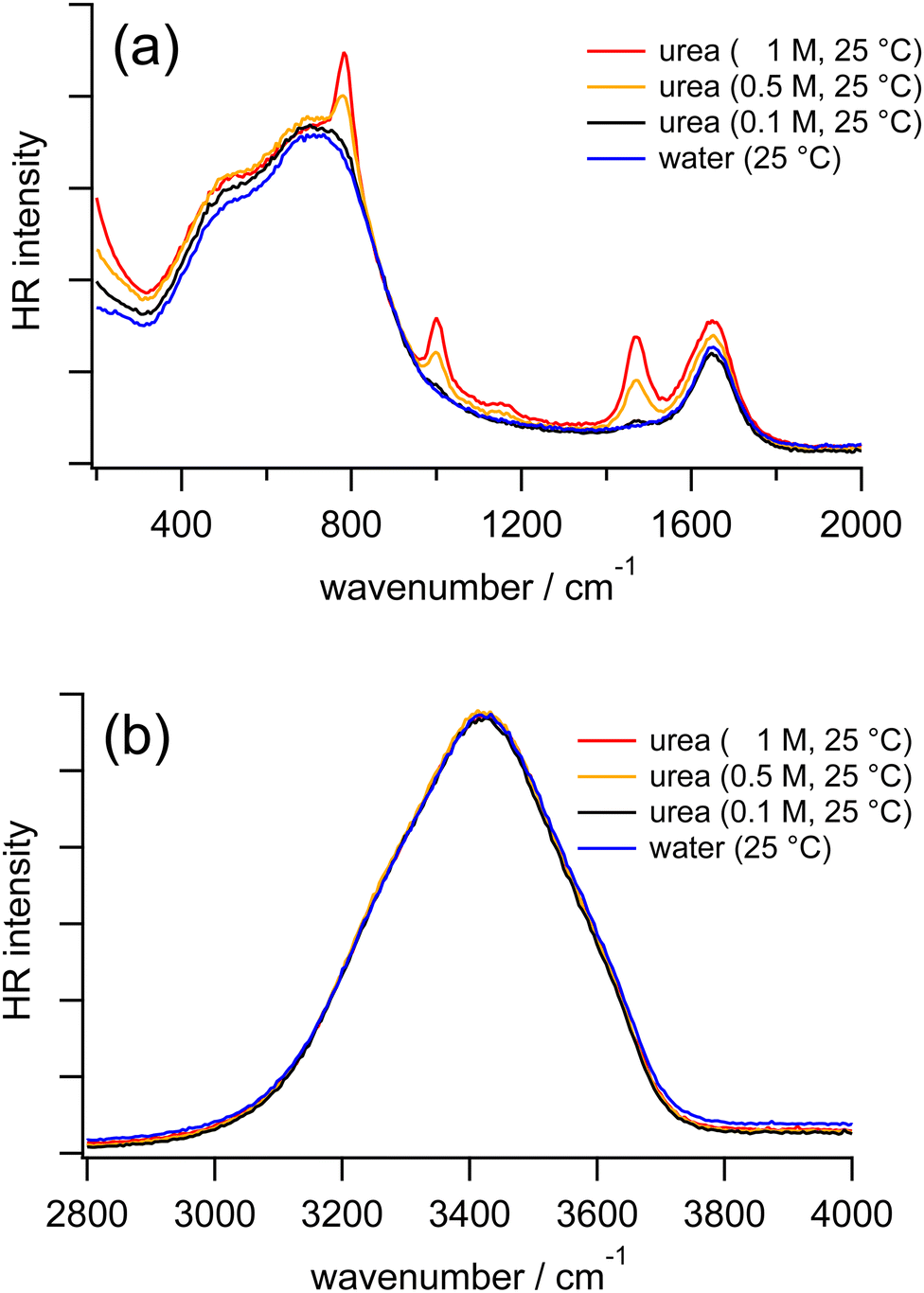 | ||
| Fig. 4 HR spectra of water (blue) and urea aqueous solutions at 0.1 M (black), 0.5 M (orange), and 1 M (red) measured at 25 °C in the low-frequency region (a) and the OH stretching region (b). | ||
Because the sharp CO wagging band of urea around 780 cm−1 is partially overlapped with the librational band L2 of water, we further carried out the measurements by replacing H2O with D2O as the solvents. In HR spectra of pure D2O and urea in D2O at various concentrations (Fig. S2 in the ESI†), the librational bands of D2O do not show distinguishable shifts as the urea concentration increases from 0 to 2 M. Based on our HR spectra of urea in H2O and D2O at different concentrations, it is confirmed that librational and stretching bands of water are not much affected by urea molecules, as we found in the measurements of TBA solutions, but different from the measurements of TMAO solutions.
