
Initial decomposition pathways of 1,1-diamino-2,2-dinitroethylene (α-FOX-7) in the condensed phase†
Komal
Yadav
a,
Yuheng
Luo
a,
Ralf I.
Kaiser
 ab and
Rui
Sun
*a
ab and
Rui
Sun
*a
aDepartment of Chemistry, University of Hawaii, Honolulu, HI 96822, USA. E-mail: ruisun@hawaii.edu
bW. M. Keck Research Laboratory in Astrochemistry, University of Hawaii, Honolulu, HI 96822, USA
First published on 12th March 2024
Abstract
The initial decomposition pathways of α-FOX-7 in the condensed phase (crystal) were investigated via density functional theory. Calculations were carried out using three FOX-7 systems with increasing complexity from 1-layer (sheet) via 2-layer (surface) to 3-layer (bulk). The encapsulated environment of the central α-FOX-7 molecule, where decomposition takes place, is reconstructed by neighbouring molecules following a crystal structure. A minimal number of neighbouring molecules that have an impact on the energetics of decomposition are identified among all surrounding molecules. The results show that the presence of intermolecular hydrogen bonds due to the encapsulated environment in the condensed phase decreases the sensitivity of α-FOX-7, i.e. it increases the barrier of decomposition, but it does not alter the initial decomposition pathways of the reaction compared to the gas phase. Moreover, increasing the complexity of the system from a single gas phase molecule via sheet and surface to bulk increases the decomposition barriers. The calculations reveal a remarkable agreement with experimental data [A. M. Turner, Y. Luo, J. H. Marks, R. Sun, J. T. Lechner, T. M. Klapötke and R. I. Kaiser, Exploring the Photochemistry of Solid 1, 1-Diamino-2, 2-Dinitroethylene (FOX-7) Spanning Simple Bon Ruptures, Nitro-to-Nitrite Isomerization, and Nonadiabatic Dynamics, J. Phys. Chem. A, 2022, 126, 29, 4747–4761] and suggest that the initial decomposition of α-FOX-7 likely takes place at the surface of the crystal.
Introduction
The development of energetic materials with low sensitivity to shock, temperature, and friction while having high detonation performance is an active area of research. In this regard, 1,1-diamino-2,2-dinitroethylene, commonly known as FOX-7 or DADNE, is a promising candidate that has attracted considerable attention over the past decades. This molecule was first synthesized by Östmark and Bemm in 1998 and features easy synthesis procedures,1–4 low sensitivity to heat,5–10 and high detonation performance.11,12 Thus, it has been considered an alternative to well-known explosives, such as 1,3,5-trinitro-2-oxo-1,3,5-triazacyclo-hexane (RDX) and 1,3,5,7-tetranitro-1,3,5,7-tetraazacyclo-octane (HMX).1,13The establishment of the interplay between the molecular structure of FOX-7 and its decomposition mechanism is essential to understanding its superior performance as an energetic material. Molecules are aligned end-to-end, facilitating a two-dimensional wave-shaped structure with extensive hydrogen-bonding in the crystal of FOX-7.14 X-ray diffraction analysis has exposed as many as four different polymorphic phases, named the α-, β-, γ-, or δ-phase, with the α-phase being the most stable at room temperature.15,16 The crystal structure plays an important role in the chemical properties of FOX-7. Gupta and coworkers reported that the FOX-7 crystal can undergo structural transformation upon heating, which leads to the strengthening of its intramolecular and intermolecular hydrogen bonds and decreases the sensitivity of FOX-7 to shock.17 Computational studies performed by Kuklja et al. suggest that the presence of internal defects in the crystal structure, such as reversed oriented molecules and shear strain defects, i.e., disruption of intermolecular hydrogen bonds decreases decomposition barriers.18 Numerous experimental studies have been attempted to establish the interplay between the chemical structure and properties of FOX-7.4,13,18–23 Jones and co-workers studied the thermal decomposition of FOX-7 in different phases with simultaneous thermogravimetry-differential thermal analysis-Fourier transform infrared spectroscopy-mass spectrometry (TG-DTA-FTIR-MS) measurements and differential scanning calorimetry. They observed a two-step decomposition process: the first step yields water (H2O), carbon dioxide (CO2), dinitrogen oxide (N2O), hydrogen cyanide (HCN), nitrogen dioxide (NO2), and isocyanic acid (HOCN), and the second step forms nitrous acid (HONO) and formic acid (HCOOH).24 The key step in the decomposition reaction, i.e., nitro-to-nitrite isomerization, was supported by differential scanning calorimetry experiments carried out by Chen et al.18 Kyncl and co-workers used a combination of laser-induced breakdown spectroscopy (LIBS) and selected ion flow tube mass spectroscopy (SIFT-MS), and observed the formation of a different set of compounds, including nitrogen oxide (NO), NO2, formaldehyde (HCHO), HCN, acetylene (C2H2), HONO, and ethanol (CH3CH2OH).19 Recently, in a series of studies of photon-induced photolysis of FOX-7, Turner et al. observed that the NO2 and amino radical (NH2) loss channels are not accessible with 355 and 532 nm photons, while nitrogen oxide (NO) was observed only with the 355 nm photon.20,22,25
Extensive computational research has also been conducted on the gas and condensed phase decomposition of FOX-7.20,26–39 In one of the earliest studies, Politzer et al. reported the C–NO2 and C–NH2 dissociation energies to be 293 and 467 kJ mol−1, respectively, using density functional theory (DFT) calculations at the B3P86/6-31+G(d,p) level of theory.26 Kimmel and colleagues reported that the decomposition of FOX-7 in the excited states features low barriers, and changes the reaction type from endothermic to exothermic (B3LYP/6-31+G(d,p)).27 The mechanism of the initial decomposition of FOX-7 was proposed by Bernstein et al., where the nitro-to-nitrite isomerization is followed by a NO dissociation (B3LYP/6-31+G(d,p)).31 Luo et al. performed an exhaustive search for the different decomposition pathways of FOX-7 in the gas phase, and suggested that the sensitivity of FOX-7 is primarily determined by the nitro group.33 Xia and co-workers investigated the impact of intermolecular forces in a dimer and bulk of FOX-7 (B3LYP with 6-311G(d,p), 6-311++G(d,p), and D95), and concluded that certain features of the bulk (such as molecular packing) can be observed in the dimer.40 Zhao and Liu investigated the initial decomposition of FOX-7 under high pressure, and found the C–NO2 bond to be the most sensitive under compression.41 The thermal decomposition of FOX-7 (at 3000 K) was studied by Zheng et al. by solving the non-SCF Harris functional approximation using local density approximation (LDA), and indicated that most of the energy is released within the first 15 ps with H2O and nitrogen molecule (N2) as the primary products.42 Michalchuk et al. studied the impact sensitivity on different polymorphs of the FOX-7 crystal using a theoretical model based on vibrational up-pumping. They observed that increasing the layer of polymorphs reduced the impact sensitivity, which was validated by the BAM fall hammer testing.10
