
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStereoelectronic interactions are too weak to explain the molecular conformation in solid state of cis-2-tert-butyl-5-(tert-butylsulfonyl)-1,3-dioxane†‡
Fátima M.
Soto-Suárez
 a,
Tania
Rojo-Portillo
a,
Eduardo H.
Huerta
a,
Alejandro
Aguilera-Cruz
a,
Alberto
Tapia-Bárcenas
a,
David Atahualpa
Contreras-Cruz
a,
Rubén A.
Toscano
a,
Beatriz
Quiróz-García
a,
Aaron
Rojas-Aguilar
b,
Fernando
Cortés-Guzmán
a,
John
Bacsa
c,
Karla
Ramírez-Gualito
d,
José Enrique
Barquera-Lozada
a and
Gabriel
Cuevas
*a
a,
Tania
Rojo-Portillo
a,
Eduardo H.
Huerta
a,
Alejandro
Aguilera-Cruz
a,
Alberto
Tapia-Bárcenas
a,
David Atahualpa
Contreras-Cruz
a,
Rubén A.
Toscano
a,
Beatriz
Quiróz-García
a,
Aaron
Rojas-Aguilar
b,
Fernando
Cortés-Guzmán
a,
John
Bacsa
c,
Karla
Ramírez-Gualito
d,
José Enrique
Barquera-Lozada
a and
Gabriel
Cuevas
*a
aUniversidad Nacional Autónoma de México, Instituto de Química, Circuito Exterior, Ciudad Universitaria, Delegación Coyoacán, C.P. 04510 Ciudad de México, Mexico. E-mail: gecgb@unam.mx
bDepartamento de Química, Centro de Investigación y de Estudios Avanzados del IPN, Av. Instituto Politécnico Nacional 2508, C.P. 072360 Ciudad de México, Mexico
cSchool of Chemistry and Biochemistry, Georgia Institute of Technology, Atlanta, Georgia 30332-0400, USA
dCentro de Nanociencias Micro y Nanotecnologías, Instituto Politécnico Nacional, Luis Enrique Erro, s/n, Nueva Industrial Vallejo, Gustavo A Madero, 07738 Ciudad de México, Mexico
First published on 14th March 2024
Abstract
cis-2-tert-Butyl-5-(tert-butylsulfonyl)-1,3-dioxane (cis-1) exhibits a high degree of eclipsing in the H–C5–S–C segment in the solid state, the origin of which remains unexplained. The eclipsed conformation that corresponds to an energetic minimum in the solid state practically corresponds to a rotational transition state in solution, which allows an approach to understand transitions states. The difference in the enthalpy of sublimation ΔsubH between cis-1 and the more stable trans-1 is 8.40 kcal mol−1, lets to consider that the intermolecular interactions in the crystalline structure must be responsible for the conformational effect observed in the solid state. The study of the experimental electron density of cis-1 in solid state allowed to establish that CH⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S intermolecular interaction is the main contribution to the observed eclipsing. The charge density analysis was also performed using the quantum theory of atoms in molecules to evaluate the nature and relevance of the intermolecular interactions in the crystal structure.
S intermolecular interaction is the main contribution to the observed eclipsing. The charge density analysis was also performed using the quantum theory of atoms in molecules to evaluate the nature and relevance of the intermolecular interactions in the crystal structure.
Introduction
In 1987, the X-ray structure of cis-2-tert-butyl-5-(tert-butylsulfonyl)-1,3-dioxane (cis-1, Scheme 1) was published1 as a result of the study that allowed determining the magnitude of the conformational free energy of the S(O)2-tert-butyl group when located at the position 5 of the 1,3-dioxane. In this case the conformation was anchored with the tert-butyl group located at position 2, and acid catalysis was used to complete the equilibrium2,3 (Scheme 1).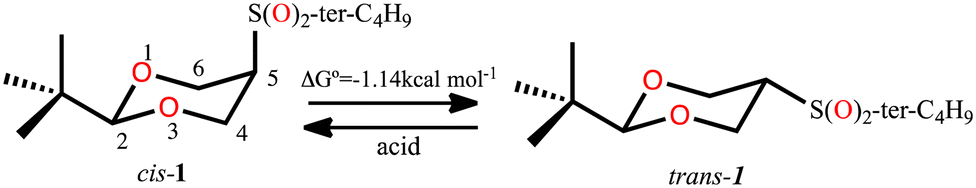 | ||
| Scheme 1 Conformational equilibrium of 2-tert-butyl-5-(tert-butylsulfonyl)-1,3-dioxane. | ||
The rigorous experimental procedure introduced by Eliel,4 required establishing the conformational composition starting from both the axial and equatorial conformer, both pure, and bringing them to equilibrium. When obtaining crystalline compounds, they were subjected to the available X-ray diffraction studies. Juaristi states that describing the almost completely eclipsed conformation of the tert-butyl-sulfonyl group was a stroke of luck5–7 and with good reason, since this conformer of minimum energy in solid phase practically corresponds to a transition state in potential energy surface in the gas phase for rotation of the HC-SO2-tert-butyl group. In this conformer, the average dihedral angle (τ) O–S–C(5)–C(4,6) (Fig. 1) is 8.25 ± 2.35°, with C(4)–C(5) S–O being 10.6° (8) and C(6)–C(5)–S–O being 5.9° (7). (A, Fig. 1). This eclipsed arrangement contrasts with alternating rotamers B and C. The absence of conformer B was explained in terms of a repulsive interaction between the neighbouring tert-butyl groups, while conformer C was ruled out by strong repulsion due to charge accumulation between oxygen atoms.
