
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDetailed quasiclassical dynamics of the F− + SiH3Cl multi-channel reaction†
Attila Á.
Dékány
and
Gábor
Czakó
 *
*
MTA-SZTE Lendület Computational Reaction Dynamics Research Group, Interdisciplinary Excellence Centre and Department of Physical Chemistry and Materials Science, Institute of Chemistry, University of Szeged, Rerrich Béla tér 1, Szeged H-6720, Hungary. E-mail: gczako@chem.u-szeged.hu
First published on 6th March 2024
Abstract
We report a detailed quasiclassical trajectory study on the F− + SiH3Cl multi-channel reaction using a full-dimensional ab initio analytical potential energy surface. Reaction probabilities, cross sections, initial attack and scattering angle distributions as well as product relative translational, internal, vibrational, and rotational energy distributions are obtained in the collision energy range of 1–40 kcal mol−1 for the following channels: SiH3F + Cl−, SiH2Cl− + HF, SiH2F− + HCl, SiH2FCl + H−, SiH2 + FHCl−, and SiHFCl− + H2. All the channels are translationally cold indicating indirect mechanisms, except proton transfer (SiH2Cl− + HF), which shows mixed direct–indirect character. The angular distributions vary depending on collision energy and inversion/retention for SiH3F + Cl−. In the case of SiH2Cl− + HF front-side/back-side attack backward–forward/forward scattering preference is found at low/high collision energy. SiH2F− + HCl is formed with isotropic scattering and the preferred angle of attack is similar to the SiH3F + Cl− channel. SiH2FCl + H−/SiH2 + FHCl− favors back-side attack and isotropic/backward scattering, whereas SiHFCl− + H2 does not show significant angular preference.
1. Introduction
The dynamics and mechanisms of bimolecular nucleophilic substitution (SN2) reactions of halide ions (X−) with methyl-halide molecules (CH3Y) have been extensively investigated,1–8 however, analogous SN2 reactions at silicon (Si) centers are less thoroughly studied. Previous theoretical work on the H−/F− + SiH4 and X− + SiH3Y systems was restricted to stationary-point characterizations9–19 and on-the-fly direct dynamics simulations20–22 until 2023 when we developed a full-dimensional coupled-cluster-quality analytical potential energy surface (PES) for the F− + SiH3Cl reaction,23 which, to the best of our knowledge, was the first PES for a Si-centered SN2 reaction. The analytical PES allows the efficient computations of a large number of quasiclassical trajectories (QCTs) providing statistically-converged cross sections and unprecedented insights into the mechanisms of the system of interest. The QCT simulations on the new PES showed that the F− + SiH3Cl SN2 reaction is not stereospecific, because the Walden-inversion and front-side attack retention pathways are competitive, especially at high collision energies.23 Besides SN2, our trajectories uncovered several other product channels such as SiH2Cl− + HF, SiH2F− + HCl, SiH2FCl + H−, SiH2 + FHCl−, SiHFCl− + H2, SiHF + H2 + Cl−, and SiH2 + HF + Cl−, showing that the dynamics of Si-centered ion–molecule reactions is even more complex than that of the analogous C-centered systems.23In our previous work23 we focused on the challenges and methodology of the PES development, the evaluation of the accuracy and performance of the analytical PES, and the atomic-level mechanisms of the different reaction channels uncovered by inspecting and analyzing several representative trajectories. In the present study we plan to report a more detailed QCT analysis of the F− + SiH3Cl reaction computing differential cross sections such as initial attack angle and scattering angle distributions as well as relative translational, internal, vibrational, and rotational energy distributions for the products. These detailed differential cross section data, for the first time, provide new insights into the dynamics and mechanisms of the different channels of the title reaction, showing the spatial preference of the reactive collisions as well as how the energy transfers from reactants to the various degrees of freedom of the products. In Section 2 we give the methodological and computational details. In Section 3 the results and discussion are given, first introducing the characteristic of the PES, then providing the reaction probabilities and cross sections of the different channels, describing the angular and post-reaction energy distributions, and comparing the present findings to those obtained for the analogous carbon- and phosphorous-centered systems. The paper ends with summary and conclusions in Section 4.
2. Methods and computational details
QCT simulations were performed on a full-dimensional analytical composite coupled-cluster-quality PES,23 which is represented by a permutationally invariant polynomial24 fitted to (CCSD-F12b + BCCD(T)−BCCD)/aug-cc-pVTZ energy points, for the SiH3Cl + F− system, across various collision energies (1, 5, and up to 40 kcal mol−1 in 5 kcal mol−1 increments) and impact parameters. The impact parameter, b, ranged from 0 to bmax, and the bmax values, where the reactive trajectories vanish, ranged from 9.5 to 25 bohr, depending on the collision energy (Ecoll). At each Ecoll and b 5000 trajectories were run.For every simulation, the initial vibrational energy was set to the zero-point energy of SiH3Cl via normal-mode sampling and its rotational energy was adjusted to zero. We arrange the initial center of mass distance between the reactants by the 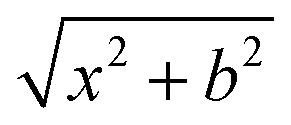 formula, where x was 40 bohr at Ecoll = 1 and 5 kcal mol−1, and 25 bohr at higher collision energies.
formula, where x was 40 bohr at Ecoll = 1 and 5 kcal mol−1, and 25 bohr at higher collision energies.
A more detailed explanation of the fitting method, the creation of the initial fitting set, the iterative PES development and the quality assessment of the final PES is given in our previous article.23 The computational details of the QCT product analysis are described below.
We define the α initial attack angle as the angle between the center-of-mass velocity vector of the SiH3Cl reactant, 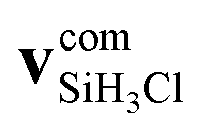 , and the vector of the spatial displacement between its Cl ligand and the central atom, xCl − xSi, obtained at the initial trajectory step, as depicted in Fig. 1a. The θ scattering angle is the angle between the center-of-mass velocity vectors of the reactant fluoride ion at the initial trajectory step,
, and the vector of the spatial displacement between its Cl ligand and the central atom, xCl − xSi, obtained at the initial trajectory step, as depicted in Fig. 1a. The θ scattering angle is the angle between the center-of-mass velocity vectors of the reactant fluoride ion at the initial trajectory step, 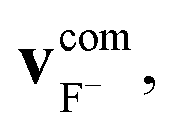 and one of the resulting products at the final step of the simulation. For each product channel, we employed the velocity coordinates of the product containing the fluorine atom to compute the scattering angles. Fig. 1b illustrates θ for the chloride substitution channel, where we track the SiH3F product. It is important to note that in the case of a product pair, owing to the law of conservation of momentum and the initial center-of-mass velocities of the reactants being set in parallel, the velocity vectors of the products are always parallel as well. Consequently, the scattering angle defined for one product complements the scattering angle defined for the other product to 180 degrees.
and one of the resulting products at the final step of the simulation. For each product channel, we employed the velocity coordinates of the product containing the fluorine atom to compute the scattering angles. Fig. 1b illustrates θ for the chloride substitution channel, where we track the SiH3F product. It is important to note that in the case of a product pair, owing to the law of conservation of momentum and the initial center-of-mass velocities of the reactants being set in parallel, the velocity vectors of the products are always parallel as well. Consequently, the scattering angle defined for one product complements the scattering angle defined for the other product to 180 degrees.
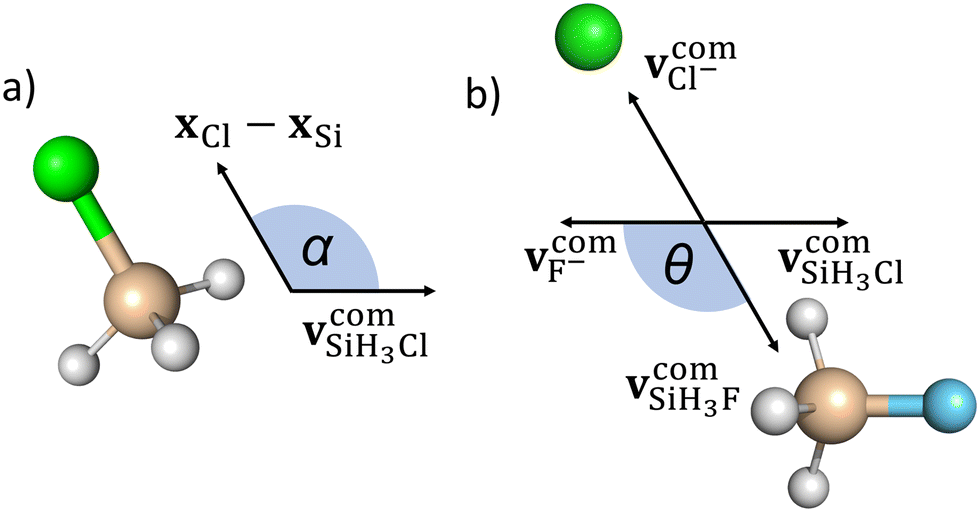 | ||
| Fig. 1 Illustration of the α initial attack angle (a) and the θ scattering angle (b). | ||
The initial attack angle distribution is obtained by partitioning the cosine of α into equally spaced bins ranging from −1 to 1. In this scheme, −1 (α = 180°) corresponds to back-side attack, where the central atom of SiH3Cl faces the F− ion as the reactants approach each other. On the other hand, when cos(α) = 1 (α = 0°), it signifies a front-side attack, indicating that the chlorine ligand of SiH3Cl faces the approaching F− ion. We follow a similar approach to obtain the scattering angle distributions by dividing cos(θ) into evenly spaced bins between −1 and 1. In this context, a value of −1 (θ = 180°) corresponds to backward scattering, while cos(θ) = 1 signifies forward scattering.