The experiment by Turner et al. offered an ideal case to probe the initial decomposition of FOX-7, as the excitation energy is barely enough to trigger the initial decomposition. After the radiation from 355 and 532 nm photons, the spectrum indicates that an overwhelming majority of the molecules in the crystal remains intact. In the same study, our group investigated the impact of the condensed phase by calculating the decomposition of FOX-7 with a small molecular cluster of the crystal structure. The calculation replaced a FOX-7 molecule in the molecular cluster with a gas-phase optimal structure (intermediates or transition states), while the neighboring molecules remained frozen. Clearly, this approach is a workaround to avert the high cost of geometry optimization of the entire molecular cluster. Nonetheless, an improved agreement (compared to the gas phase calculation) with the experiment was achieved, which indicates that the encapsulated environment plays an important role in the initial decomposition.
In this study, we demonstrate the effect of the condensed phase environment on the initial decomposition of FOX-7. The DFT method was validated by previous experiment and high-level calculations on this specific system.33 The initial decomposition barriers for FOX-7 were computed in three different environments: (i) sheet, (ii) surface, and (iii) bulk. In each scenario, the FOX-7 molecule at the center of the system was considered the reactive site, and the influence of neighbouring molecules on the decomposition of the reactive site was analyzed. We have also identified a minimal list of neighbouring molecules that directly affect the decomposition. Our computation indicates that the decomposition is likely to first take place at the surface of the FOX-7 crystal, is validated by the experiments of Turner and coworkers.20
Methodology
The initial decomposition of FOX-7 was studied using DFT. The stationary points on the potential energy profile were optimized at the M06-2X-D3/6-31G(d) level of theory.43–45 The selected level of theory employed a rather small basis set to balance the computational cost and the accuracy of the calculations for a system of considerable size (especially for the surface and the bulk systems). However, it was carefully benchmarked with a previous study by Luo et al.,33 where a much larger basis set (def2-TZVPP46) was employed (e.g., the root mean square error, RMSE between our values and those reported by Luo et al. was found to be ∼9.5 kJ mol−1), and the results agree well with the experiments. It is important to note that including dispersion corrections is necessary to get an accurate description of the intermolecular (van der Waals) interaction within energetic materials. Dispersion effects have a significant impact on the precision of theoretical reaction thermodynamics, and are also crucial for obtaining a good understanding of non-covalent and intermolecular interactions. Empirical dispersion is an approach that takes into account the fact that DFT generally underestimates the van der Waals interaction in molecular systems. Among the different variants available, Grimme's D3 correction has proven to provide accurate results as the dispersion coefficients are geometry-dependent and hence are adjusted based on the local geometry. Therefore, Grimme's D3 correction was used in our calculations.33,44,47–50 The intermediates and transition states are verified by their number of imaginary frequencies. For a partially optimized geometry (i.e., frozen second solvation shell) such as the one reported in the manuscript, the Hessian matrix components corresponding to frozen atoms are set to zero. The 3(N–m) normal modes are calculated, where N and m are the total number of atoms in the system and the number of frozen atoms, respectively. The intrinsic reaction coordinate (IRC) calculations were carried out to verify the connection between transition states and intermediates.51 To further validate the accuracy of the M06-2X-D3/6-31G(d) level of theory, the single-point energies of the stationary points optimized at the M06-2X-D3/6-31G(d) level of theory in the sheet system were calculated at the ωb97X-D52/6-31G(d) level of theory. The double-hybrid DFT functional ωb97X-D accurately modeled the dispersion interaction, which is important in the condensed phase system like the current one. The energies computed with M06-2X-D3/6-31G(d) and ωb97X-D/6-31G(d) are almost identical. In addition, both M06-2X-D3 and ωb97X-D DFT functionals have previously been reported to successfully model reactions involving radicals. Moreover, these methods provide a reasonable balance between the computational cost and chemical accuracy, which is crucial in studying large molecular clusters like the ones reported in this manuscript.33,53–61 All calculations were performed using the Gaussian16 software package.62As noted in the introduction, the crystal of FOX-7 possesses a wave-shaped structure (Fig. 1). This research focuses on the energetics of the initial decomposition of α-FOX-7, the most stable crystal among all forms at room temperature. Hence, to understand the effect of the encapsulated environment on the initial decomposition, an additive approach with increasing complexity is adapted to mimic the initial decomposition of FOX-7 in the crystal (gas → sheet → surface → bulk, Fig. 2). The initial structures of these systems were obtained from the α-FOX-7 crystal structure provided by Bemm and Östmark.63 The decomposition occurs at the central molecule. During geometry optimization, the central molecule and its neighbouring molecules that have direct contact with the reaction site are allowed to fully relax, while the rest of the molecules are held fixed to maintain the wave-shaped structure of the FOX-7 crystal. A promising approach is to wisely choose the neighbouring molecules that most significantly impact the energetics of the decomposition of the central molecule to minimize the number of degrees of freedom in geometry optimization. A detailed discussion of how these molecules are selected is provided in the following sections.