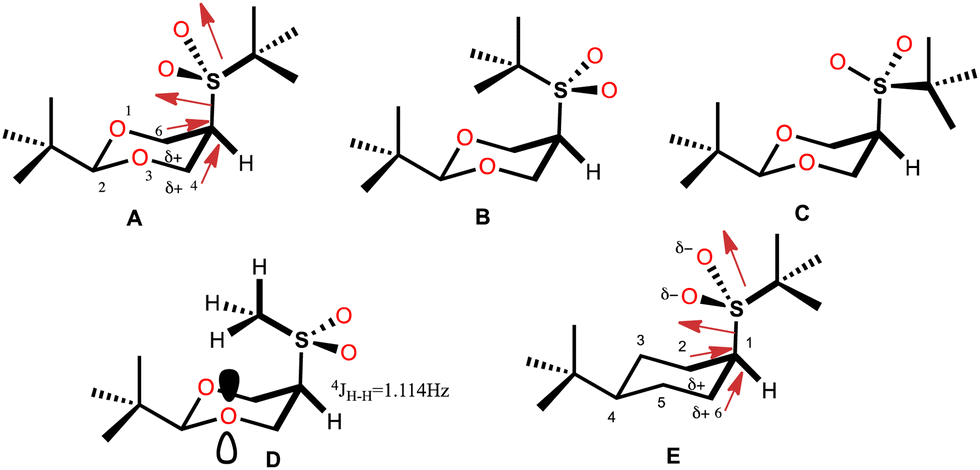 | ||
| Fig. 1 Rotamers of cis-1 (A)–(C) and observed rotamer of cis-2 (D) and cis-3 (E). | ||
This arrangement of the tert-butyl-sulfonyl group contrasts with that adopted by the 5-methylsulfonyl group (D, Fig. 1), in which the methyl group is oriented alternately and pointing towards the center of the 1,3-dioxane ring.8 This arrangement was established because it is possible to determine a large long-range coupling constant of 4JH–H = 1.14 ± 0.02 Hz produced by the W coupling between the proton at position 5 and the hydrogen atom of the anti-periplanar methyl group,9,10 rotamer in which the repulsion between oxygen atoms is avoided and a possible attractive nO/H–C interaction is incorporated.11
Molecular mechanics calculations supported the proposal for molecular stabilization through two dipole-induced dipole interactions between sulfone oxygen atoms and the carbons 4,6 polarized by the electronegativity of the endocyclic oxygen atoms (A, Fig. 1). Stabilization through the syn-periplanar 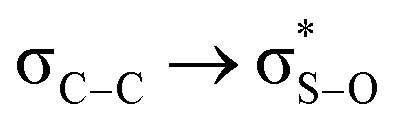 interaction was also proposed, which was not very efficient with respect to the antiperiplanar, recognizing a contribution of no more than 0.6–0.7 kcal mol−1 and an additional quantum mechanical effect.12
interaction was also proposed, which was not very efficient with respect to the antiperiplanar, recognizing a contribution of no more than 0.6–0.7 kcal mol−1 and an additional quantum mechanical effect.12
The finding that the tert-butylsulfonyl group also adopts the near-eclipsing conformation when substituting in the cyclohexane is very interesting. The O–S–C1–C2 angle in 4-tert-butyl-1-(tert-butylsulfonyl)-cyclohexane is 7.8° (3, E Fig. 1),2 a fact that makes it possible to establish the low relevance of the endocyclic oxygen atoms in conformational preference. However, the same electrostatic interaction model as with dioxane was proposed, even though the electronegativity of carbon does not allow it to host a significant change of the distribution of charge of the neighboring atoms.
Calculations at the B3LYP/6-311++G(d,p) level allowed estimating the evolution of energy according to the rotation of the C5–S bond in 1, followed by NBO analysis. The conclusion is that the energy associated to the 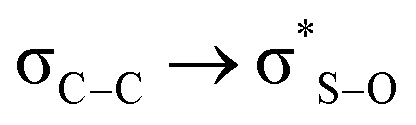 syn-periplanar interaction is negligible with respect to the anti; briefly commenting and without providing evidence that crystal packing may probably be responsible for the observed conformation.12
syn-periplanar interaction is negligible with respect to the anti; briefly commenting and without providing evidence that crystal packing may probably be responsible for the observed conformation.12
Results and discussion
At the M06-2X/6-311++G(2d,2p) level that allows the correct description of long-range interactions, we find that, in gas phase, according to experimental observations, the trans conformer is more stable than cis by ΔG° = 2.87 kcal mol−1. The trans configuration minimum, the most stable of the rotamer set, occurs when the H–C–S–C (τ) angle is 24.2°, exhibiting a small degree of eclipsing. By allowing the rotation of the τ angle in the 0 to 180° segment, trans-1 generates two additional minima, one at τ = 59.1 (ΔG° = 0.92 kcal mol−1) and another one at τ = 163.5 (ΔG° = 3.80 kcal mol−1). Furthermore, it presents 4 stationary states of maximum energy (transition states) at 0, 50, 117.8, and 179.9°. Following the Boltzmann partition scheme, the conformational population is made up of 82.45, 17.42 and 0.13% of these conformers respectively (Fig. S1, ESI‡).The conformational analysis of cis-1 isomer shows three minima, at τ = 20.1 (ΔG° = 2.35 kcal mol−1), τ = 73.9 (ΔG° = 4.08 kcal mol−1), and τ = 182.3° (ΔG° = 0.0 kcal mol−1), two of them are far from being considered alternating and show certain degree of eclipsing. The conformer in which the cyclic C–O bonds are eclipsed with the S–O bonds, τ = 0.0 (ΔG° = 3.40 kcal mol−1), corresponds to a conformational transition state. The conformational population would be made up of 1.87, 0.0, 98.13% of each minima, respectively. Both profiles show a flattened region at the potential energy surface (PES) (Fig. S2, ESI‡).
The dipole moment for the cis isomer is maximum for τ = 0° (μ = 5.7 Debye) and minimum for τ = 180°, μ = 3.9 Debye; but for the trans isomer the trend is opposite, thus for τ = 0°, μ = 3.2 Debye and for τ = 180°, μ = 5.0 Debye (Fig. S3, ESI‡). It is essential to highlight that in no case do the calculations predict the rotamer that is observed in the solid state of each isomer.
To understand this, the published synthesis3 was replicated and the obtained crystals were subjected to X-ray diffraction. In our hands, the angle for both cis and trans isomer was found to be 6.1 and 41.8°, respectively. These angles do not correspond to calculated stationary states and are far from being considered alternating.
So far, there is no reported evidence of the conformation of both isomers in solution. Therefore, NOESY-1D experiments were performed as qualitative proof. Fig. 2 shows the results of the NOESY-1D experiments performed in cis-1 dissolved in chloroform. As can be seen for cis-1 isomer (Fig. 2), when inverting the signal of the tert-butyl group attached to the sulfur atom a notable increase in the signal of the tert-butyl group in equatorial position 2 (1.97%) is observed. In agreement with the presented calculations, since the conformer with the tert-butyl group pointing towards the center of the ring is the most abundant (rotamer iii in Fig. S2, ESI‡). For comparison, the compound that is an intermediate in the synthesis of sulfone 1, the cis-2-tert-butyl-5-(tert-butylsulfinyl)-1,3-dioxane (4) was subjected to a similar study. In Fig. 2, the result of inverting the S-tert-butyl group is observed, showing a very small increase in the signal of the C2-tert-butyl group (0.27%). From our calculations (Fig. S4, ESI‡), we know that the conformational population of cis-4 is made up of 4 rotamers where the one with a tert-butyl group pointing towards the center of the ring has a population of 2.46%, which generates a small signal. On the other hand, the most abundant rotamer corresponds to the one containing the S-tert-butyl group outside the ring and distant from the tert-butyl group at position 2, which agrees with the substantial increase of the signal corresponding to the equatorial hydrogen at position 5. Thus, compound 1 in solution is alternating, keeping the two tert-butyl groups close, showing that steric repulsion is a minor contribution in agreement with calculations.