Comparison of TMAO aqueous solutions and neat water
Fig. 5 summarizes the peak frequencies of the librational and the OH stretching bands of water in the HR spectra of the TMAO aqueous solutions as the concentration increases from 0 to 1 M. First, we notice that the frequencies of the librational bands shift much more than those of the OH stretching band in this concentration range as we explained in the previous section. There is a clear difference between the shifts of the librational and the OH stretching bands as the TMAO concentration increases to 1 M; the librational bands upshift more than 15 cm−1, while the OH stretching band only shows a small downshift, <5 cm−1. This contrast suggests that collective rotational motions of water molecules, detected in the librational bands, are strongly hindered by added TMAO molecules with tiny changes in the local hydrogen bonding network reflected in the OH stretching bands. In the reported IR spectra of TMAO aqueous solutions at high concentrations (>5 M), a significant downshift of the OH stretching band has been emphasized, implying that hydrogen bonds between water and TMAO molecules are much enhanced.29 In contrast, in the HR spectra of TMAO aqueous solutions at submolar concentrations close to the real concentrations in living organisms, the OH stretching band does not give remarkable downshifts. It is considered that at submolar concentrations, only a small fraction of water molecules can directly interact with TMAO molecules through hydrogen bond formation, while TMAO molecules influence other water molecules in their vicinity, giving rise to the large shifts of the librational modes of water.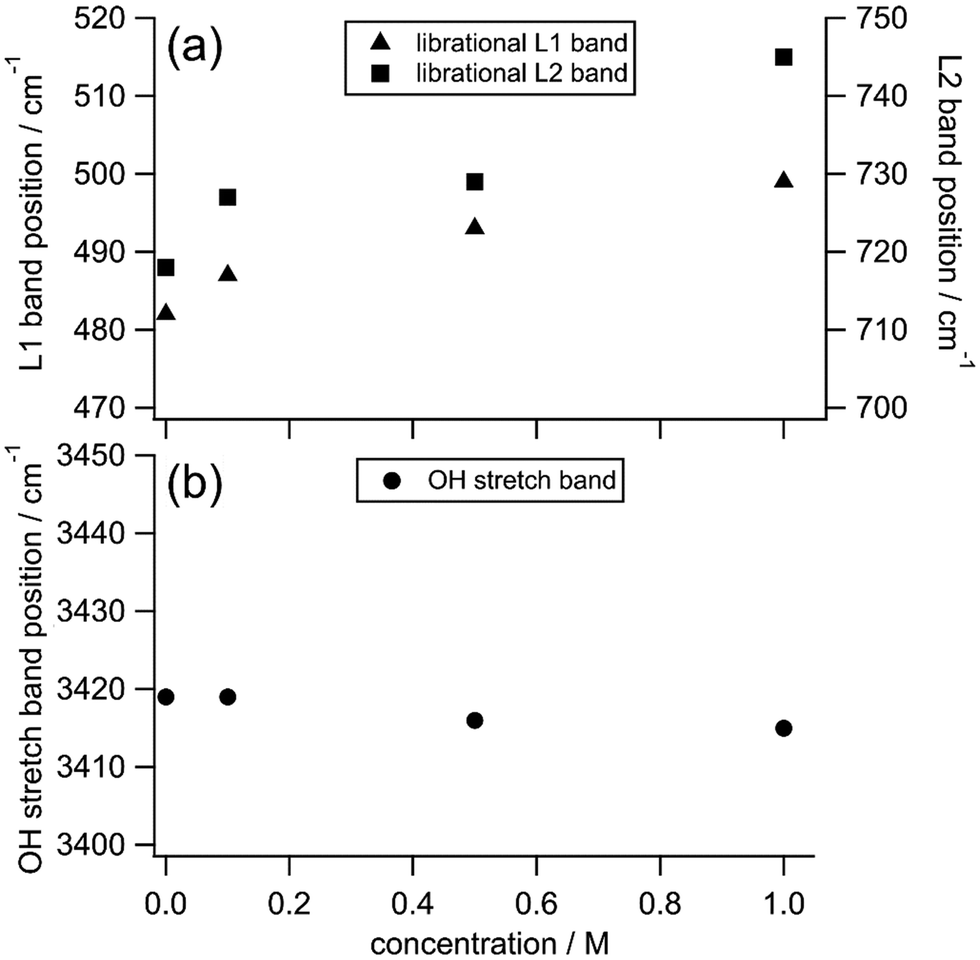 | ||
| Fig. 5 The peak positions of the librational L1 bands (triangles, a), the librational L2 bands (squares, a), and the OH stretching bands (circles, b) originating from water molecules in the HR spectra of TMAO aqueous solutions at different concentrations. | ||
In the reported femtosecond mid-infrared spectra of the TMAO and TBA aqueous solutions, it is found that part of the water molecules displays much slower orientation dynamics than the water molecules in the bulk.14 Furthermore, in the reported dielectric spectra of the TMAO aqueous solutions, based on the water relaxation modes, water molecules are classified into three types; the bulk-like water, the slow water, and the bound water, respectively.5 The bulk-like water is also observable in neat water, the slow water is located in the hydration shell of TMAO, and the bound water is confined in the TMAO–water complexes such as TMAO·2H2O and TMAO·3H2O depending on the TMAO concentration.5,14 The experimentally observed effective dipole moments of the TMAO–water complexes are much larger than the calculated dipole moment of an isolated TMAO molecule, showing stronger interactions between hydrated complexes and water molecules.5,18 Besides, those hydrated species are long-lived because the orientation dynamics of surrounding water molecules show an obvious slowdown even at low concentrations.4,5 In the TMAO aqueous solutions, fraction ratios of the bound water, the slow water, and the bulk-like water have been estimated to be about 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7:90 