 | ||
| Fig. 1 (a) Molecular structure of FOX-7 in the gas phase; (b) unit cell of α-FOX-7; (c) top view of α-FOX-7 with intramolecular (black) and intermolecular (cyan) H-bonds highlighted. | ||
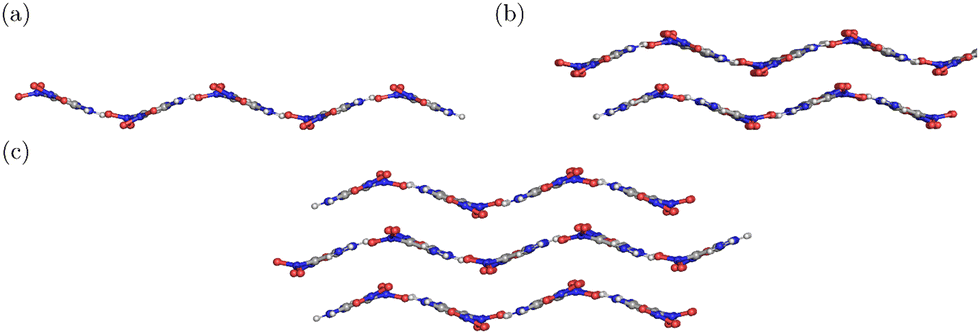 | ||
| Fig. 2 Representation of α-FOX-7 (a) one-layer (sheet), (b) two-layer (surface), and (c) three-layer (bulk) used for the calculations. | ||
Results
1. Initial decomposition of FOX-7 in the gas phase
The potential energy profile for the initial decomposition of FOX-7 in the gas phase is given in Fig. 3. A total of four pathways resulting in three gas-phase products are reported: (i) a nitro (–C–NO2) to nitrite (–C–ONO) isomerization via a cyclic 3-membered transition state ts1 (271.6 kJ mol−1) leads to the formation of an intermediate int1 (−19.4 kJ mol−1). It is interesting to note that int1 can be formed via another transition state, ts1′ (296.5 kJ mol−1), of a higher energy. The ts1 is lower in energy than ts1′ due to the intramolecular hydrogen-bond between the oxygen atom of the isomerizing –NO2 group and the neighbouring –NH2 group (Fig. S1 in the ESI†). A torsion about the C–O bond in int1 forms another intermediate int2 (−25.1 kJ mol−1) via transition state ts2 (13.2 kJ mol−1), which is followed by the C–O bond dissociation, leading to the formation of int4 and NO. The reaction energy of this pathway is slightly exothermic (−15.5 kJ mol−1). The second (ii) pathway involves an H-atom migration from the –NH2 group to the neighbouring C-atom containing the –NO2 moiety via the cyclic four-membered transition state ts3 (210.9 kJ mol−1) to form intermediate int3. The H-atom shift causes a change in the hybridization of the C-atom containing the –NO2 groups from sp2 to sp3. The intermediate int3 then undergoes a C–NO2 bond dissociation, producing int5 and NO2. Alternatively, intermediate int5 and NO2 can also be formed via pathway (iii), which starts with the dissociation of one of the C–NO2 bonds, leading to the formation of int6 and NO2 (321.5 kJ mol−1). This is followed by an H-atom migration of the –NH2 group to the carbon atom of –C–NO2 to form intermediate int5viats4 (436.7 kJ mol−1). The formation of int5 and NO2 is an endothermic (+222.6 kJ mol−1) process. The fourth (iv) pathway involves the dissociation of one of the C–NH2 bonds to form int7 + NH2 (474.5 kJ mol−1). This pathway is the most endothermic among the three initial gas-phase products.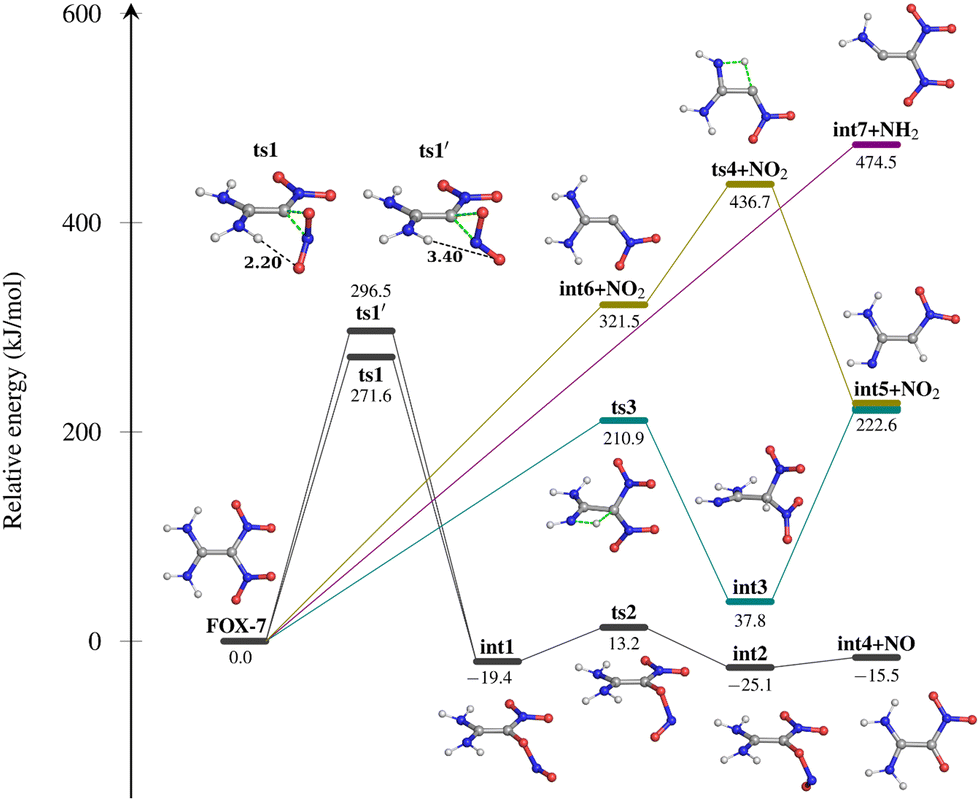 | ||
| Fig. 3 Potential energy profile (in kJ mol−1, zero-point energy included) for the initial decomposition pathways of FOX-7 in the gas-phase at the M06-2X-D3/6-31G(d) level of theory. Pathway (i), black; pathway (ii), green; pathway (iii), olive; pathway (iv), purple. | ||
2. Initial decomposition of FOX-7 on a sheet