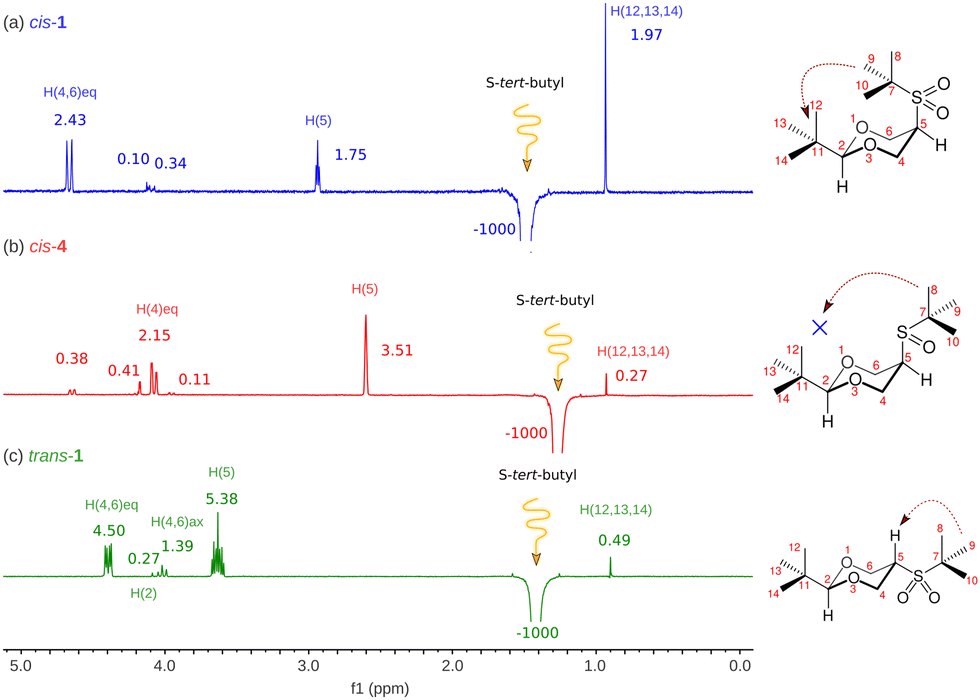 | ||
| Fig. 2 Conventional selective NOESY-1D experiments, (a) for cis-1, (b) for cis-4 and (c) trans-1, resulting from saturating (irradiating) protons of S-tert-butyl group at δ (ppm) 1.495, 1.268 and 1.420 for cis-1, cis-4, and trans-1 respectively. | ||
To make a rigorous comparison, we studied compound trans-1, in which the proximity of the referred tert-butyl groups is impossible. In this case, the growth of the signal of interest is minimal, which confirms previous observations.
In search of whether some quantum effect could be responsible for the observed conformational arrangement, we performed NBO analysis of the systems studied here at the M06-2X/6-311++G(2d,2p) level. In this partition scheme, we determine the energy of the 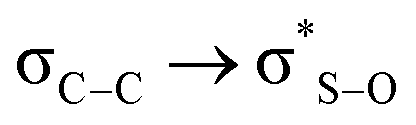 interaction for both the axial and equatorial isomers, in eclipsed conformation (which corresponds to transition states since this conformation is only a minimum in the solid state PES), and in the antiperiplanar arrangement (real minima). The interaction
interaction for both the axial and equatorial isomers, in eclipsed conformation (which corresponds to transition states since this conformation is only a minimum in the solid state PES), and in the antiperiplanar arrangement (real minima). The interaction 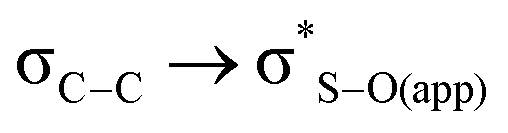 is about 0.6 kcal mol−1 while
is about 0.6 kcal mol−1 while 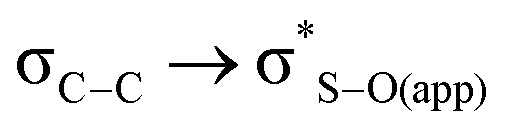 is weaker, finding similar energy in all cases, and in agreement with previous data,12 so it cannot be the origin of the observed difference.
is weaker, finding similar energy in all cases, and in agreement with previous data,12 so it cannot be the origin of the observed difference.
Rather than looking for an additional quantum mechanical effect2 one must look at the properties of the crystal, for example the intermolecular interactions as those responsible for stabilizing the eclipsed configuration corresponding to a transition state in the gas phase. To do this, we determined the enthalpy of sublimation by thermogravimetry of the two isomers of 1 (see ESI‡). Enthalpy of sublimation is of major importance given that is directly related to the intermolecular cohesion energy in the crystalline arrangement, providing insight about the energetics and structure of the solid. The ΔsubH (cis-1,298.15K) has a value of 34.4 ± 1.2 kcal mol−1, while ΔsubH (1-trans,298.15K) has a value of 26.0 ± 0.1 kcal mol−1. As can be seen, the enthalpy of sublimation of cis-1 is 8.40 kcal mol−1 higher than that of trans. This suggests that crystalline lattice of the cis-1 includes more intense intermolecular interactions than in the crystalline arrangement of trans-1.
Characterizing crystal structure would allow us to understand the intermolecular interaction responsible for producing the eclipsing and rationalize the phenomenon observed. In addition, the findings could be used in crystal engineering to construct crystal lattices. Therefore, the study of the experimental electron density of cis-1 was carried out.