The number of bins of the distributions of both attack and scattering angles, as well as energy distributions was chosen based on the reaction probabilities. We utilized 20 bins for chloride substitution and proton abstraction, 10 bins for HCl production, and 5 bins for the less likely hydride substitution, molecular hydrogen production, and bihalide ion production channels.
3. Results and discussion
3.1 Potential energy surface
Fig. 2 illustrates the reaction channels of the SiH3Cl + F− system, alongside the classical (adiabatic) energies of the stationary points and the products obtained on the PES, as reported in ref. 23. The PES describes six SiH3Cl + F− → A + B and one SiH3Cl + F− → A + B + C type reactions (SiHF + H2 + Cl−) with reasonable accuracy. Chloride substitution occurs through two primary mechanisms: the barrierless Walden inversion, featuring a minimum (WMIN), and the retention channel with a deep double-well of a transition state (FSTS), a reactant-like minimum (FSPreMIN) and a product-like minimum (FSPostMIN). Stationary points characteristic of chloride substitution can also decay by losing a hydride ion, leading to SiH2FCl + H−. Proton abstraction by the fluoride ion is a barrierless process, which can be followed by the formation of hydrogen-bonded complexes, anti- or syn-FBHMIN, preceding the bihalide formation (SiH2 + FHCl−). The anionic products of the substitution reactions, the chloride and hydride ions, can perform proton abstractions, leading to SiH2F− + HCl and SiHFCl− + H2, respectively. Molecular hydrogen can be formed through a more complex mechanism as well, featuring the formation of a dihydrogen-bonded complex (ClDHBMIN) originating from SiH2F− and HCl, followed by the decomposition of this complex into three fragments, SiHF + H2 + Cl−, and ultimately, the recombination of SiHF with the chloride ion. There are two additional minima (anti- and syn-ClBHMIN) and two transition states (FDITS and ClDITS), the geometries of which can be optimized on the energy surface. However, these four stationary points do not play significant roles in the formation of any of the main products.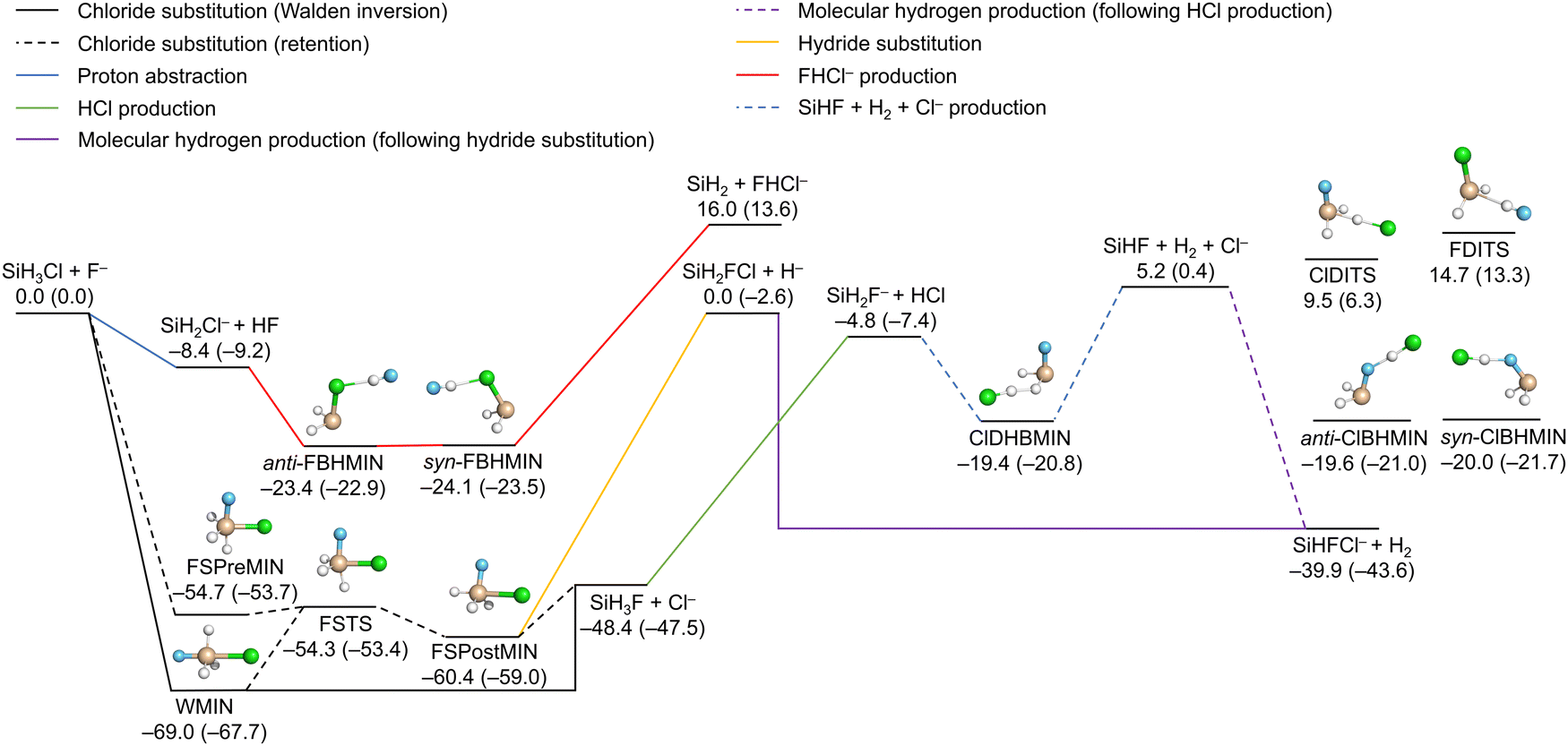 | ||
| Fig. 2 Schematic potential energy surface of the SiH3Cl + F− system featuring the main reaction channels as well as the classical (adiabatic) energies of the minima, transition states and the products relative to the reactants in kcal mol−1.23 | ||
3.2 Reaction probabilities
Fig. 3 displays the opacity functions, i.e., the reaction probabilities as a function of impact parameter, for the SiH3Cl + F− → A + B reactions obtained at collision energies between 1 and 40 kcal mol−1.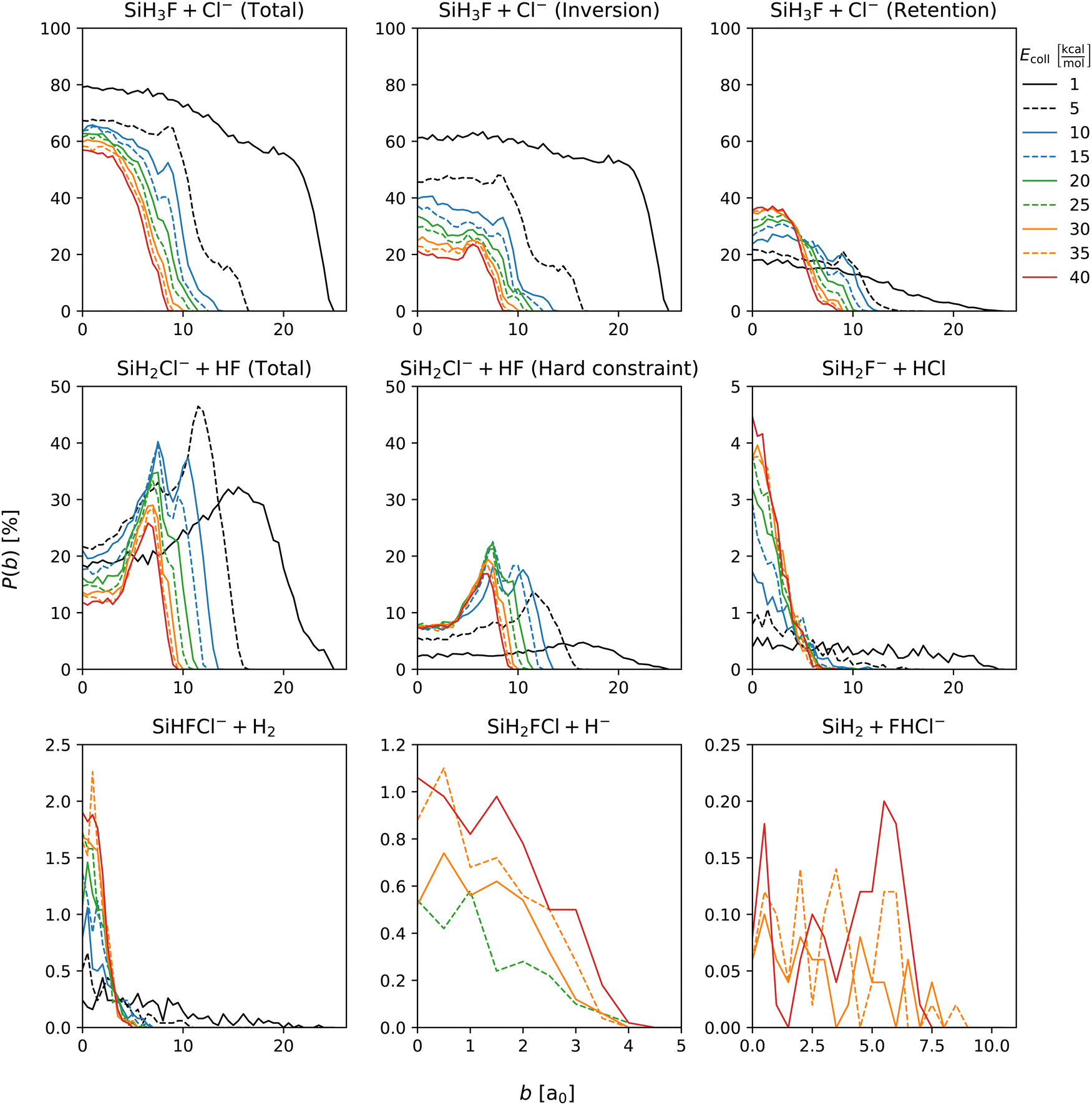 | ||
| Fig. 3 Reaction probabilities of the SiH3Cl + F− → A + B reactions as a function of impact parameter and collision energy. | ||
Dividing the chloride substitution into inversion and retention channels, the b = 0 reaction probability decreases from 60% to 20% in case of inversion and increases from around 20% to approximately 35% for retention as the collision energy increases. The combined effect leads to a drop in the b = 0 reaction probability from 80% to 55%. The maximum reactive impact parameters are also decreasing from b = 25 bohr to 8 bohr both for retention and inversion. In the case of proton abstraction, the maximum reactive impact parameters are consistent with those of the chloride substitution across all collision energies. The peaks of these probabilities occur at higher impact parameters, 33% and 26% at b = 16 and 6.5 bohr at Ecoll = 1 and 40 kcal mol−1, respectively. The most pronounced peak is around 45%, observed at Ecoll = 5 kcal mol−1 and b = 12 bohr. When considering the hard vibrational energy constraint, which means that both SiH2Cl− and HF products must possess a vibrational energy at least equal to their zero-point vibrational energies, reaction probabilities significantly reduce, but the maximum reactive impact parameters remain unaffected. The b = 0 reaction probability of proton abstraction with the hard vibrational constraint remains constant 8% at the collision energy range between 10 and 40 kcal mol−1. HCl production displays an almost constant reaction probability across the impact parameter range at Ecoll = 1 kcal mol−1, slightly