The crystalline structure of FOX-7 indicates that a single unit cell consists of four FOX-7 molecules,63i.e., 56 atoms. The molecules in the crystal are aligned end-to-end, facilitating a two-dimensional wave-shaped structure with extensive intermolecular H-bonding within a layer and van der Waals interaction between layers. These interactions stabilize the FOX-7 molecule.50 It is also important to note that the structure of FOX-7 in the crystal deviates from its optimal gas phase structure (Fig. S2 in the ESI†), thus destabilizing the molecule. The exact chemistry of the initial decomposition of FOX-7 depends on the balance between these two competing factors. It is important to note that our previous calculations for a tetramer of FOX-7 did not maintain the wave-shaped structure of the FOX-7 crystal. However, the experiments showed that the wave-shaped structure of the crystal is preserved during the initial decomposition reaction. Therefore, to replicate the experimental setup accurately, we have performed a restrained optimization of the sheet/surface/bulk by keeping the molecules in the second solvation shell frozen. For the sheet model (Fig. 4a), the central FOX-7 (where the decomposition takes place) is surrounded by two solvation shells. The six FOX-7 molecules in the first solvation shell are allowed to fully relax during the optimization, while the 12 FOX-7 molecules in the second solvation shell are frozen to maintain the wave-shaped structure of the system. The sheet model consists of a total of 19 FOX-7 molecules. Since the product molecules in Fig. 3 have been shown to agree with the experiments well,20–22,25 the focus of the condensed phase study is on how the encapsulated environments alternate the energetics. It should be noted that in the current setup, only the central FOX-7 molecule in the sheet is set to be reactive - in the initial structure for geometry optimization, only its structure deviates from the crystal structure. Considering that almost all FOX-7 molecules remain intact after the radiation from 355 and 532 nm lasers, this setup mimics the initial decomposition.22 | ||
| Fig. 4 A representation of the neighbouring molecules to the central FOX-7 molecule (enclosed in an orange circle) for FOX-7 (left) and ts1 (right). Panel (a) shows all the molecules in the first solvation FOX-7, and panel (b) shows the minimum number of neighbouring molecules for ts1. | ||
The barrier (ts1, the –NO2 to –ONO isomerization) of the pathway (i) slightly increased from 271.6 kJ mol−1 in the gas phase to 287.6 kJ mol−1 in the sheet due to the restriction of the intermolecular hydrogen bonds (Fig. 5b and c). Interestingly, the transition state for the other torsional isomer for the –NO2 to –ONO isomerization, ts1′, slightly decreased from 296.5 kJ mol−1 in the gas phase to 291.9 kJ mol−1. As a result, the two pathways of –NO2 to –ONO have comparable barriers (ΔΔEts1′–ts1 = 4.6 kJ mol−1) as compared to the difference in the gas phase of ∼25 kJ mol−1. The decrease in energy difference between ts1 and ts1′ can be explained by the presence of intermolecular H-bonding among the FOX-7 molecules in the sheet model. As Fig. 5c shows, the isomerizing –NO2 group in ts1′ is stabilized by an intermolecular H-bond of O–H distance of 2.1 Å. Although the isomerizing –NO2 group in ts1 also forms an intermolecular H-bond, this interaction is much weaker (e.g., O–H distance is 2.3 Å). The second (ii) pathway accounts for an H-atom migration from –NH2 to the C-atom containing the –NO2 groups via the cyclic four-membered transition state ts3.