Experimental electron density
Hirshfeld atom refinement (HAR) and multipolar refinement were employed to gain insight of the non-covalent interactions that allows the stabilization of the near eclipsed structure of cis-1. The HAR refinement allows us to fully refine the hydrogen positions and their anisotropic displacement parameters (ADPs). This refinement yields C–H bond distances within the expected range. Within the crystal structure, the cis-1 molecule is surrounded by six other molecules. Due to symmetry, only three out of the six (cis-1)–(cis-1) interactions are distinct, labeled as A, B, and C in Fig. 3. Each unique interaction is duplicated within the cluster surrounding the single molecule. All interactions occur along the longest sides of the molecules, and in two of these binary interactions, the oxygen atoms of the sulfone group appear to play a crucial role. In interaction A, oxygen from the sulfone group interacts with the H of C5 atom (Scheme 1, interaction length, 2.25 Å), which is the most acidic H atom since it is located in the α position from the sulfone group. Moreover, there is also a short contact between an O atom of the dioxane moiety and a C–H (2.41 Å) in position α to a dioxane O atom and β to the sulfone group (methylene groups at position 4 and 6, Scheme 1). In interaction B, the other sulfone O atom is interacting with an equatorial H–C4,6 group α to a O atom of the dioxane moiety and β to the sulfone group (2.30 Å). In the third pair there is no C–H⋯O interactions but there is a short contact of the type H⋯H between hydrogens of two methyl groups (2.21 Å).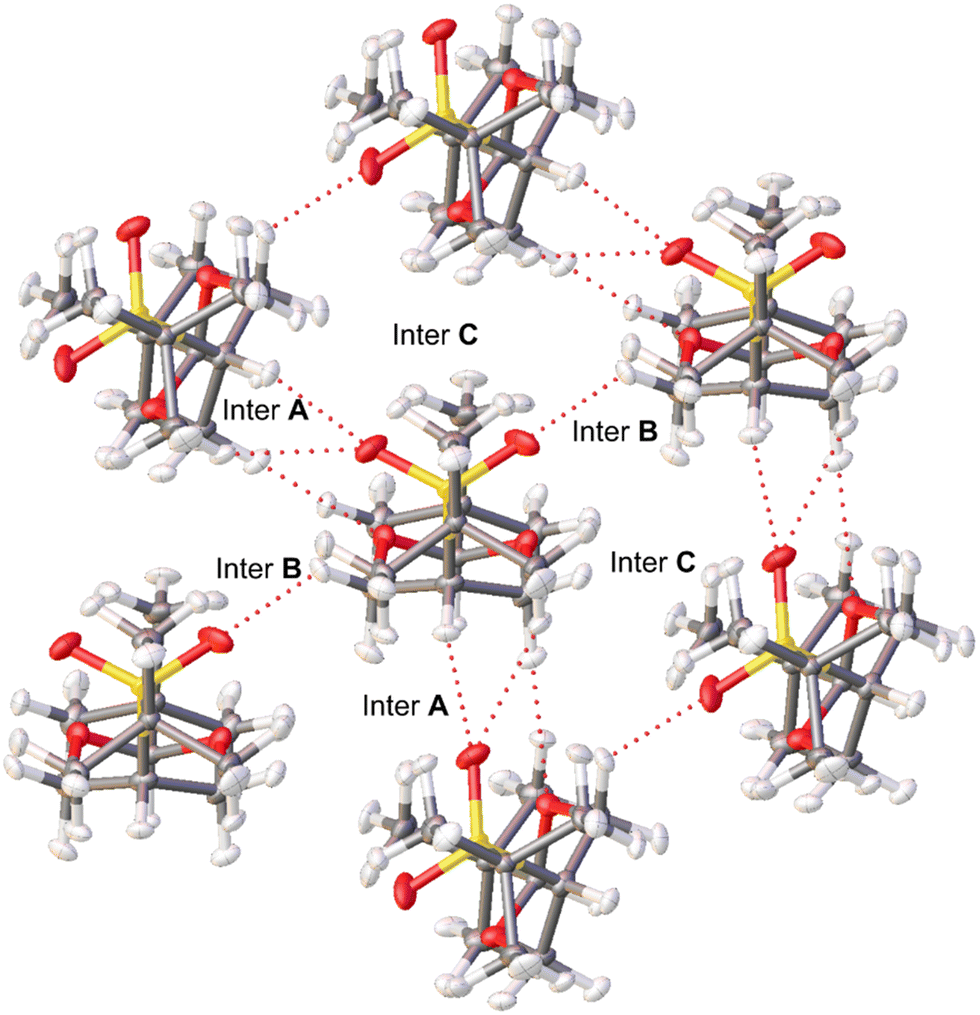 | ||
| Fig. 3 First neighbors in the crystal structure of cis-1. | ||
We perform a charge density analysis in the frame of the quantum theory of atoms in molecules (QTAIM)13 to evaluate the nature and the importance of the intermolecular interactions in the crystal. The following analysis is performed with the experimental electron density obtained from the multipolar refinement. The reduced density gradient (RDG) intermolecular isosurface (0.5) colored by the virial (V(r)) and the Lagrangian kinetic energy density (G(r)) (Fig. 4) and the electron density (ρ(r)) at the intermolecular bond critical points (BCP) show that the pair A is the most relevant. The non-covalent interactions (NCI) maps (the density multiplied by the sign of the second eigenvalue of its Hessian, sign![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) λ2ρ, plotted over RDG isosurfaces)14 were also analysed but the colored isosurfaces shows hardly any difference between the three interactions (see ESI‡). It has been shown that V(r) and G(r) fields offer a more comprehensive understanding of the electronic nature of weak interactions compared to signλ2ρ.15,16G(r) maps reveal that the strongest (indicated by highly positive values, red) closed shell interactions are in pair A (Fig. 4b). Moreover, the V(r) maps (Fig. 4a) show that this pair has also important dispersive interactions (indicated by highly negative values, blue). It is expected that V(r) is more negative for closer contacts. According to the RDG maps and the values of ρ(r) at the BCP extracted from the experimental density, the two C–H⋯O interactions discussed in the previous paragraph are the main interactions in pair A. In one of these interactions, an oxygen atom from the sulfone group (O(3)) establishes three bond paths (see Fig. 4 for numbering). The first path involves H(3), with a density at the BCP of 0.070 e Å−3—the second-highest value for intermolecular interactions within the crystal. The other two paths connect with hydrogen atoms bonded to C(11) and C(12), the value of ρ(r) at BCP for these two are 0.043 and 0.034 e Å−3, respectively. In the other interaction an O atom of the dioxane moiety, label as (O(1)), forms a bond path with H(4b). The ρ(r) value at BCP is 0.055 e Å−3, the third-largest value for an intermolecular interaction. On the other hand, in pair B, there is only one important interaction that is between the other O atom of the sulfone (O(4)) and a H(2b) group (see Fig. 4 for numbering). The V(r) and G(r) maps (Fig. 4c and d) show that only this interaction has an important closed shell and dispersive character. Moreover, ρ(r) at its BCP (0.079 e Å−3) is significantly larger than the rest, the following value is (0.025 e Å−3). Finally in pair C, there are several interactions, although not as strong as the C–H⋯O contacts of the other two pairs. The interaction that seems to be the most important, according to V(r) and G(r) maps (Fig. 4e and f) and ρ(r) at BCP, is C–H⋯H–C interaction between two methyl groups. The electron density at the H(6a)⋯H(7a) BCP is 0.047 e Å−3. The remaining two interactions within pair C, as revealed by the RDG maps, are considered weak but not insignificant. These involve C(4) of the dioxane moiety with H(7c) of one methyl group, and C(12) of the S-tert-butyl group with H(10a) of another methyl group, exhibiting ρ(r) values at BCP are 0.033 and 0.029 e Å−3, respectively. Consequently, pair A boasts two notably significant intermolecular interactions, whereas pair B has only one, and pair C, while lacking a predominant interaction, features three interactions that cannot be deemed negligible. It is evident that pair A should exhibit the strongest interaction energy, yet the relative energetic ranking between the other two pairs remains uncertain.