below 0.5%. Increasing the collision energy, the maximum reactive impact parameter decreases substantially, while reaction probabilities at smaller impact parameters increase rapidly as the b = 0 probability reaches 4.5%. The reaction probability profile for the H2 production channel is similar to that of the HCl production. At the smallest collision energy this product is observable at high impact parameters, beyond b = 20 bohr. As the collision energy increases, the maximum reactive impact parameter drops, and the peak of the reaction probability at low impact parameters rises from 0.2% at the smallest to 2% at the highest collision energy. In the case of hydride substitution, the maximum reactive impact parameter is roughly b = 4 bohr between Ecoll = 25 and 40 kcal mol−1, and the reaction probability at b = 0 grows with increasing collision energy reaching 1%. For the bihalide production channel, between Ecoll = 30 and 40 kcal mol−1, the maximum reactive impact parameter is between 9.5 and 7.5 bohr. The reaction probability of the bihalide ion production does not exceed 0.2%. Due to the small number of the bihalide production observations its opacity functions are less defined.
As seen above, vibrational energy constraint is only applied for the proton-abstraction channel due to different reasons. In the case of the SN2 reaction, the zero-point energy violation is negligible partly owing to its high exothermicity. Hydride substitution is also not affected by the vibrational energy constraint. For the other minor channels, the constraint may significantly increase the statistical error because of their low probability.
3.3 Integral cross sections
Integral cross sections of the SiH3Cl + F− → A + B reactions are shown by Fig. 4. In the case of chloride substitution, the cross sections decrease with increasing collision energy, for both retention and inversion channels. The combined ICS exceeds 1000 bohr2 and drops below 70 bohr2 at the lowest and highest collision energy, respectively. At Ecoll = 1 kcal mol−1 the ICS of the inversion channel is almost 900 bohr2, meanwhile the contribution of the retention is only 120 bohr2. However, at the highest collision energies their cross sections become comparable. The cross section of proton abstraction also shows a monotonic decrease, from 350 bohr2 to around 50 bohr2 in the applied collision energy range, if zero-point energy violating trajectories are also included. By excluding the energy constraint violating trajectories, the cross section is drastically reduced to 50 and 25 bohr2 at the lowest and highest collision energy, respectively. A maximum appears at around 10 kcal mol−1 collision energy, 65 bohr2. The integral cross sections of HCl production and H2 production have similar shapes, maxima at the smallest collision energy, minima at around 10 kcal mol−1 and slight increase at higher collision energies. The cross section of the HCl and H2 productions at the lowest collision energy are 5 and 1.2 bohr2, respectively. In case of hydride substitution and the bihalide production channels, their cross sections monotonically increase as the collision energy being increased. However, the cross section of bihalide production is almost negligible at collision energies below 25 kcal mol−1. In case of hydride substitution, a monotonic increase can be observed starting from the smallest collision energy. The cross section of the hydride substitution and bihalide production reaches 0.25 bohr2 and 0.15 bohr2 at the highest collision energy, respectively.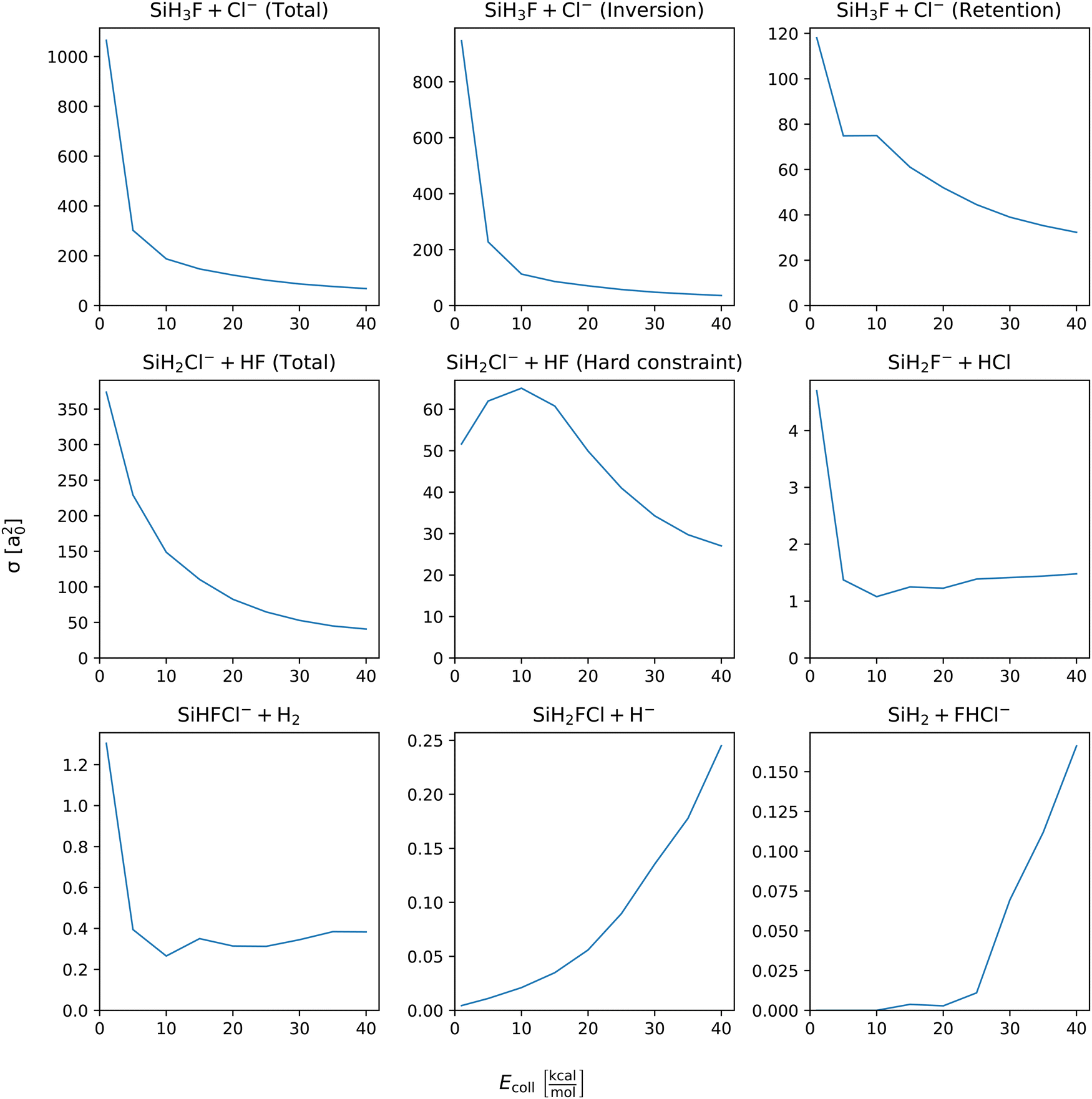 | ||
| Fig. 4 Integral cross sections of the SiH3Cl + F− → A + B reactions as a function of collision energy. | ||
Branching ratios as a function of Ecoll for the different channels and pathways of the title reaction are given in Fig. S1 of the ESI.† It is clearly seen that the SN2 and proton-abstraction channels dominate with ∼60% and 30–40%, respectively. The fraction of the SN2 inversion reactivity decreases with Ecoll, whereas the retention becomes more and more dominant as Ecoll increases. The branching ratios of the other channels, such as SiH2F− + HCl, SiHFCl− + H2, SiH2FCl + H−, and SiH2 + FHCl−, are usually small (<1%) in the Ecoll range of 1–40 kcal mol−1 and steeply increase with collision energy.