 | ||
| Fig. 5 The restructures of important intra-(black) and intermolecular (cyan) hydrogen bonds of (a) FOX-7, (b) ts1, (c) ts1′, and (d) ts3. | ||
Compared to the gas phase, the same procedure in the sheet must break one intermolecular hydrogen bond before the H-atom migration (Fig. 5d). Indeed, the barrier for H-migration viats3 increased to 252.8 kJ mol−1 (41.9 kJ mol−1 higher than ts3 in the gas phase). Pathway (iii) involves the dissociation of the C–NO2 bond to form NO2 and int6. Once formed, NO2 is assumed to have left the sheet. Thus, two intermolecular hydrogen bonds are broken, resulting in a 20.2 kJ mol−1 increase in int6. Both pathways (ii) and (iii) eventually form int5 and NO2, whose energy is also increased from the gas phase by ∼72 kJ mol−1 due to the breaking of three intermolecular hydrogen bonds (two from the escaping NO2 molecule, and one from the migrated hydrogen from the –NH2 group). The fourth (iv) pathway involves the highly endothermic C–NH2 dissociation, which also leads to the breaking of two intermolecular hydrogen bonds, resulting in a 70.2 kJ mol−1 increase in the energy of the dissociation product, int7 and NH2.
As given in Fig. 5, the decomposition of FOX-7 in the condensed phase involves reconstruction of intermolecular hydrogen bonds with its neighboring molecules, which results in different energetics as compared to the same process in the gas phase. Therefore, it is reasonable to expect that only those FOX-7 molecules that form the hydrogen bonds with the central (reactive) molecule make a significant contribution to such a difference (Fig. 4b). It is of interest to investigate whether there exists a minimal number of neighbouring molecules, with which the calculations reproduce the ones with the full first solvation shell. This is particularly important for the study of the decomposition at the surface and in the bulk, where allowing a minimal number of neighbouring molecules will play an essential role in the feasibility of the calculation. A comparison of the energetics computed from relaxing the entire first solvation shell and relaxing only a minimal number of neighbouring molecules is given in Table 1. As the results show, relaxing only a minimal number of neighbouring molecules provides a near-identical representation of the energetics obtained from considering the entire first solvation shell. In fact, the maximum energy difference between the two cases was found to be only ∼4 kJ mol−1. With a much smaller number of degrees of freedom to optimize, a significant number of computational resources can be saved. The general rule is that all molecules that form hydrogen bonds with the reactive site of the central molecule (either –NH2 or –NO2) need to be included in the minimal number of neighbouring molecules. However, for ts3 and int5, which involve changes in both C–(NO2)2 and C–(NH2)2 groups, all molecules in the first solvation shell of the reactive FOX-7 play a significant role and must be included in the calculation. From this point on, the geometry optimizations to study the decomposition of FOX-7 will only relax a minimal number of neighbouring molecules in the first solvation shell with the rest, and molecules in the second solvation shell remain frozen.