λ2ρ, plotted over RDG isosurfaces)14 were also analysed but the colored isosurfaces shows hardly any difference between the three interactions (see ESI‡). It has been shown that V(r) and G(r) fields offer a more comprehensive understanding of the electronic nature of weak interactions compared to signλ2ρ.15,16G(r) maps reveal that the strongest (indicated by highly positive values, red) closed shell interactions are in pair A (Fig. 4b). Moreover, the V(r) maps (Fig. 4a) show that this pair has also important dispersive interactions (indicated by highly negative values, blue). It is expected that V(r) is more negative for closer contacts. According to the RDG maps and the values of ρ(r) at the BCP extracted from the experimental density, the two C–H⋯O interactions discussed in the previous paragraph are the main interactions in pair A. In one of these interactions, an oxygen atom from the sulfone group (O(3)) establishes three bond paths (see Fig. 4 for numbering). The first path involves H(3), with a density at the BCP of 0.070 e Å−3—the second-highest value for intermolecular interactions within the crystal. The other two paths connect with hydrogen atoms bonded to C(11) and C(12), the value of ρ(r) at BCP for these two are 0.043 and 0.034 e Å−3, respectively. In the other interaction an O atom of the dioxane moiety, label as (O(1)), forms a bond path with H(4b). The ρ(r) value at BCP is 0.055 e Å−3, the third-largest value for an intermolecular interaction. On the other hand, in pair B, there is only one important interaction that is between the other O atom of the sulfone (O(4)) and a H(2b) group (see Fig. 4 for numbering). The V(r) and G(r) maps (Fig. 4c and d) show that only this interaction has an important closed shell and dispersive character. Moreover, ρ(r) at its BCP (0.079 e Å−3) is significantly larger than the rest, the following value is (0.025 e Å−3). Finally in pair C, there are several interactions, although not as strong as the C–H⋯O contacts of the other two pairs. The interaction that seems to be the most important, according to V(r) and G(r) maps (Fig. 4e and f) and ρ(r) at BCP, is C–H⋯H–C interaction between two methyl groups. The electron density at the H(6a)⋯H(7a) BCP is 0.047 e Å−3. The remaining two interactions within pair C, as revealed by the RDG maps, are considered weak but not insignificant. These involve C(4) of the dioxane moiety with H(7c) of one methyl group, and C(12) of the S-tert-butyl group with H(10a) of another methyl group, exhibiting ρ(r) values at BCP are 0.033 and 0.029 e Å−3, respectively. Consequently, pair A boasts two notably significant intermolecular interactions, whereas pair B has only one, and pair C, while lacking a predominant interaction, features three interactions that cannot be deemed negligible. It is evident that pair A should exhibit the strongest interaction energy, yet the relative energetic ranking between the other two pairs remains uncertain.
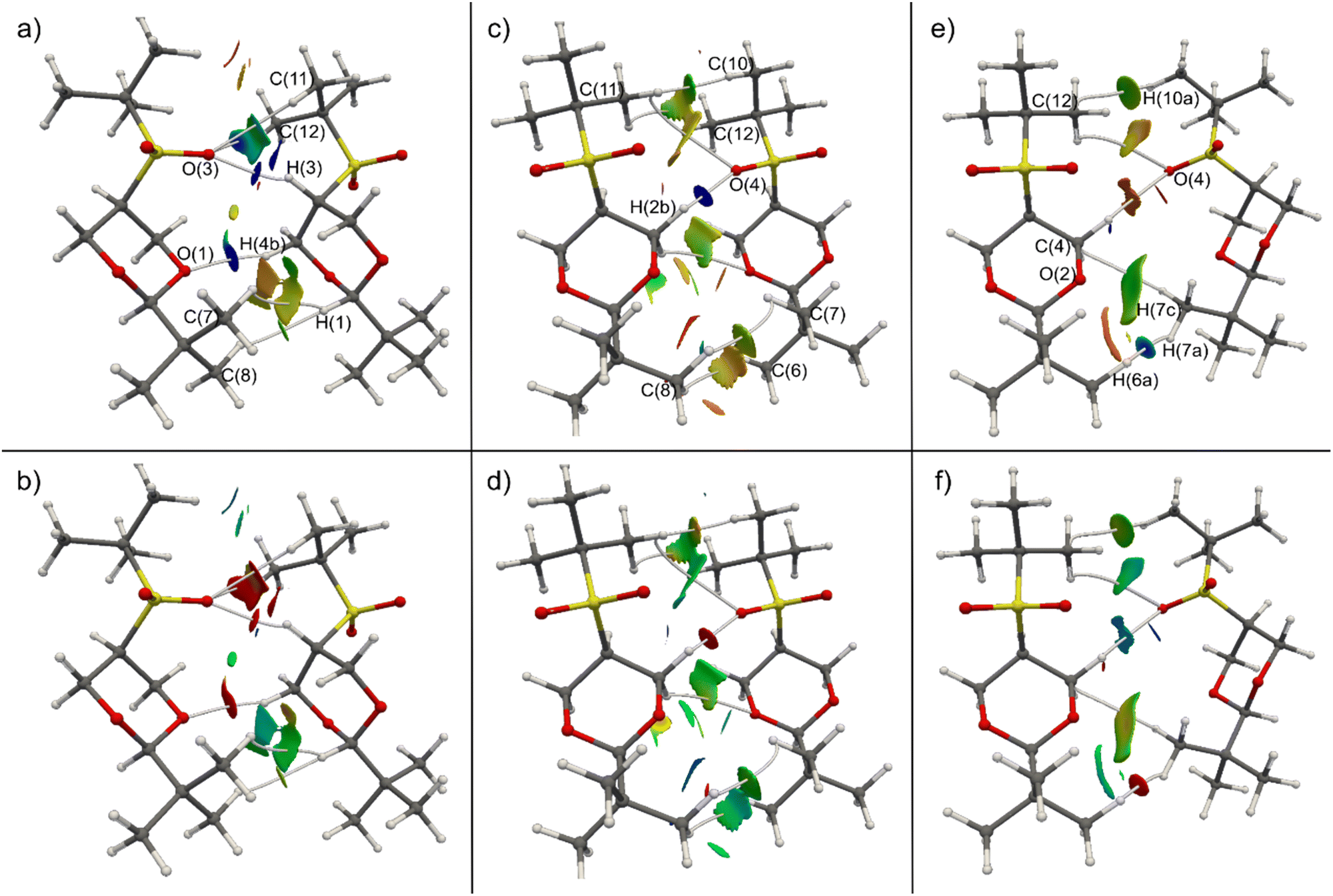 | ||
| Fig. 4 Intermolecular 0.5 RDG isosurface of cis-1 pairs A (a) and (b), B (c) and (d) and C (e) and (f). The isosurfaces are colored by V(r) (top) and G(r) (bottom) scalar fields. V(r) color code: ≤−1.0 × 10−1 e Å−5 (dark blue), −5.0 × 10−2 e Å−5 (green) and 0.0 e Å−5 (red). G(r) color code: ≥1.0 × 10−1 e Å−5 (red), 5.0 × 10−2 e Å−5 (green) and 0.0 e Å−5 (dark blue). | ||
DFT interaction energy calculations (see Theoretical methods section for details) help to clarify which of the three interactions is the most energetic. The interaction energy accounts for all the interactions within each dimer defined from the crystal structure (see Fig. 3, for the definition of dimers A, B and C). According to them, pair A is by far the strongest one (11.82 kcal mol−1). As mentioned, this large energy could be due to the contribution in a similar extent of two important interactions (a CH⋯OS and a CH⋯O(C)2). On the other hand, pair B only have one important C–H⋯O interaction which accounts for its lower energy value (5.72 kcal mol−1). The interaction energy of pair C was somewhat unexpected (5.73 kcal mol−1) but aligns with the picture depicted by the RDG maps. This energy is noteworthy since, base solely in the crystal structure it was expected a larger energy for pair B, this pair has a very short intermolecular contact between O(4) and H(2b) and pair C lack any such interaction. However, while in pair C there is not an interaction that stands out, the combined strength of the three C–H⋯H–C interactions is comparable to the C–H⋯O interaction in pair B. This piece of information is relevant because it allows to adequately ponder the properties of the C–H⋯H–C interaction, and its contribution to the stability of the crystal frame, generally considered as very weak and even responsible for the steric effect.17