3.4 Initial attack angle and scattering angle distributions
Fig. 5 shows the initial attack angle distributions. Chloride substitution exhibits a slight preference for back-side attack at low collision energies. However, as the collision energy increases, there is a shift towards a preference for cos(α) at around −0.5 (α ≈ 120°). The distribution of α for HCl production has similar shape as that of the chloride substitution channel across all collision energies. In the inversion channel of chloride substitution, there is a pronounced preference for back-side attack at low collision energies, and as the collision energy increases, the distribution becomes isotropic. On the other hand, the retention channel favors the front-side attack at low collision energy, while at high energy, there is a peak in the distribution around α = 120°. This significant change in the distribution of the retention channel aligns with our previous observations regarding the collision energy dependence of the mechanism. At the lowest collision energy, the formation of WMIN precedes chloride substitution. However, depending on the impact parameter, energy transfer to the F–Si–Cl bending can occur, leading to the formation of a front-side complex that decomposes into retention products. By examining the cos(α) distributions, we can conclude that the initial attack angle, in addition to the impact parameter, plays a crucial role in determining the formation of front-side complexes. For proton abstraction, the preference changes from front-side attack at the lowest collision energy to back-side attack beyond Ecoll = 5 kcal mol−1. The shape of the distributions is not affected by considering the vibrational constraint. The front-side attack preference of proton abstraction at the lowest collision energy can be explained by the extremely competitive nature of WMIN formation at back-side attack which mostly results in chloride substitution. The H2 production channel exhibits nearly isotropic cos(α) distribution. The hydride substitution channel shows a preference for back-side attack. The bihalide ion production, which only observed at higher collision energies, also prefers back-side attack. Since this reaction follows the proton abstraction it can be expected that their initial attack angle preference coincides.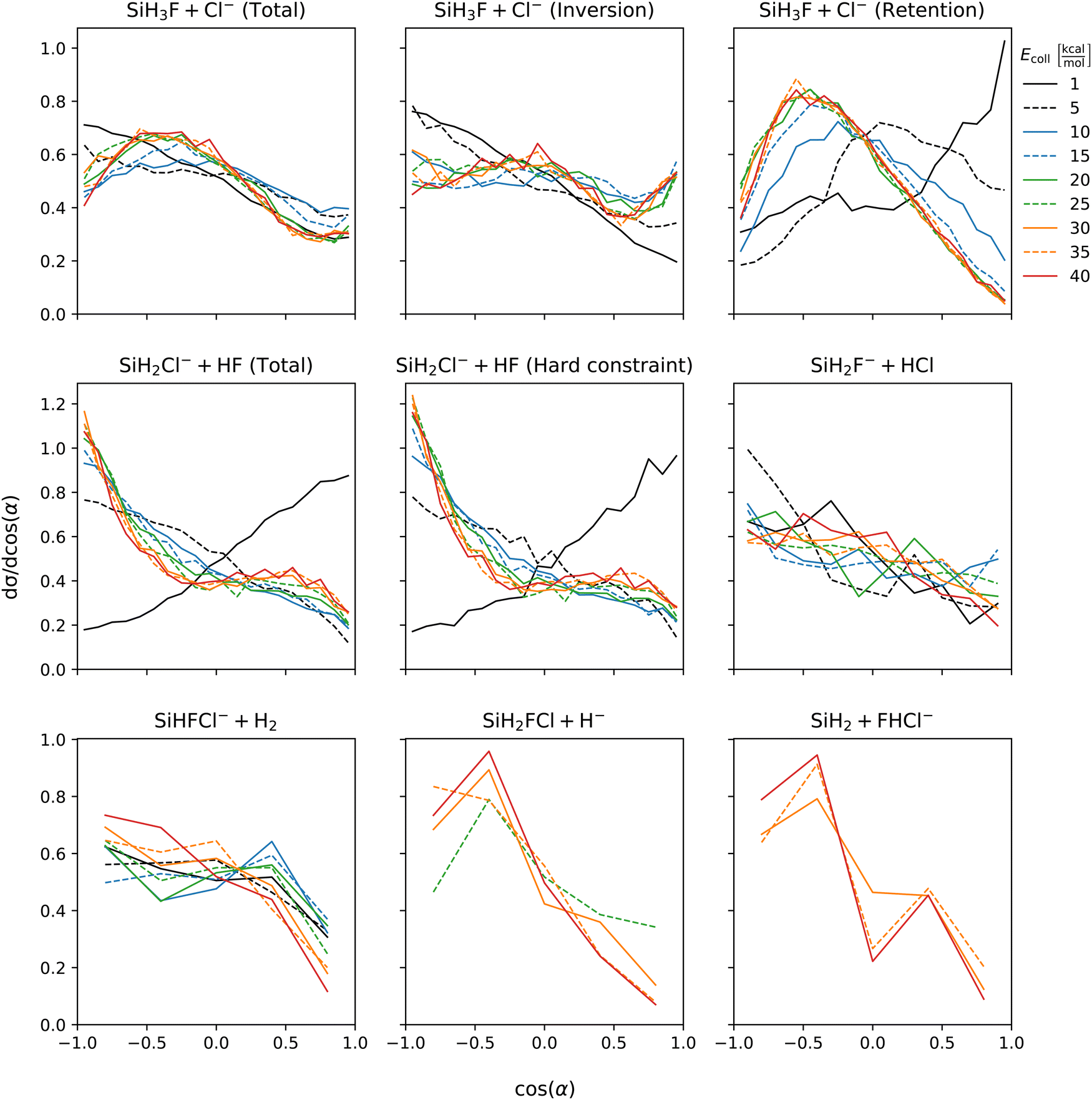 | ||
| Fig. 5 Initial attack angle distributions of the SiH3Cl + F− → A + B reactions obtained at different collision energies. | ||
Scattering angle distributions are shown at Fig. 6. At low collision energy chloride substitution exhibits backward–forward symmetry. However, as the collision energy increases, forward scattering becomes dominant. Separating the inversion and retention channels, we find that the preference for forward scattering at high collision energies is primarily attributed to the inversion mechanism. In contrast, the retention channel shows a slight preference for forward scattering at low collision energies, which gradually becomes nearly isotropic at the highest collision energies. Proton abstraction displays symmetric backward–forward scattering at low collision energy. At high collision energies, a preference for forward scattering emerges and the distribution is unaffected by considering vibrational energy constraint. Isotropic scattering angle distributions are observed for both the HCl and H2 production channels, which is in accordance with the post-reaction-interaction-induced multistep mechanisms, i.e., SiH3Cl + F− → SiH3F + Cl− → SiH2F− + HCl and SiH3Cl + F− → SiH2FCl + H− → SiHFCl− + H2, respectively, involved in the formation of these products (see Fig. 2). Of course, here the products of the first step are not completely separated and the ion–dipole and/or hydrogen-bond interactions between them induce the second reaction. Hydride substitution also exhibits an isotropic scattering pattern, possibly due to the ease with which the hydride ion can be deflected due to its extremely small mass. The bihalide production channel, despite its multistep mechanism (see Fig. 2), displays a preferred direction for scattering, which is the backward direction. This contrasts with the forward scattering dominance observed in the proton abstraction reaction at high collision energies. This discrepancy is unexpected since the FHCl− production follows proton abstraction, and at high collision energies, most of the HF species scatter forward.