at 0.5 m (0.5 mol kg−1, ∼0.5 M) and 4:8:88 at 1 m (1 mol kg−1, ∼0.9 M).5 In this submolar concentration range, the summation of the bound water and the slow water accounts for around 10% of all water molecules in the TMAO solutions, and the rest 90% of water molecules behave like those in pure water.5 Our HR spectral results in the submolar concentration range show shat the librational bands of water show obvious shifts only in the TMAO solutions but not in the TBA or urea solutions. Because of the unique complex formation of the TMAO–water complexes, surrounding water molecules in the vicinity of the hydrated species alter their collective motions, namely, librational modes of water molecules. Therefore, we conclude that the HR shift of the librational bands is ascribed to the bound water and the slow water whose rotation is strongly hindered due to the addition of TMAO, approximately 10% of all water molecules in the TMAO solutions at submolar concentrations.
:7:90 at 0.5 m (0.5 mol kg−1, ∼0.5 M) and 4:8:88 at 1 m (1 mol kg−1, ∼0.9 M).5 In this submolar concentration range, the summation of the bound water and the slow water accounts for around 10% of all water molecules in the TMAO solutions, and the rest 90% of water molecules behave like those in pure water.5 Our HR spectral results in the submolar concentration range show shat the librational bands of water show obvious shifts only in the TMAO solutions but not in the TBA or urea solutions. Because of the unique complex formation of the TMAO–water complexes, surrounding water molecules in the vicinity of the hydrated species alter their collective motions, namely, librational modes of water molecules. Therefore, we conclude that the HR shift of the librational bands is ascribed to the bound water and the slow water whose rotation is strongly hindered due to the addition of TMAO, approximately 10% of all water molecules in the TMAO solutions at submolar concentrations.
To investigate how differently the librational motions of water are associated with osmolytes and temperature, we also measured the HR spectra of osmolyte solutions at 0.5 M and pure water at various temperatures (Fig. S3 in the ESI†). The strength and geometry of hydrogen bonds in liquid water intrinsically vary as temperature changes.57,58 Those hydrogen bonds are further enhanced by amphiphilic solutes such as alcohols with hydroxyl groups.59–61 Compared to hydrogen bonds in the bulk water, hydrogen bonds in the TMAO–water complexes are much enhanced, in which two to three water molecules are captured at one end of the molecules and then temporarily immobilized by the head group of TMAO molecules.5,14,25 Here we focus on the results of TMAO that shows the extraordinary upshifts of the librational bands in our concentration-dependent measurements. Fig. 6 shows the frequencies of the librational bands originating from water in the HR spectra of pure water upon temperature variation and those in the HR spectrum of the TMAO solution at 0.5 M measured at 25 °C. Over a temperature range between 10 and 60 °C, the slopes of the librational band shift per degree Celsius by fitting with linear functions are −0.5 cm−1 °C−1 for the librational L1 band and −0.6 cm−1 °C−1 for the librational L2 band, respectively.