The analysis of intermolecular interactions is crucial to explain phenomena of the eclipsed conformation that molecule cis-1 exhibits in the crystal structure. This conformation grants that sulfone O atoms can interact with other molecules, which allow the formation of two strong C–H⋯O interactions. Moreover, this elongated conformation of cis-1 enables the formation of several van der Waals interactions along the molecule. The energy gained by these intermolecular interactions is enough to overcome the low rotational barrier and can explain the difference in the heat of sublimation of 8.40 kcal mol−1 measured for the cis-1 isomer respect to the trans-1. The entropies of sublimation computed from the heats of sublimation at 298.15 K (ΔsubSm = ΔsubHm/T) also support this interpretation. A higher sublimation entropy (115.5 cal K−1 mol−1) in the cis-1 isomer implies that during the phase change more intense C–H⋯O and a larger number of dispersion interactions are broken in the crystal of this isomer respect to the trans-1 (ΔsubSm = 87.2 cal K−1 mol−1).
As indicated in the introduction, the 4-tert-butyl-1-(tert-butylsulfonyl)-cyclohexane (cis-3) shows eclipsing like cis-1, in which angle τ has a value of 14.1°. The SO⋯H–C interaction must be relevant too and the eclipsing in both molecules has the same origin.
Conclusions
Computational methods in gas phase are not able to correctly describe the eclipsed conformation in the HCSC segment present in the cis-1 compound, but they correctly predict the conformation in chloroform solution. Thus, the origin of the studied effect is not intrinsic to the molecule and hence we explore the intermolecular interactions as responsible of the observed effect. The difference in sublimation enthalpy between ΔsubH cis-1 and trans-1 is 8.40 kcal mol−1 allows to distinguish that the stabilization energy in the cis-1 isomer is greater than in the trans-1 isomer. The experimental electron density of the first compound allowed establishing the existence of CH⋯OS interactions (dependent on the acidity of the involved protons), CH⋯O(C)2 and CH⋯HC interactions in the crystal lattice as the stabilizing factors. The eclipsed conformation of cis-1 in the crystal allows maximizing the CH⋯OS interactions. The characterization of this interaction allows its implementation in crystal engineering for the construction of crystalline systems.
Experimental methods
X-Ray analysis and refinement
A high-quality crystal with dimensions 0.209 × 0.196 × 0.192 mm was used for a high-resolution X-ray experiment. The data were measured on a Rigaku XtaLAB Synergy diffractometer with a HyPix-6000HE detector and a PhotonJet Mo X-ray microsource (λ = 0.71073 Å). The data integration, scaling and merging was performed with the CrysAlisPro program. The data were collected up to (sinθ)/λ of 1.31 Å−1. The structure was solved by direct methods using SHELXT18 and it was subsequently refined with the independent atom model with Olex2.refine.19
The final Hirshfeld atom refinement was performed with Olex2 1.5,19 Orca 4.2.120 and NoSpherA221 software. The ω-B97X/Def2-TZVP level of theory was used for this refinement using a small cluster of 7 molecules, a central molecule and its six closest neighbours. The positions of the atoms do not change significantly from the refinement using a single molecule. The positions and the anisotropic displacement parameters (ADPs) of all atoms, H atoms included, were refined. Anharmonic parameters were refined solely for the S atom.
The multipole refinement was performed using XD2016 software using the Clementi and Roetti core and spherical valence densities.22 The initial atomic positions and ADPs were obtained from the Hirshfeld refinement. The R–H distances were fixed at the distance found in the Hirshfeld refinement. The spherical harmonics were used up to hexadecapoles for heavy atoms and up to dipoles for H atoms. The heavy atoms were refined anisotropically, while the H atoms isotropically. The anharmonic parameters of the S atoms were also refined.
The RDG isosurfaces and the QTAIM properties were calculated from the experimental electron density obtained after the multipole refinement. The TOPXD algorithm23 that is part of XD2016 was used for this purpose. For the calculation of G(r), V(r), and K(r), we use the approximation of Kirzhnitz24 that is a good approximation in the interatomic regions.25 We also calculate G(r), V(r), and K(r) without approximations from a theoretical calculation (ω-B97XD/Def2-TZVP level of theory). This intermolecular RDG isosurfaces were very similar from the experimental isosurfaces (see ESI‡).