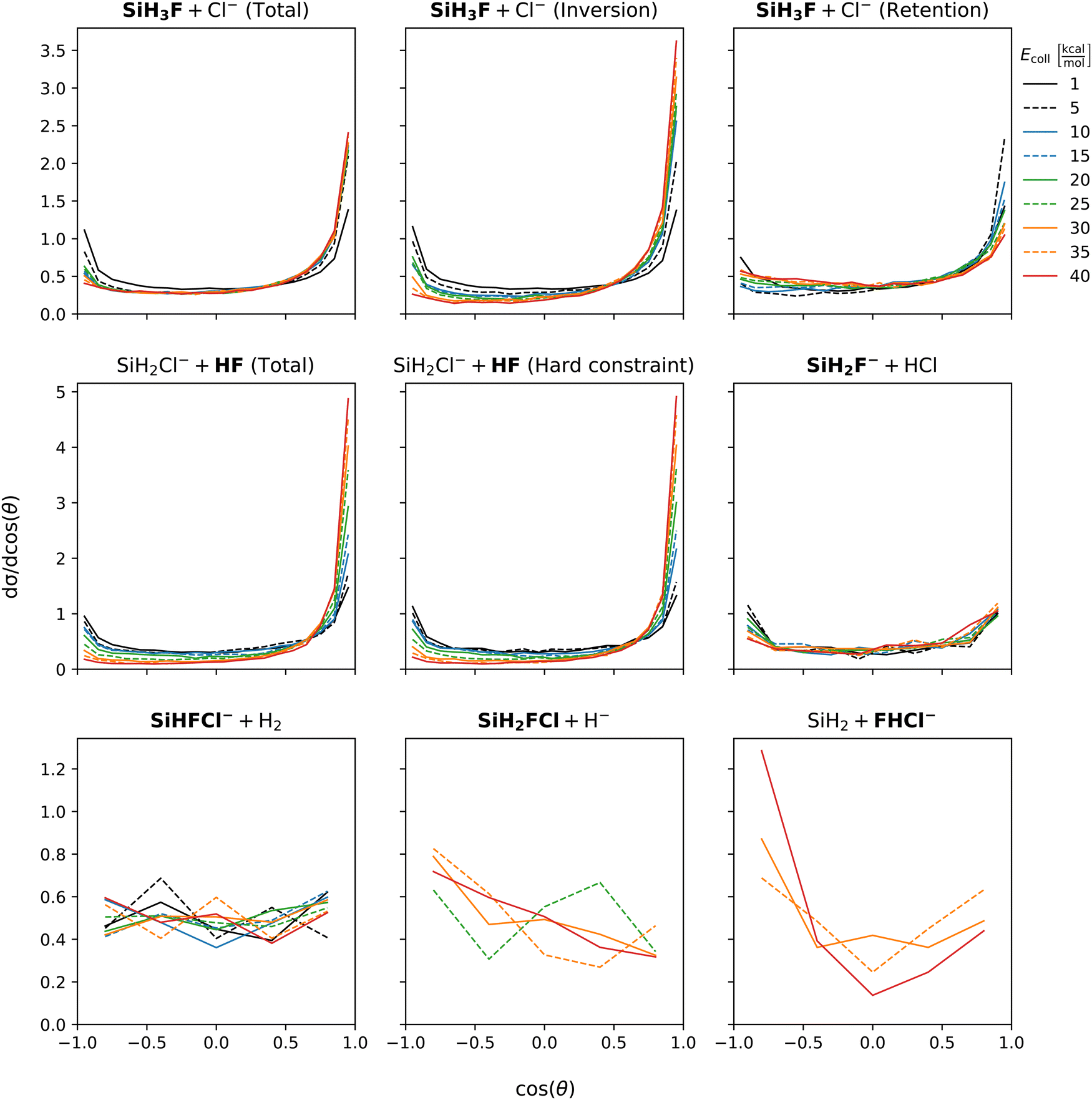 | ||
| Fig. 6 Scattering angle distributions of the SiH3Cl + F− → A + B reactions obtained at different collision energies. Scattering angles are defined as the angle between the center-of-mass velocities of the F− reactant and the fluorine atom containing product (in bold). | ||
3.5 Post-reaction energy distributions
The distribution of both internal and translational energies offers valuable insights into the directness of product formation. For an indirect mechanism, complex formation takes place, leading to the excitation of internal modes of motion: rotation and vibration. In contrast, during a direct reaction, no complexes form and most of the energy from the reactant collision is directly transferred into the translational energy of the resultant products. Throughout our simulations the total energy remains constant. It can be expressed as the sum of the zero-point vibrational energy of SiH3Cl, the collision energy, and the negative of the reaction energy. The reaction energies are the relative classical energies of the product formations, illustrated in Fig. 2.Fig. 7–10 show the translational (Etrans), internal (Eint), vibrational (Evib) and rotational (Erot) energy distributions of the SiH3Cl + F− → A + B reactions, respectively. In the ESI,† we provide the distributions of the different energy fractions (E/Etot) as well, where E denotes Etrans, Eint, Evib, and Erot.
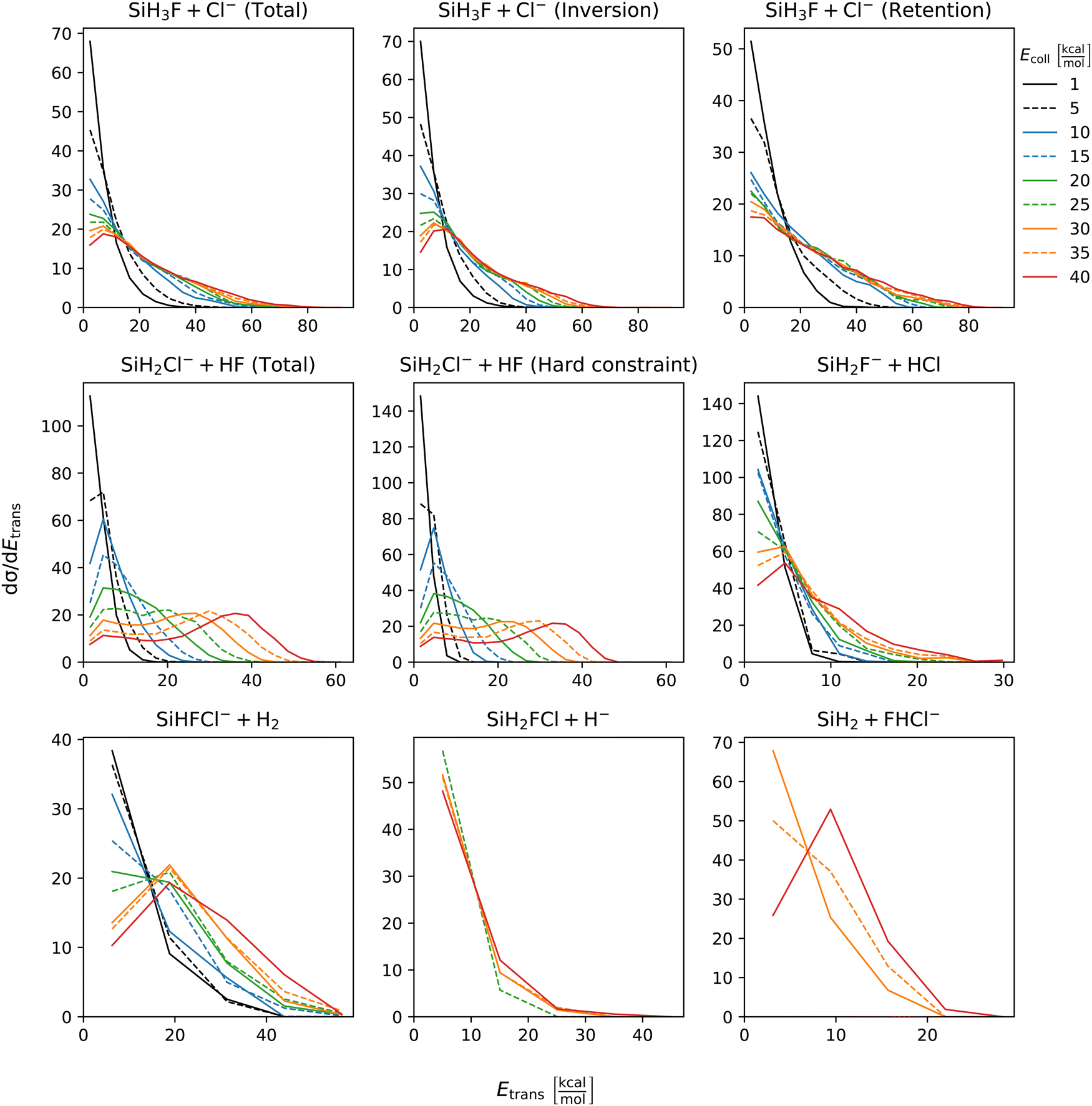 | ||
| Fig. 7 Translational energy distributions of the SiH3Cl + F− → A + B reactions as a function of collision energy. | ||
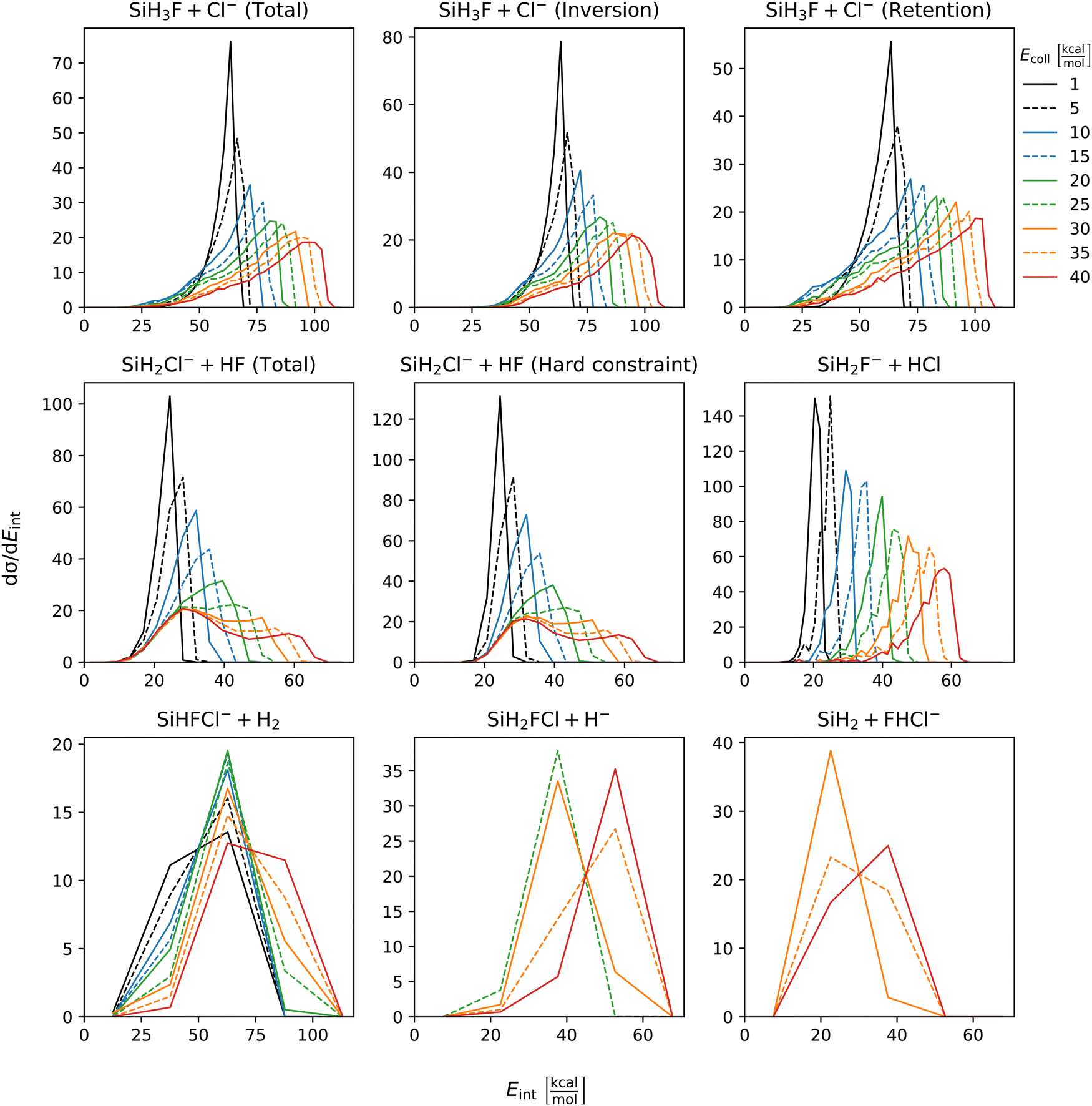 | ||
| Fig. 8 Internal energy distributions of the SiH3Cl + F− → A + B reactions as a function of collision energy. | ||
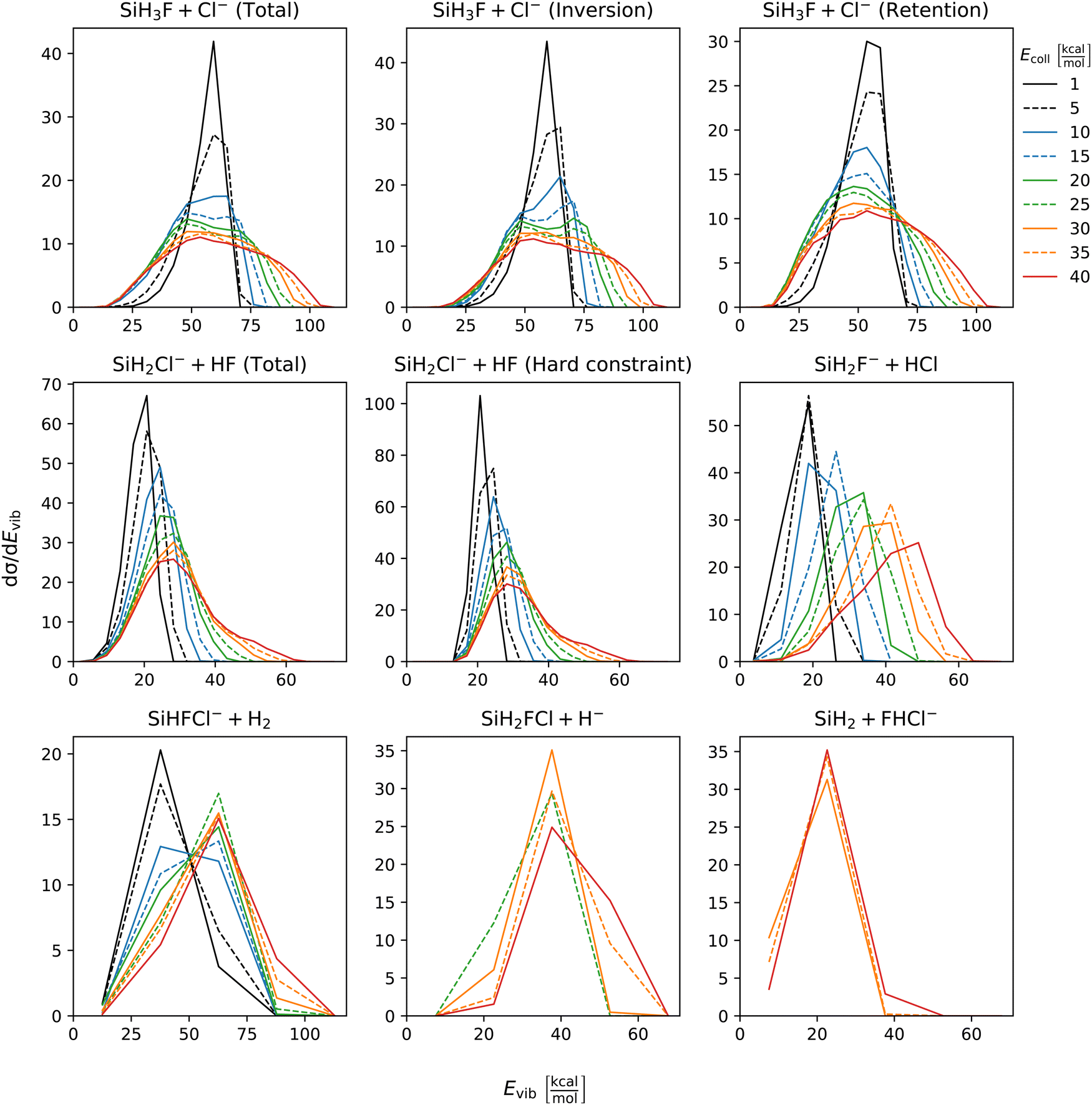 | ||
| Fig. 9 Vibrational energy distributions of the SiH3Cl + F− → A + B reactions as a function of collision energy. | ||
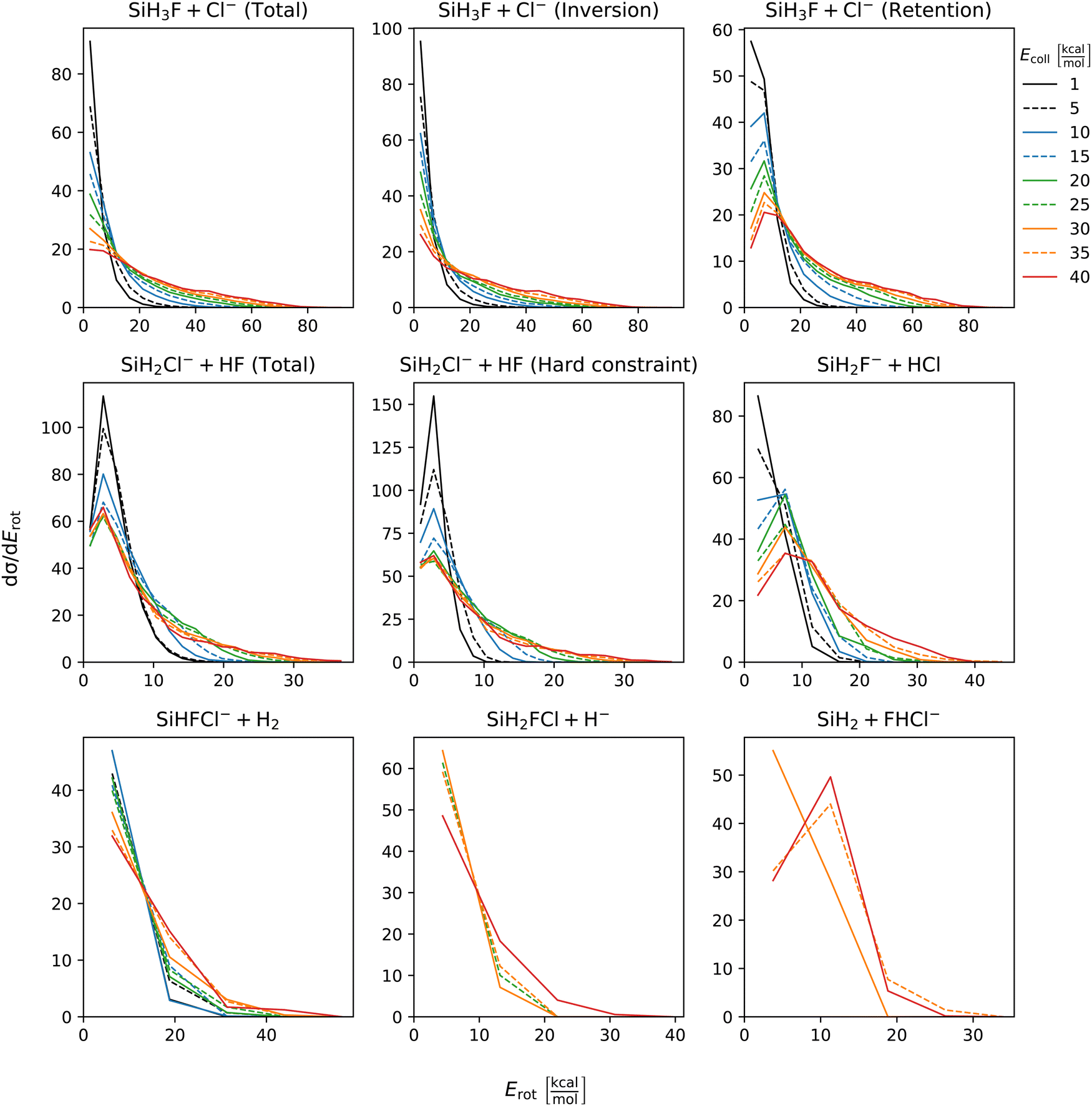 | ||
| Fig. 10 Rotational energy distributions of the SiH3Cl + F− → A + B reactions as a function of collision energy. | ||
For chloride substitution the total available energy is 66.2 and 105.2 kcal mol−1 at Ecoll = 1 and 40 kcal mol−1, respectively. The translational energy distribution of chloride substitution, including both inversion and retention channels, peaks at low energies, specifically below 10 kcal mol−1, across all collision energy ranges. As collision energy increases, this distribution only broadens. Only a minor fraction of the chloride substitution products possesses more than half of the available energy as translation. Conversely, the internal energy distribution, as depicted in Fig. 8, has peaks near the maximum available energy. These observations are consistent with indirect mechanisms characterized by complex formation. According to Fig. 9, the vibrational contribution consistently hovers around 60 kcal mol−1 across all collision energies and the most probable contribution from rotation is less than 20 kcal mol−1 (Fig. 10). At high collision energies the peak of the Evib/Etot distribution is around 50%. The Etrans/Etot and Erot/Etot distributions are comparable, meaning that at high collision energy half of the available energy is approximately evenly shared between rotation and translation (Fig. S2, S6 and S9, ESI†).