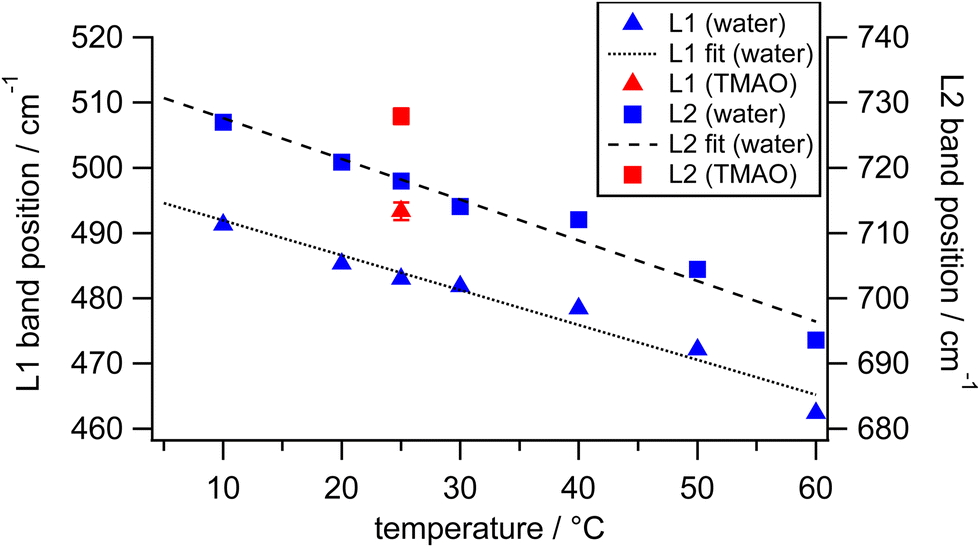 | ||
| Fig. 6 The peak positions of the librational L1 bands (triangles) and the librational L2 bands (squares) originating from water in the HR spectra of water (blue) and the TMAO aqueous solution at 0.5 M (red) at different temperatures. Black dashed lines are the fits of the librational band positions. | ||
From the frequency shifts of librational bands of water by minor TMAO molecules at room temperature, we try to estimate the corresponding temperature of pure water that gives a spectral response similar to TMAO aqueous solution. At 25 °C, the librational band positions in our HR spectra of pure water are 483 and 718 cm−1. As the TMAO concentration increases from 0 to 0.5 M, the two librational bands simultaneously show upshifts of about 10 cm−1. On the other hand, the decrease of the temperature of pure water from 25 °C to around 5 °C induces the equivalent shifts. As discussed above, the upshifts of the librational bands of water upon the addition of TMAO are contributed by approximately 10% of water molecules in the solution at 0.5 M. Those minor water molecules around TMAO molecules at room temperature nominally behave like bulk water molecules at much lower temperatures due to the strong hydrogen bonds in the vicinity of TMAO. Suppose we can exclusively measure the librational bands of the minor water molecules under the influence of TMAO molecules, which give upshifts of about 10 cm−1 in the HR spectra of the whole TMAO solution at 0.5 M. In this case, since the minor water molecules account for about 10% of all water molecules in the TMAO solution, the librational bands of the minor water perturbed by TMAO may actually upshift ten times more than those observed from the TMAO solution, that is, about 100 cm−1. In the reported HR spectra of ice, the librational bands, L1 and L2, are located at 515 and 825 cm−1, respectively.38 In other words, when liquid water at 25 °C is cooled down to solid-state ice at 0 °C, the L1 band upshifts about 30 cm−1, and the L2 band upshifts more than 100 cm−1. Thus, we infer that TMAO molecules effectively immobilize water molecules; their collective motions are much more restricted, close to water molecules confined in ice crystals. It should be noticed that the OH stretching band does not shift much (<5 cm−1), verifying that in the submolar TMAO solutions, TMAO molecules induce changes in the collective nature of the hydrogen bonding networks.