| Stationary point | One-layer (sheet) | |
|---|---|---|
| 1st solvation shell | Nearest neighbours | |
| ts1 | 291.9 [292.0] | 293.0 [293.6] |
| ts1′ | 287.6 [285.9] | 289.1 [285.8] |
| ts3 | 252.8 [263.3] | 252.8 [263.3] |
| int5 + NO2 | 291.0 [279.2] | 291.0 [279.2] |
| int6 + NO2 | 341.7 [330.6] | 345.6 [333.9] |
| int7 + NH2 | 511.6 [535.2] | 513.7 [536.2] |
3. Decomposition of FOX-7 on a surface
To mimic the decomposition of FOX-7 on a surface, a two-layered FOX-7 system is constructed (Fig. 2) by adding another layer directly beneath the sheet following the crystal structure. The second layer consists of 12 FOX-7 molecules, among which the central three molecules situated directly below the reaction site are bound to play an important role in the decomposition reaction. Hence, during the search for stationary points, these three molecules are added to the list of the minimal number of neighbouring molecules identified in the previous section and are allowed to relax. The rest of the molecules are frozen to maintain the structure of the system (Fig. S5, ESI†). The additional sheet complicates the decomposition pathways. Unlike the decomposition in the sheet structure, the gas phase product (and the transition states leading to it) can be shed off, facing either the vacuum or the bulk. An example of the vacuum-facing and bulk-facing transition state (e.g., ts1 in the pathway (i), the nitro-to-nitrite isomerization leading to the formation of NO) is given in Fig. 6a and b. The energy of the vacuum-facing barrier, ts1v, is 314.9 kJ mol−1, slightly lower than the energy of the bulk-facing barrier ts1b of 325.4 kJ mol−1. As the figure shows, the steric hindrance caused by the underlying layer leads to the bond elongation towards the vacuum in both cases. The steric hindrance also leads to both transition states possessing a higher energy than its counterpart in the sheet (287.6 kJ mol−1). The nitro-to-nitrite isomerization can also occur viats1′, facing either the bulk or vacuum. Interestingly, ts1v′ (292.5 kJ mol−1) is very similar in energy compared to ts1′ (293.0 kJ mol−1) obtained in the sheet model; ts1b′ is about 15 kJ mol−1 higher in energy compared to ts1′ (Fig. 6c and d). Similar to the arguments made in the previous section, the low energy of ts1v′ can be explained by looking at the O (in –NO2 of the reacting molecule)–H (in –NH2 of the neighbouring molecule) distance, which is 2.0 and 2.8 Å for ts1v′ and ts1b′, respectively. In other words, ts1v′ is stabilized by an H-bonding to a greater extent than ts1b′. Similarly, the H-atom transfer viats3 (pathway (ii), leading to the formation of NO2) could also take place in the direction facing the vacuum (ts3v, 292.0 kJ mol−1) or the bulk (ts3b, 308.6 kJ mol−1). In ts3v (Fig. 6e), the CC(NH2) plane is twisted to a 63.5° angle to the (approximate) plane containing (NO2)2CC, while in ts3b (Fig. 6f), the CC(NH2) plane is more tilted (43.1°) to minimize the steric hindrance from the layer beneath. It should be noted that the upper layer of FOX-7 is slightly displaced vertically (the distance between the layers increases) after optimization. This perpendicular expansion can be a result of the breaking of the weak van der Waals interaction present between the layers, which is consistent with the experimental observations made by Thompson and coworkers.64 Once again, both ts3v and ts3b are higher in energy than their counterpart in the sheet (252.8 kJ mol−1).50 | ||
| Fig. 6 The structure of ts1, i.e., nitro-to-nitrite isomerization occurring facing the (a) vacuum, ts1v, and (b) bulk, ts1b; ts1′ facing the (c) vacuum, ts1v′ and (d) facing bulk, ts1b′; and the H-transfer transition state (ts3) facing the (e) vacuum, ts3v, and (f) bulk, ts3b. | ||
In the two-layer surface model, the gas phase products (NO2, NH2, and NO) are assumed to dissipate eventually from the surface without major energy barriers. Therefore, these gaseous products formed with int5, int6, and int7 are optimized with the rest of the neighbouring molecules. Formation of these intermediates (int5, int6, and int7) in the surface model results not only in the breaking of different inter- and intramolecular H-bonds as reported in the sheet, but also loss of van der Waals interaction between the leaving group and the FOX-7 molecules of the basal layer. Hence, the energy required for these pathways is expected to be higher as compared to the sheet model. Specifically, the reaction energy of int5 and NO2 is 348.7 kJ mol−1, higher than its counterpart in the sheet (291.0 kJ mol−1) and gas phase (222.6 kJ mol−1). Similarly, the energy of NO2 and int6 (377.9 kJ mol−1) is approximately 32 and 56 kJ mol−1 higher compared to the sheet and gas phase, respectively. Finally, the most endothermic initial decomposition product, int7 (543.3 kJ mol−1) and NH2, also increased by 68.8 and 29.6 kJ mol−1 compared to the decomposition in the gas phase and sheet model, respectively.
Among the reaction pathways discussed above, another potential pathway where a FOX-7 molecule first desorbs from the surface, subsequently dissociating in the gas phase, cannot be ruled out. Assuming no transition state is involved in the desorption, the energy required for the first FOX-7 molecule to escape the surface (i.e., creating a vacant site) is 348.3 kJ mol−1. It takes only 225.6 kJ mol−1 to remove a second FOX-7 molecule that is neighbouring the vacant site from the surface, as the crystal structure has been undermined. A pictorial representation of the desorption process is shown in the ESI† (Fig. S7 and S8). The desorbed FOX-7 molecule can dissociate in the gas phase following the pathways, as discussed in the earlier section of the manuscript. Therefore, the desorption-decomposition pathway is energetically unfavourable compared to decomposition in the condensed phase.