Analogous to chloride substitution, the peak of the Etrans distribution in the hydride substitution channel remains consistent across varying collision energies, peaking at the lowest energy region, implying complex formation. The total available energy for hydride substitution at Ecoll = 40 kcal mol−1 is 56.8 kcal mol−1. Fig. S3 (ESI†) shows that almost all hydride substitution products allocate more than half of this energy in internal motion, predominantly as vibration (Fig. S6, ESI†).
In the case of proton abstraction, Etot = 65.2 kcal mol−1 at the highest collision energy. This reaction shows a compound translational energy distribution consistent with a mixture of direct and indirect mechanisms, as there is a peak near the smallest translational energy across all collision energies, and a second peak which shifts according to the collision energy increase. At low collision energies, most of the total available energy goes into vibration as the Evib/Etot distribution shows a maximum at around 70%. However, at the highest collision energy the peak shifts towards 40%.
The formation of HCl is marked by a low translational energy. The peak of this distribution remains static, even with increasing collision energy, indicating indirect mechanism. During HCl production, the proton abstraction by the leaving ion is more favored when the preceding substitution products move slowly. Interestingly, the partial energy distributions remain consistent during the collision energy increase. The Evib/Etot and Erot/Etot distributions peak at 75% and around 15–20%, respectively. A significant portion of the vibrational energy is localized at SiH2F−, as shown in Fig. S7 (ESI†).
The peak of the translational energy distribution of the H2 production shifts with increasing collision energies, approximately with half the increase. As depicted in Fig. S7 (ESI†), at lower collision energies, most of the available energy is channeled into the vibration of SiHFCl−. However, at higher collision energies, this proportion reduces to around half.
In case of bihalide production, the maximum of the translational energy distribution shifts by the collision energy increase. According to the Etrans/Etot, Erot/Etot and Evib/Etot distributions, about half of the available energy goes into vibration and the remaining half is shared equally between rotation and translation. About two thirds of the vibrational energy goes into the FHCl− ion.
In order to obtain additional insight into the directness of the title reaction, we have plotted the integration time distributions of the different channels in Fig. S12 of the ESI.† As Ecoll increases all the distributions shift toward shorter times, however, at the highest Ecoll range of 30–40 kcal mol−1 the collision energy dependence is not significant. The minor channels, especially SiH2F− + HCl, SiHFCl− + H2, and SiH2 + FHCl−, are the most indirect, i.e., their distributions peak at the longest time values at a given Ecoll, but the indirect character (time scale) of the SN2 and proton-abstraction channels is also substantial.