Conclusion
We have demonstrated how TMAO molecules in aqueous solutions influence the librational motions of surrounding water molecules, as revealed by the vibrational bands of water in the low-frequency region of the HR spectra. Simultaneously, we have also compared TMAO to other common osmolytes, TBA and urea. In the previous spectroscopic studies on TMAO aqueous solutions, enhancement of the hydrogen bonding network among water and TMAO in very concentrated solutions (>5 M) and the existence of the unique TMAO–water complexes have been reported. Moreover, in addition to the water molecules directly interacting with TMAO molecules, it was observed that the surrounding water molecules are also influenced as their orientation dynamics slow down. In this study, we found that in the submolar concentration region, the librational bands originating from collective motions of water molecules upshift as the TMAO concentration increases, while the OH stretching band reflecting hydrogen bonding gives a negligible downshift. Interestingly, such upshifts of the librational bands of water were not observable in the HR spectra of TBA or urea aqueous solutions. Different from TBA and urea, TMAO exhibits its distinct ability to enhance hydrogen bonds through the formation of the hydrated complexes in its aqueous solutions. In addition to the water molecules partially and temporarily confined in the TMAO–water complexes, HR spectroscopy enables us to observe that even at submolar concentrations, certain water molecules are undoubtedly affected by TMAO molecules as their collective motions are distinct from those in pure water. We demonstrated that TMAO molecules can further influence water molecules in the vicinity in an indirect way. At present, the spectral analysis is limited to the frequency shifts because of the overlapping intermolecular vibrational bands and the hyper-Rayleigh signals in the low-frequency region. It would be promising to estimate the number of water molecules perturbed by the added TMAO molecules based on intensity and band shape changes of the librational bands. We expect to provide a supplementary approach to understand how TMAO stabilizes protein structures in specific living organisms containing natural osmolytes at submolar concentrations.Abbreviations
| HR spectroscopy | Hyper-Raman spectroscopy |
| TMAO | Trimethylamine N-oxide |
| TBA | tert-Butyl alcohol |
| HB | Hydrogen bond |
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was partially funded by JSPS KAKENHI (grant number 21KK0091 and 21K04975), YAMADA Science Foundation, and JST FOREST (grant number JPMJFR211K).References
- P. H. Yancey, Organic osmolytes as compatible, metabolic and counteracting cytoprotectants in high osmolarity and other stresses, J. Exp. Biol., 2005, 208(15), 2819–2830 CrossRef CAS PubMed.
- M. T. Velasquez, A. Ramezani, A. Manal and D. S. Raj, Trimethylamine N-Oxide: The Good, the Bad and the Unknown, Toxins, 2016, 8(11), 326 CrossRef PubMed.
- H. Laurent, T. G. A. Youngs, T. F. Headen, A. K. Soper and L. Dougan, The ability of trimethylamine N-oxide to resist pressure induced perturbations to water structure, Commun. Chem., 2022, 5, 116 CrossRef CAS PubMed.
- K. L. Munroe, D. H. Magers and N. I. Hammer, Raman Spectroscopic Signatures of Noncovalent Interactions Between Trimethylamine N-oxide (TMAO) and Water, J. Phys. Chem. B, 2011, 115(23), 7699–7707 CrossRef CAS PubMed.
- J. Hunger, K.-J. Tielrooij, R. Buchner, M. Bonn and H. J. Bakker, Complex Formation in Aqueous Trimethylamine-N-oxide (TMAO) Solutions, J. Phys. Chem. B, 2012, 116(16), 4783–4795 CrossRef CAS PubMed.
- Y. L. A. Rezus and H. J. Bakker, Effect of urea on the structural dynamics of water, Proc. Natl. Acad. Sci. U. S. A., 2006, 103(49), 18417–18420 CrossRef CAS PubMed.
- P. Ganguly, P. Boserman, N. F. A. van der Vegt and J.-E. Shea, Trimethylamine N-oxide Counteracts Urea Denaturation by Inhibiting Protein–Urea Preferential Interaction, J. Am. Chem. Soc., 2018, 140(1), 483–492 CrossRef CAS PubMed.
- M. Nasralla, H. Laurent, D. L. Baker, M. E. Ries and L. Dougan, A study of the interaction between TMAO and urea in water using NMR spectroscopy, Phys. Chem. Chem. Phys., 2022, 24, 21216–21222 RSC.
- L. Hua, R. Zhou, D. Thirumalai and B. J. Berne, Urea denaturation by stronger dispersion interactions with proteins than water implies a 2-stage unfolding, Proc. Natl. Acad. Sci. U. S. A., 2008, 105(44), 16928–16933 CrossRef CAS PubMed.
- W. K. Lim, J. Rösgen and S. W. Englander, Urea, but not guanidinium, destabilizes proteins by forming hydrogen bonds to the peptide group, Proc. Natl. Acad. Sci. U. S. A., 2009, 106(8), 2595–2600 CrossRef CAS PubMed.
- A. D. Michele, M. Freda, G. Onori and A. Santucci, Hydrogen Bonding of Water in Aqueous Solutions of Trimethylamine-N-oxide and tert-Butyl Alcohol: A Near-Infrared Spectroscopy Study, J. Phys. Chem. A, 2004, 108(29), 6145–6150 CrossRef.