4. Decomposition of FOX-7 in bulk
To study the impact of an encapsulated environment on the initial decomposition in bulk, an additional layer was added above the surface model described in the previous section (Fig. 6). Following the crystal structure, the top and bottom layers are mirror images of each other. Same as the bottom layer, the central three molecules on the top layer are situated directly on the top of the reactive molecule, and they were also added to the list of minimal neighbouring molecules to relax during geometry optimization. It is expected that the initial decomposition of FOX-7 will be further hindered due to an increase in steric repulsion from the layers above. Fig. 7(a) shows the nitro-to-nitrite isomerization via transition state ts1 in bulk. Compared to its counterparts on the surface (Fig. 6a and b), the largest difference lies in the elongated bond in the –NO2 moiety. Instead of stretching out of the crystal plane and to the vacuum, the –NO2 moiety rotates with respect to the C–N bond to minimize the interaction with neighbouring layers. The energy of ts1 is 350.8 kJ mol−1, the highest among ts1 found in all environments. In addition to ts1, the isomer ts1′ also has the highest barrier (323.9 kJ mol−1) as compared to its counterparts in gas (296.5 kJ mol−1), sheet (293 kJ mol−1), and surface models (292.5 kJ mol−1 for ts1v′ and 308.4 kJ mol−1 for ts1b′). Compared to ts3 on the surface, the additional top layer pushes the CC(NH2) plane to become more parallel to the (approximate) (NO2)CC plane – the angle between them is only 36.4° compared to 43.1° and 63.5° in ts3b and ts3v, respectively. In this scenario, the barrier for the H-transfer pathway viats3 is found to be 346.9 kJ mol−1, greater than the ts3 energetics on the surface (292.0 kJ mol−1 for ts3v and 308.6 kJ mol−1 for ts3b), on a sheet (252.8 kJ mol−1), and in the gas phase (210.9 kJ mol−1). The physical meaning of the energetics of int5, int6, and int7 is less clear, as it is somewhat invalid to assume the accompanying gas-phase products have left the reaction site (diffusing through at least one layer of FOX-7) without significant barriers. Nonetheless, their energies were computed (374.8, 406.3, and 561.4 kJ mol−1 for int5, int6, and int7, respectively). Compared to their counterparts on the surface, int5, int6, and int7 in bulk have even higher energies for the same reason argued in the previous section. | ||
| Fig. 7 Side view of the transition states obtained using the bulk model (a) ts1, (b) ts1′, and (c) ts3. | ||
Discussion
A comparison of the barriers involved in the initial decomposition of α-FOX-7 in the gas phase, sheet, surface, and bulk is given in Table 2. As discussed in the previous section, although the condensed phase environment theoretically has competing effects on the decomposition energetics (e.g., destabilizing the molecule by deviating its geometry from the gas-phase optimized structures, and stabilizing the molecule by forming an intermolecular hydrogen bond), the results clearly show that the latter outweighs the former in all cases. A more direct comparison can be found in Fig. 8, where the relative energy of species in the gas phase is set to zero, and only the differences of the same species in various environments are plotted. There are four key observations:| Stationary point | Gas phase | One-layer (sheet) | Two-layers (surface) | Three-layers (bulk) |
|---|---|---|---|---|
| The notations v and b indicate that the reaction takes place facing the vacuum and bulk, respectively. | ||||
| ts1 (→ NO) | 271.6 | 289.1 | 314.9v, 325.4b | 350.8 |
| ts1′ (→ NO) | 296.5 | 293.0 | 292.5v, 308.4b | 323.9 |
| ts3 (→ NO2) | 210.9 | 252.8 | 292.0v, 308.6b | 346.9 |
| int5 + NO2 | 222.6 | 291.0 | 348.7v | 374.8 |
| int6 + NO2 | 321.5 | 345.6 | 377.9v | 406.3 |
| int7 + NH2 | 474.5 | 513.7 | 543.3v | 561.4 |
(1) As the environment gets more encapsulated (gas → sheet → surface → bulk), the barrier of decomposition increases in all cases except for ts1′.
(2) The amount of increase in the decomposition barrier is directly related to the number of H-bonds broken during the reaction. For example, those pathways that lead to the breaking of three intermolecular hydrogen bonds (ts3 and int5) possess a larger increase in energy compared to those that involve the breaking of only two intermolecular hydrogen bonds (ts1, int6, and int7).
(3) In the only two cases where the decomposition barrier decreases (ts1′, gas → sheet; sheet → surface (vacuum facing)), the transition state is stabilized by an intermolecular hydrogen with a neighbouring molecule.
(4) The condensed phase alters the reaction pathway. For example, ts1 is replaced by ts1′ as the energetically favoured pathway for nitro-to-nitrate isomerization.