Enthalpies of sublimation by thermogravimetry
The sublimation enthalpy of each isomer of sulfone 1 were measured by thermogravimetric analysis (TGA) and applying the Langmuir equation,26 which relates the mass loss of a sample with its vapour pressure during the sublimation process: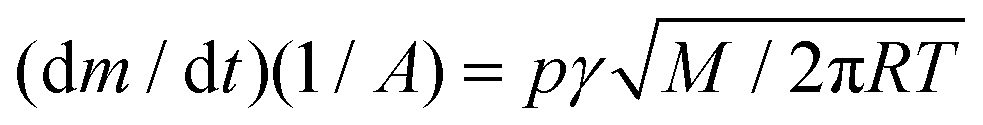 | (1) |
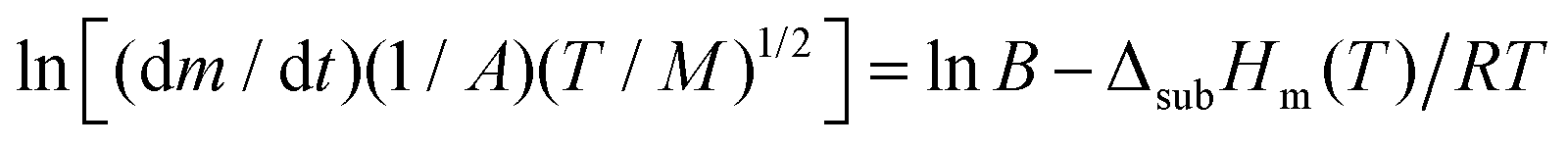 | (2) |
υ vs. I/T is linear, and its slope is ΔsubHm(T)/RT. The reliability of this methodology for deriving accurate results of sublimation enthalpy has already been shown.27,28
The mass loss rate (dm/dt) to substitute in eqn (2), was computed from data of the respective thermogravimetric derivative curve obtained by heating a sample of each sulfone in a thermogravimetric device. The sensitive element in this instrument is a dual-beam thermobalance with a 200-mg sample capacity and 0.1 μg sensitivity. A thermocouple in each beam measures the temperature of the samples located on it with an uncertainty of ±0.01 °C, and the beams operate inside of a furnace with a temperature control of ±1.0 °C. The heating rate and the flow of the purge gas in the furnace can be controlled with a sensitivity of 0.1 °C min−1 and 1.0 cm3 min−1 respectively. The TGA/DSC device was calibrated for mass measurement with a standard mass traceable to NIST of (315.1620 ± 0.0048) mg. The temperature scale was calibrated by analyzing the melting temperature of NIST 2232 Indium.
The thermogravimetric and derivative (dm/dt) curves were generated using the Universal Analysis® software of the SDT Q600® device. Detailed experimental conditions and data for each of the sublimation experiments are provided in the ESI.‡
Theoretical methods
The calculation of the interaction energy of the three unique pairs extracted from the crystallographic structure were performed with the ω-B97XD/Def2-TZVP level of theory, which was the same used for the Hirshfeld refinement. For these calculations, the geometries of each pair of molecules were extracted from the crystallographic structure (Fig. 3) and only the positions of the H atoms were optimized, the position of all the other atoms was taken from the Hirshfeld refinement. A Counterpoise correction was applied for the calculation of the interaction energies. Therefore, the interaction energy was calculated with the formula Eint = EAB(AB) − EA(AB) − EB(AB), where EAB(AB) is the energy of the dimer, EA(AB) and EB(AB) are the energies of the monomers calculated with the basis of the dimer.Conformational analysis for all compounds in gas phase was performed at M06-2X/6-311++G(2d,2p) level of theory. All the calculations were performed with Gaussian 16.29
Author contributions
Fátima M. Soto-Suarez, performed the synthesis, purification, structural elucidation, and computational calculations, Tania Rojo-Portillo, Eduardo H. Huerta, Alejandro Aguilera-Cruz, Alberto Tapia-Bárcenas, David Atahualpa Contreras-Cruz, worked on the synthesis, purification, structural elucidation, Rubén A. Toscano carried out the previous X-ray diffraction studies, Beatriz Quiróz-García carried out the NMR studies. Aaron Rojas-Aguilar performed the calorimetry studies, Fernando Cortés-Guzmán and John Bacsa performed high-resolution X-ray diffraction, Karla Ramírez-Gualito, NMR nuclear Overhauser Effect, José Enrique Barquera-Lozada and Gabriel Cuevas, proposal and financing of the project, theoretical calculations and study of experimental theoretical density. All authors primarily wrote the manuscript, provided suggestions to improve and modify the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the referees for useful comments, DGAPA-UNAM via Grants IN-217523, IN-205620 and IN-208623, to CONACYT for support of F. M. S.-S. (PhD scholarship numbers 289329) and financial support via Grant No. 252589 to Dirección General de Cómputo y de Tecnologías de Información y Comunicación (LANCAD-UNAM-DGTIC-094 and LANCAD-UNAM-DGTIC-304), and to CONACYT via grants 104299 CB-2008 and 286452 CB-2016. We are grateful to Dr Javier Pérez Flores, M. en A. Gladys Edith Cortés Romero, M. en C. Lucero Mayra Ríos Ruiz, and M. I. A. María Magdalena Aguilar Araiza for technical assistance.Notes and references
- E. Juaristi, R. Martínez, R. Méndez, R. A. Toscano, M. Soriano-García, E. L. Eliel, A. Petsom and R. S. Glass, Conformational Analysis of 1,3-dioxanes with sulfide, sulfoxide sulfone substitution at C(5). Finding an eclipsed conformation in cis-2-tert-Butyl-5-tert-butylsulfonyl-1,3-dioxane, J. Org. Chem., 1987, 52, 3806–3811, DOI:10.1021/jo00226a015.
- B. Gordillo, E. Juaristi, R. Martínez, R. A. Toscano, P. S. White and E. L. Eliel, Conformational Analysis of 5-Substituted-1,3-Dioxanes. 5. Bond Eclipsing in tert-Buthylsulfonyl-Substituted 1,3-Dioxanes and Cyclohexanes. X-ray Diffraction Studies, MMP2 Calculations, and Interpretation, J. Am. Chem. Soc., 1992, 114, 2157–2162, DOI:10.1021/ja00032a033 (See pg. 2159, column B and pg. 2161 column B).
- E. Juaristi, B. Gordillo, R. Martínez and R. A. Toscano, The Use of Precise Structural Information for the Understanding of the Conformational Behavior of cis-5-(tert-butylsulfonyl)- and cis-5-(tert-butylsulfinyl)-2-tert-butyl-1,3-dioxane, J. Org. Chem., 1989, 54, 5963–5967, DOI:10.1021/jo00286a032.
- E. L. Eliel and S. H. Wilen, Stereochemistry Organic Compoounds, Wiley-Intersciense Pub., New York, 1994, pp. 578–580, ISBN: 978-0-471-01670-0 Search PubMed.
- E. Juaristi, Conformational Analysis of Six-membered Sulfur-Containing Saturated Heterocycles, Acc. Chem. Res., 1989, 22, 357–364, DOI:10.1021/ar00166a003.
- E. Juaristi, Looking for Treasure in Stereochemistry-Land. A Path Marked by Curiosity, Obstinacy and Serendipity, J. Org. Chem., 2012, 77, 4861–4884, DOI:10.1021/jo300195m.