- S. Paul and G. N. Patey, Why tert-Butyl Alcohol Associates in Aqueous Solution but Trimethylamine-N-oxide Does Not, J. Phys. Chem. B, 2006, 110(21), 10514–10518 CrossRef CAS PubMed.
- F. Meersman, D. Bowron, A. K. Soper and M. H. J. Koch, Counteraction of Urea by Trimethylamine N-Oxide Is Due to Direct Interaction, Biophys. J., 2009, 97(9), 2559–2566 CrossRef CAS.
- C. Petersen, A. A. Bakulin, V. G. Pavelyev, M. S. Pshenichnikov and H. J. Bakker, Femtosecond midinfrared study of aggregation behavior in aqueous solutions of amphiphilic molecules, J. Chem. Phys., 2010, 133, 164514 CrossRef PubMed.
- L. Comez, L. Lupi, M. Paolantoni, F. Picchiò and D. Fioretto, Hydration properties of small hydrophobic molecules by Brillouin light scattering, J. Chem. Phys., 2012, 137, 114509 CrossRef CAS PubMed.
- A. Fiore, V. Venkateshwaran and S. Garde, Trimethylamine N-Oxide (TMAO) and tert-Butyl Alcohol (TBA) at Hydrophobic Interfaces: Insights from Molecular Dynamics Simulations, Langmuir, 2013, 29(25), 8017–8024 CrossRef CAS PubMed.
- L. Comez, M. Paolantoni, S. Corezzi, L. Lupi, P. Sassi, A. Morresi and D. Fioretto, Aqueous solvation of amphiphilic molecules by extended depolarized light scattering: the case of trimethylamine-N-oxide, Phys. Chem. Chem. Phys., 2016, 18, 8881–8889 RSC.
- T. Ohto, J. Hunger, E. H. G. Backus, W. Mizukami, M. Bonn and Y. Nagata, Trimethylamine-N-oxide: its hydration structure, surface activity, and biological function, viewed by vibrational spectroscopy and molecular dynamics simulations, Phys. Chem. Chem. Phys., 2017, 19, 6909–6920 RSC.
- G. Stirnemann, E. Duboué-Dijon and D. Laage, Ab Initio Simulations of Water Dynamics in Aqueous TMAO Solutions: Temperature and Concentration Effects, J. Phys. Chem. B, 2017, 121(49), 11189–11197 CrossRef CAS PubMed.
- W. J. Xie, S. Cha, T. Ohto, W. Mizukami, Y. Mao, M. Wagner, M. Bonn, J. Hunger and Y. Nagata, Large Hydrogen-Bond Mismatch between TMAO and Urea Promotes Their Hydrophobic Association, Chem, 2018, 4(11), 2615–2627 CAS.
- S. G. Zetterholm, G. A. Verville, L. Boutwell, C. Boland, J. C. Prather, J. Bethea, J. Cauley, K. E. Warren, S. A. Smith, D. H. Magers and N. I. Hammer, Noncovalent Interactions between Trimethylamine N-Oxide (TMAO), Urea, and Water, J. Phys. Chem. B, 2018, 122(38), 8805–8811 CrossRef CAS.
- V. Agieienko and R. Buchner, Urea hydration from dielectric relaxation spectroscopy: old findings confirmed, new insights gained, Phys. Chem. Chem. Phys., 2016, 18, 2597–2607 RSC.
- F. Meersman, D. Bowron, A. K. Soper and M. H. J. Koch, An X-ray and neutron scattering study of the equilibrium between trimethylamine N-oxide and urea in aqueous solution, Phys. Chem. Chem. Phys., 2011, 13, 13765–13771 RSC.
- C. J. Sahle, M. A. Schroer, I. Juurinen and J. Niskanen, Influence of TMAO and urea on the structure of water studied by inelastic X-ray scattering, Phys. Chem. Chem. Phys., 2016, 18, 16518–16526 RSC.
- C. J. Sahle, M. A. Schroer, J. Niskanen, M. Elbers, C. M. Jeffries and C. Sternemann, Hydration in aqueous osmolyte solutions: the case of TMAO and urea, Phys. Chem. Chem. Phys., 2020, 22, 11614–11624 RSC.
- Y. Sasaki, Y. Horikawa, T. Tokushima, K. Okada, M. Oura and M. Aida, Hydration structure of trimethylamine N-oxide in aqueous solutions revealed by soft X-ray emission spectroscopy and chemometric analysis, Phys. Chem. Chem. Phys., 2016, 18, 27648–27653 RSC.