3.6 The effect of replacing the central atom
In a theoretical study of gas-phase ion–molecule reactions, it is worth comparing analogous systems to place our observations in a larger context. Our title system, SiH3Cl + F−, can be compared to the carbon- and the phosphorous-analogues: CH3Cl + F− and PH2Cl + F−, respectively. These comparisons are justified by the fact that carbon and silicon are in the same main group, while phosphorus and silicon are in neighboring groups within the same period. The CH3Cl + F− system has been widely studied,1,3,6–8,25 meanwhile for PH2Cl + F−, our research group recently published a QCT study.26In these three systems, the most important reactions are the SN2 chloride ion substitutions and the proton abstractions. The analogous chloride substitutions are all exothermic reactions.25,26 However, from a stereochemical perspective, we observe significant differences in the substitution reactions, largely attributable to the relative energies of the stationary points.
SN2 by Walden inversion is favorable in all systems, as evidenced by the submerged stationary points. A notable difference between the carbon analogous inversion and the others is that, in case of carbon, the potential shape is a double well, whereas for SiH3Cl + F− and PH2Cl + F−, single well potentials can be found. Front-side attack resulting in retention SN2 products is characteristic of both SiH3Cl + F− and PH2Cl + F−, as the stationary points of the corresponding reactions all possess negative energies. The result is a stereochemical mixture of inversion and retention SN2 products at all collision energies. Meanwhile, for CH3Cl + F−, the front-side attack transition state leading to retention has high positive energy, limiting retention product formation at low collision energy.
However, retention SN2 becomes possible at collision energies lower than the relative energy of the corresponding front-side attack transition state, thanks to a recently-discovered compound mechanism known as double inversion.25 In the double-inversion chloride substitution of CH3Cl + F−, the attacking nucleophile fails to detach the proton from the methyl chloride molecule. Instead, it creates a transition state with an inverted configuration by moving the proton to the opposite side of the carbon atom. This transition state then rearranges into an inverted Walden transition state, which decomposes via a second inversion into the retention SN2 products. The discovery of the double-inversion mechanism was significant in explaining the unexpectedly high probability of SN2@C retention at low collision energy.25 For systems with phosphorus and silicon central atoms, investigating both the possibility and significance of double inversion is worthwhile.