- E. Juaristi, Stable eclipsed conformations, in Encyclopedia of computational chemistry, ed. N. L. Allinger and P. V. R. Schleyer, Wiley, New York, 1998 DOI:10.1002/0470845015.CSA022.
- E. Juaristi, Introduction to Stereochemistry & Conformational Analylis, Wiley & Sons, New York, 1991, pp. 271–280, ISBN: 978-0-471-54411-1 Search PubMed.
- M. K. Kaloustrian, N. Dennis, S. Mager, S. A. Evans, F. Alcudia and E. L. Eliel, Conformational analysis. XXXI. Conformational equilibria of 1,3-dioxanes with polar substituents at C-5, J. Am. Chem. Soc., 1976, 98, 956–965, DOI:10.1021/ja00420a015.
- E. L. Eliel and S. A. Evans, Unusual strong intramolecular interaction between the sulfone or sulfoxide and the alkoxide functions, J. Am. Chem. Soc., 1972, 94, 8587–8589, DOI:10.1021/ja00779a054.
- Y. Gu, T. Kar and S. Scheiner, Fundamental properties of the CH⋯O interaction: Is it a true hydrogen bond?, J. Am. Chem. Soc., 1999, 121, 9411–9422, DOI:10.1021/ja991795g.
- E. Juaristi and R. Notario, Computational reexamination of the eclipsed conformation in cis-2-tert-butyl-5-(tert-butylsulfonyl)-1,3-dioxane, Struct. Chem., 2013, 24, 1855–1862, DOI:10.1007/s11224-013-0236-y.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Clarendon Press, 1994 Search PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, Revealing noncovalent interactions, J. Am. Chem. Soc., 2010, 132, 6498–6506, DOI:10.1021/ja100936.
- D. Morales-Pumarino and J. E. Barquera-Lozada, Electron density and its reduced density gradient in the study of π–π interactions, Int. J. Quantum Chem., 2022, e27051, DOI:10.1002/qua.27051.
- G. Saleh, C. Gatti and L. Lo Presti, Energetics of non-covalent interactions from electron and energy density distributions, Comput. Theor. Chem., 2015, 1053, 53–59, DOI:10.1016/j.comptc.2014.10.011.
- V. Duarte-Alaníz, T. Rocha-Rinza and G. Cuevas, Assessment of hydrophobic Intercations and their Contributions through the analysis of the Methane Dimer, J. Comput. Chem., 2015, 36, 361–375, DOI:10.1002/jcc.23798.
- G. M. Sheldrick, SHELXT-Integrated space-group and crystal-structure determination, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8, DOI:10.1107/S2053273314026370.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. J. Puschmann, OLEX2: a complete structure solution, refinement and analysis program, Appl. Crystallogr., 2009, 42, 339–341, DOI:10.1107/S0021889808042726.
- F. Neese, Software update: the ORCA program system, version 4.0, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327, DOI:10.1002/wcms.1327.
- F. Kleemiss, O. V. Dolomanov, M. Bodensteiner, N. Peyerimhoff, L. Midgley, L. J. Bourhis, A. Genoni, A. Malaspina, D. Jayatilaka, J. L. Spencer, F. White, B. Grundkötter-Stock, S. Steinhauer, D. Lentz, H. Puschmann and S. Grabowsky, Accurate crystal structures and chemical properties from NoSpherA2, Chem. Sci., 2021, 12, 1675–1692, 10.1039/D0SC05526C.
- A. Volkov, P. Macchi, L. J. Farrugia, C. Gatti, P. Mallinson, T. Richter and T. Koritsanszky XD2016 – A Computer Program Package for Multipole Refinement, Topological Analysis of Charge Densities and Evaluation of Intermolecular Energies from Experimental and Theoretical Structure Factors, 2016.
- A. Volkov, C. Gatti, Y. Abramov and P. Coppens, Evaluation of net atomic charges and atomic and molecular electrostatic moments through topological analysis of the experimental charge density, Acta Cryst., 2000, A56, 252–258, DOI:10.1107/S0108767300001628.
- A. Kirzhnitz, Quantum Corrections to the Thomas-Fermi Equation, Soviet Phys. JETP, 1957, 5, 64 Search PubMed.
- Y. A. Abramov, On the Possibility of Kinetic Energy Density Evaluation from the Experimental Electron-Density Distribution, Acta Cristallogr. Sect. A Found Crystallogr., 1997, 53, 264, DOI:10.1107/S010876739601495X.
- I. Langmuir, The vapor pressure of metallic tungsten, Phys. Rev., 1913, 2, 329–342, DOI:10.1103/PhysRev.2.329.
- T. Sánchez-Bulás, O. Cruz-Vásquez, J. Hernández-Obregón and A. Rojas, Enthalpies of fusion, vaporisation and sublimation of crown ethers determined by thermogravimetry and differential scanning calorimetry, Thermochim. Acta, 2017, 650, 123–133, DOI:10.1016/j.tca.2016.03.013.
- M. Martínez Herrera, M. Campos, L. A. Torres and A. Rojas, Enthalpies of Sublimation of Fullerenes by Thermogravimetry, Thermochim. Acta, 2015, 622, 72–81, DOI:10.1016/j.tca.2015.09.001.
- M. J. Frisch; G. W. Trucks; H. B. Schlegel; G. E. Scuseria; M. A. Robb; J. R. Cheeseman; G. Scalmani; V. Barone; G. A. Petersson; H. Nakatsuji; X. Li; M. Caricato; A. V. Marenich; J. Bloino; B. G. Janesko; R. Gomperts; B. Mennucci; H. P. Hratchian; J. V. Ortiz; A. F. Izmaylov; J. L. Sonnenberg; D. Williams-Young; F. Ding; F. Lipparini; F. Egidi; J. Goings; B. Peng; A. Petrone; T. Henderson; D. Ranasinghe; V. G. Zakrzewski; J. Gao; N. Rega; G. Zheng; W. Liang; M. Hada; M. Ehara; K. Toyota; R. Fukuda; J. Hasegawa; M. Ishida; T. Nakajima; Y. Honda; O. Kitao; H. Nakai; T. Vreven; K. Throssell; J. A. Montgomery Jr.; J. E. Peralta; F. Ogliaro; M. J. Bearpark; J. J. Heyd; E. N. Brothers; K. N. Kudin; V. N. Staroverov; T. A. Keith; R. Kobayashi; J. Normand; K. Raghavachari; A. P. Rendell; J. C. Burant; S. S. Iyengar; J. Tomasi; M. Cossi; J. M. Millam; M. Klene; C. Adamo; R. Cammi; J. W. Ochterski; R. L. Martin; K. Morokuma; O. Farkas; J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
Footnotes |
| † This paper is dedicated to Prof. Dr Joaquín Tamariz Mascarúa who retired after a successful career. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2302528. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cp04914k |
| This journal is © the Owner Societies 2024 |