- F. Lehmkühler, Y. Forov, M. Elbers, I. Steinke, C. J. Sahle, C. Weis, N. Tsuji, M. Itou, Y. Sakurai, A. Poulain and C. Sternemann, Temperature dependence of the hydrogen bond network in trimethylamine N-oxide and guanidine hydrochloride–water solutions, Phys. Chem. Chem. Phys., 2017, 19, 28470–28475 RSC.
- L. Knake, G. Schwaab, K. Kartaschew and M. Havenith, Solvation Dynamics of Trimethylamine N-Oxide in Aqueous Solution Probed by Terahertz Spectroscopy, J. Phys. Chem. B, 2015, 119(43), 13842–13851 CrossRef CAS.
- K. A. Sharp, B. Madan, E. Manas and J. M. Vanderkooi, Water structure changes induced by hydrophobic and polar solutes revealed by simulations and infrared spectroscopy, J. Chem. Phys., 2001, 114, 1791–1796 CrossRef CAS.
- A. Idrissi, P. Bartolini, M. Ricci and R. Righini, Time resolved optical Kerr effect analysis of urea–water system, J. Chem. Phys., 2001, 114, 6774–6780 CrossRef CAS.
- A. Idrissi, P. Bartolini, M. Ricci and R. Righini, Temperature dependence of the reorientational dynamics and low-frequency response of aqueous urea solutions investigated by femtosecond optical Kerr-effect spectroscopy and molecular-dynamics simulation, Phys. Chem. Chem. Phys., 2003, 5(20), 4666–4671 RSC.
- K. Mazur, I. A. Heisler and S. R. Meech, THz Spectra and Dynamics of Aqueous Solutions Studied by the Ultrafast Optical Kerr Effect, J. Phys. Chem. B, 2011, 115(11), 2563–2573 CrossRef CAS PubMed.
- N. Samanta, D. D. Mahanta and R. K. Mitra, Does Urea Alter the Collective Hydrogen-Bond Dynamics in Water? A Dielectric Relaxation Study in the Terahertz-Frequency Region, Chem. – Asian J., 2014, 9(12), 3457–3463 CrossRef CAS PubMed.
- S. Imoto, H. Forbert and D. Marx, Aqueous TMAO solutions as seen by theoretical THz spectroscopy: hydrophilic versus hydrophobic water, Phys. Chem. Chem. Phys., 2018, 20, 6146–6158 RSC.
- T. Seki, K.-Y. Chiang, C.-C. Yu, X. Yu, M. Okuno, J. Hunger, Y. Nagata and M. Bonn, The Bending Mode of Water: A Powerful Probe for Hydrogen Bond Structure of Aqueous Systems, J. Phys. Chem. Lett., 2020, 11(19), 8459–8469 CrossRef CAS PubMed.
- G. E. Walrafen, Raman Spectral Studies of Water Structure, J. Chem. Phys., 1964, 40, 3249–3256 CrossRef CAS.
- G. E. Walrafen, Raman Spectral Studies of the Effects of Temperature on Water Structure, J. Chem. Phys., 1967, 47, 114–126 CrossRef CAS.
- V. Korepanov, C.-C. Yu and H. Hamaguchi, Hyper-Raman investigation of intermolecular vibrations of water and ice, J. Raman Spectrosc., 2018, 49(11), 1742–1746 CrossRef CAS.
- C.-I. Wen and H. Hiramatsu, The 532-nm-excited hyper-Raman spectroscopy of globular protein and aromatic amino acids, J. Raman Spectrosc., 2020, 51(2), 274–278 CrossRef CAS.
- M. Asakura and M. Okuno, Hyper-Raman Spectroscopic Investigation of Amide Bands of N-Methylacetamide in Liquid/Solution Phase, J. Phys. Chem. Lett., 2021, 12(20), 4780–4785 CrossRef CAS PubMed.
- M. Okuno, Hyper-Raman spectroscopy of alcohols excited at 532 nm: Methanol, ethanol, 1-propanol, and 2-propanol, J. Raman Spectrosc., 2021, 52(4), 849–856 CrossRef CAS.
- C.-I. Wen, C.-N. Yu, N. Thirumalaivasan and H. Hiramatsu, 532-nm-excited hyper-Raman spectroscopy of amino acids, J. Raman Spectrosc., 2021, 52(3), 641–654 CrossRef CAS.
- C.-N. Yu and H. Hiramatsu, Resonance Hyper-Raman Spectroscopy of Nucleotides and Polynucleotides, J. Phys. Chem. Lett., 2022, 126, 9309–9315 CrossRef CAS PubMed.