In the SiH3Cl + F− system, chloride substitution with retention makes a significant contribution even at the lowest collision energies, as predicted by the schematic PES and evidenced by QCT simulations. Therefore, the retention SN2@Si can be explained by the simple, straightforward front-side attack mechanism. On the other hand, it is of great theoretical interest to explore if the analogue of double inversion is even possible in silicon-centered systems, as it elucidates the atomistic dynamics of this mechanism type.
After examining several SN2 trajectory animations in the SiH3Cl + F− system, in which at least the Si–H–F bond is present or proton abstraction is one of the partial steps of the entire mechanism, we have observed that double inversion described by Szabó and Czakó25 is atypical here because of the following reasons. (1) Unlike at the analogous C-entered reaction, in the present case the attacking nucleophile easily detaches the proton due to the exothermicity of the HF formation channel. (2) Substitution via retro-proton-abstraction leads to a stereochemical mixture because the formation of both inversion and retention SN2 complexes are allowed at low energies. It appears that in double inversion, not only the existence of the DI transition state and its energy being lower than that of the front-side attack transition state are relevant. It is favorable for the mechanism if the reaction energy of proton abstraction is also higher than the relative energy of DITS; otherwise, proton abstraction occurs instead of the partial abstraction and stereochemical rearrangement.
It is also noteworthy that vibrationally highly-excited SiH3F can invert due to its flexibility. In carbon-centered systems, post-reaction SN2 product inversion is usually not observed, though in the case of the NH2− + CH3I reaction retention can occur via the formation of a [CH3⋯NH2] complex in which a rotation of the CH3 group may cause a second inversion.27
In the analogous proton abstraction reactions, the process is endothermic in the CH3Cl + F− system, accessible only at high collision energies. Conversely, proton abstraction in both SiH3Cl + F− and PH2Cl + F− is exothermic and a high-probability reaction at all collision energies.
In addition to chloride substitution and proton abstraction in PH2Cl + F−, various low-probability channels analogous to reactions in the silicon-centered system are observed.26 PHF− + HCl is the third most probable reaction, similar to how HCl production is the third most dominant in SiH3Cl + F−. Molecular hydrogen, bihalide ion, as well as hydride ion production reactions are observed with low probability as well, which are also characteristic of the silicon-containing system. None of these product formations are significant in CH3Cl + F−.
4. Summary and conclusions
We have computed the first differential cross sections such as various angular and product energy distributions, for a Si-centered SN2 reaction, F− + SiH3Cl. The QCT simulations have utilized our recently-developed full-dimensional analytical ab initio PES,23 which allows uncovering even the low-probability channels such as SiH2F− + HCl, SiH2FCl + H−, SiH2 + FHCl−, SiHFCl− + H2, SiHF + H2 + Cl−, and SiH2 + HF + Cl−, besides the major SN2 and proton-transfer channels leading to SiH3F + Cl− and SiH2Cl− + HF, respectively. The following conclusions can be summarized based on the differential cross sections obtained for the different two-fragment product channels:(a) The cold translational and hot internal energy distributions indicate that the SN2 reaction, SiH3F + Cl− channel, is mainly indirect, however, the angular distributions show spatial preferences depending on collision energy as well as inversion/retention.
(b) The angular distributions of the proton-abstraction channel, SiH2Cl− + HF, show front-side attack backward–forward scattering preference at low Ecoll, which shifts toward back-side attack and forward scattering as Ecoll increases. The energy distributions indicate a mixed direct–indirect proton-transfer mechanism.
(c) The attack angle distributions of the SiH2F− + HCl channel are similar to those of the SN2 reaction. The SiH2F− + HCl products are formed with isotropic scattering and translationally cold due to the indirect mechanism of this channel.
(d) The SiH2FCl + H− channel prefers back-side attack and scatters isotropically with low translational energy indicating an indirect pathway.
(e) Even if SiH2 + FHCl− has a multistep mechanism, its angular distributions show back-side attack and backward scattering preference.
(f) The angular distributions of the SiHFCl− + H2 channel are mostly isotropic and the product translational energy is cold, in consistent with its indirect mechanism.
Finally, we note that the above-described two-fragment channels produce different ionic products, thus, their selective experimental detection may be straightforward. We hope that the present work motivates such experimental investigations as well as further theoretical studies.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank the National Research, Development and Innovation Office-NKFIH, K-125317 and K-146759, project no. TKP2021-NVA-19, provided by the Ministry of Innovation and Technology of Hungary from the National Research, Development and Innovation Fund, financed under the TKP2021-NVA funding scheme, and the Momentum (Lendület) Program of the Hungarian Academy of Sciences for financial support.References
- P. Manikandan, J. Zhang and W. L. Hase, J. Phys. Chem. A, 2012, 116, 3061 CrossRef CAS PubMed.
- J. Xie, R. Otto, J. Mikosch, J. Zhang, R. Wester and W. L. Hase, Acc. Chem. Res., 2014, 47, 2960 CrossRef CAS PubMed.
- P. Liu, D. Wang and Y. Xu, Phys. Chem. Chem. Phys., 2016, 18, 31895 RSC.
- C. Hennig and S. Schmatz, Phys. Chem. Chem. Phys., 2016, 18, 19668 RSC.
- J. Xie and W. L. Hase, Science, 2016, 352, 32 CrossRef CAS PubMed.
- Y. Li, Y. Wang and D. Wang, J. Phys. Chem. A, 2017, 121, 2773 CrossRef CAS PubMed.
- I. Szabó and G. Czakó, J. Phys. Chem. A, 2017, 121, 9005 CrossRef PubMed.
- R. Wester, Mass Spectrom. Rev., 2022, 41, 627 CrossRef PubMed.
- A. P. Bento, M. Solà and F. M. Bickelhaupt, J. Comput. Chem., 2005, 26, 1497 CrossRef CAS PubMed.
- A. P. Bento and F. M. Bickelhaupt, J. Org. Chem., 2007, 72, 2201 CrossRef CAS PubMed.
- S. C. A. H. Pierrefixe, C. Fonseca Guerra and F. M. Bickelhaupt, Chem. – Eur. J., 2008, 14, 819 CrossRef PubMed.
- M. A. van Bochove and F. M. Bickelhaupt, Eur. J. Org. Chem., 2008, 649 CrossRef CAS.
- Y. Ren, X. Wang, S.-Y. Chu and N.-B. Wong, Theor. Chem. Acc., 2008, 119, 407 Search PubMed.
- Z.-Z. Yang, Y.-L. Ding and D.-X. Zhao, J. Phys. Chem. A, 2009, 113, 5432 CrossRef CAS PubMed.
- Y.-L. Ding, J.-R. Mu and L.-D. Gong, J. Chin. Chem. Soc., 2013, 60, 327 CrossRef CAS.
- T. A. Hamlin, M. Swart and F. M. Bickelhaupt, ChemPhysChem, 2018, 19, 1315 CrossRef CAS PubMed.
- M. Fugel, A. Dittmer, F. Kleemiss and S. Grabowsky, J. Phys. Chem. A, 2021, 125, 4070 CrossRef CAS PubMed.
- T. H. Dunning, Jr., L. T. Xu and J. V. K. Thompson, J. Phys. Chem. A, 2021, 125, 7414 CrossRef PubMed.
- A. Á. Dékány, G. Z. Kovács and G. Czakó, J. Phys. Chem. A, 2021, 125, 9645 CrossRef PubMed.
- T. Taketsugu and M. S. Gordon, J. Phys. Chem., 1995, 99, 8462 CrossRef CAS.
- T. Taketsugu, T. Yanai, K. Hirao and M. S. Gordon, J. Mol. Struct.: THEOCHEM, 1998, 451, 163 CrossRef CAS.
- T. Matsubara and T. Ito, J. Phys. Chem. A, 2016, 120, 2636 CrossRef CAS PubMed.
- A. Á. Dékány and G. Czakó, J. Chem. Phys., 2023, 158, 224303 CrossRef PubMed.
- B. J. Braams and J. M. Bowman, Int. Rev. Phys. Chem., 2009, 28, 577 Search PubMed.
- I. Szabó and G. Czakó, Nat. Commun., 2015, 6, 5972 Search PubMed.
- A. Giricz, G. Czakó and D. Papp, Chem. – Eur. J., 2023, 29, e202302113 CrossRef CAS PubMed.
- D. A. Tasi and G. Czakó, J. Chem. Phys., 2022, 156, 184306 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Branching ratios as well as Etrans/Etot, Eint/Etot, Evib/Etot, and Erot/Etot fraction and integration-time distributions for the different channels of the title reaction. See DOI: https://doi.org/10.1039/d4cp00048j |
| This journal is © the Owner Societies 2024 |