- I. Ohmine, Liquid Water Dynamics: Collective Motions, Fluctuation, and Relaxation, J. Phys. Chem., 1995, 99(18), 6767–6776 CrossRef CAS.
- I. Ohmine and S. Saito, Water Dynamics: Fluctuation, Relaxation, and Chemical Reactions in Hydrogen Bond Network Rearrangement, Acc. Chem. Res., 1999, 32(9), 741–749 CrossRef CAS.
- Y. Tong, T. Kampfrath and R. K. Campen, Experimentally probing the libration of interfacial water: the rotational potential of water is stiffer at the air/water interface than in bulk liquid, Phys. Chem. Chem. Phys., 2016, 18, 18424–18430 RSC.
- W. T. S. Cole and R. J. Saykally, Hydrogen bond network rearrangement dynamics in water clusters: Effects of intermolecular vibrational excitation on tunneling rates, J. Chem. Phys., 2017, 147, 064301 CrossRef PubMed.
- S. Ramos and J. C. Lee, Water bend–libration as a cellular Raman imaging probe of hydration, Proc. Natl. Acad. Sci. U. S. A., 2023, 120(42), e2313133120 CrossRef CAS PubMed.
- J. L. Skinner, Raman imaging of water in biological cells, Proc. Natl. Acad. Sci. U. S. A., 2023, 120(45), e2316387120 CrossRef CAS PubMed.
- K. Inoue and M. Okuno, Hyper-Raman Study of Hydrated Excess Protons in Water: Measurement of Concentrated HCl Solution, J. Raman Spectrosc., 2022, 53(10), 1679–1685 CrossRef CAS.
- K. Inoue, Y. Litman, D. M. Wilkins, Y. Nagata and M. Okuno, Is Unified Understanding of Vibrational Coupling of Water Possible? Hyper-Raman Measurement and Machine Learning Spectra, J. Phys. Chem. Lett., 2023, 14, 3063–3068 CrossRef CAS PubMed.
- W. B. Bosma, L. E. Fried and S. Mukamel, Simulation of the intermolecular vibrational spectra of liquid water and water clusters, J. Phys. Chem., 1993, 98, 4413–4421 CrossRef CAS.
- H. R. Zelsmann, Temperature dependence of the optical constants for liquid H2O and D2O in the far IR region, J. Mol. Struct., 1995, 350, 95–114 CrossRef CAS.
- J. Korppi-Tommola, Tert-butyl alcohol—matrix i.r. spectra and vibrational assignment, Spectrochim. Acta, Part A, 1978, 34(11), 1077–1085 CrossRef.
- A. Panuszko, P. Bruździak, J. Zielkiewicz, D. Wyrzykowski and J. Stangret, Effects of Urea and Trimethylamine-N-oxide on the Properties of Water and the Secondary Structure of Hen Egg White Lysozyme, J. Phys. Chem. B, 2009, 113(44), 14797–14809 CrossRef CAS PubMed.
- R. Keuleers, H. O. Desseyn, B. Rousseau and C. V. Alsenoy, Vibrational Analysis of Urea, J. Phys. Chem. A, 1999, 103(24), 4621–4630 CrossRef CAS.
- K. A. T. Silverstein, A. D. J. Haymet and K. A. Dill, The Strength of Hydrogen Bonds in Liquid Water and Around Nonpolar Solutes, J. Am. Chem. Soc., 2000, 122(33), 8037–8041 CrossRef CAS.
- K. Modig, B. G. Pfrommer and B. Halle, Temperature-Dependent Hydrogen-Bond Geometry in Liquid Water, Phys. Rev. Lett., 2003, 90, 075502 CrossRef PubMed.
- L. Evangelisti and W. Caminati, Internal dynamics in complexes of water with organic molecules. Details of the internal motions in tert-butylalcohol–water, Phys. Chem. Chem. Phys., 2010, 12, 14433–14441 RSC.
- J. Andersen, J. Heimdal and R. W. Larsen, The influence of large-amplitude librational motion on the hydrogen bond energy for alcohol–water complexes, Phys. Chem. Chem. Phys., 2015, 17, 23761–23769 RSC.
- D. Mihrin, J. Andersen, P. W. Jakobsen and R. W. Larsen, Highly localized H2O librational motion as a far-infrared spectroscopic probe for microsolvation of organic molecules, Phys. Chem. Chem. Phys., 2019, 21, 1717–1723 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp01025f |
| This journal is © the Owner Societies 2024 |