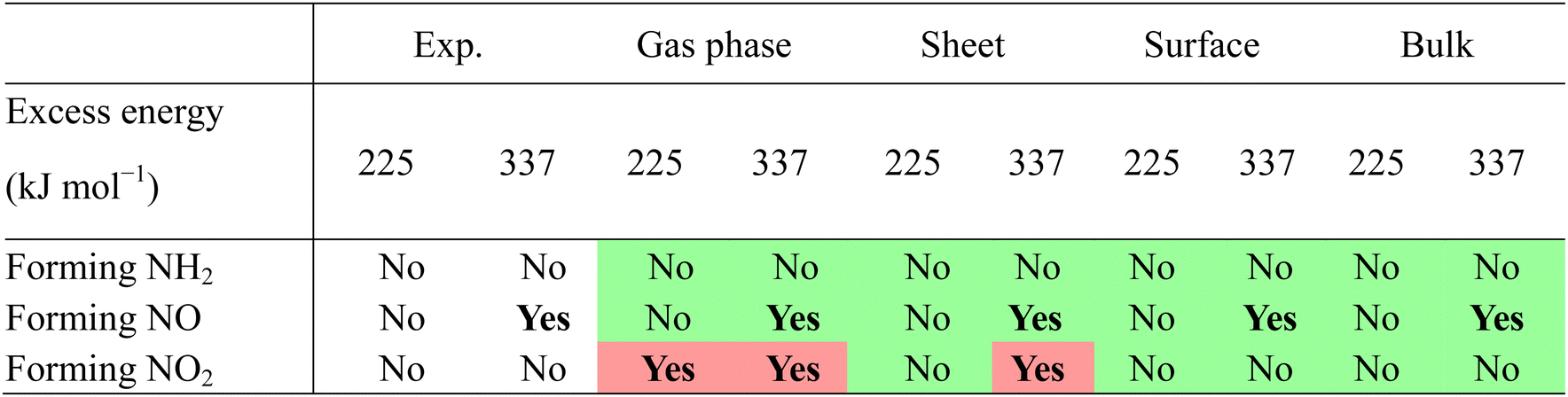
As this study represents the most comprehensive computational account to date for the initial decomposition of α-FOX-7 in the condensed phase, it is interesting to compare with the experiments by Turner and co-workers where the system was given just enough excess energy to observe the initial decomposition.20 In the study reported by Turner and co-workers, α-FOX-7 was excited at two levels: 225 kJ mol−1 (532 nm) and 337 kJ mol−1 (355 nm), and the results that are relevant to this study can be summarized as follows:
(1) The NO loss channel (through ts1 and/or ts1′) is not accessible at 225 kJ mol−1, but is accessible at 337 kJ mol−1.
(2) Neither NO2 nor NH2 were formed at both excitation levels.
(3) The structure of α-FOX-7 had negligible change after the excitation; thus, an overwhelming majority of the molecules in the crystal remained intact.
The comparison between the computations and experiment can be found in Table 3. The comparison was made based on the assumption that the excess energy of the system equals to the energy of the photon, which determines whether a reaction channel is accessible. The most exothermic product, NH2, cannot be formed in any of the computed environments with either level of excitation, in good agreement with the experiment. Regarding the NO product, it is quite interesting to note that the computations in all environments can reproduce the experimental result (NO not formed at 225 kJ mol−1, NO formed at 337 kJ mol−1). These results suggest that although the gas phase calculation is a grotesque estimation of the condensed phase system, errors from all sources cancel out. To further distinguish the performance of the calculations in different environments, more experiments with excitation energy between 225 and 337 kJ mol−1 are required. The formation of NO2via path (i) seems unfavourable on a surface and bulk using the photons of both energies. In contrast, the formation of NO2 highlights that a realistic environment is key in comparison with the experiment. The barrier (ts3) and reaction energies (NO2 + int5/int6) computed in the gas phase are severely underestimated; thus, the formation of NO2 should be allowed at both 225 and 337 kJ mol−1. The barrier and reaction energies computed in the sheet are a step in the right direction in mimicking the decomposition in the condensed phase, and predict that NO2 would only be formed at 337 kJ mol−1. It indeed takes a realistic model (the surface and bulk) to correctly characterize the energy profile of this decomposition pathway that prevents the formation of NO2 at neither 225 nor 337 kJ mol−1. As Table 3 shows, the decomposition of FOX-7 using the surface and bulk model was found to be in great agreement with the experimental observations.
|
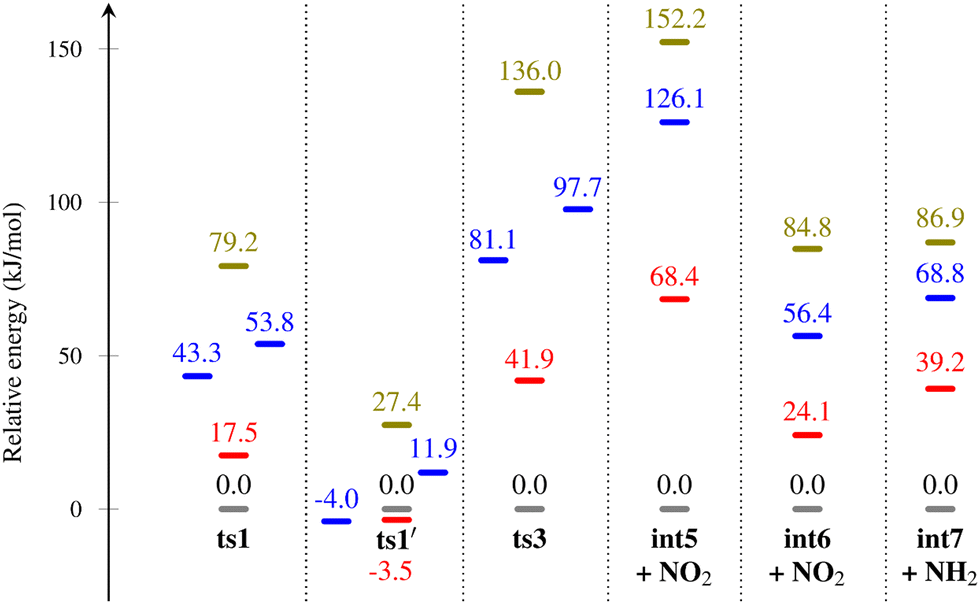 | ||
| Fig. 8 Change in relative energy for different species optimized in various environments: gas-phase (grey); sheet (red); surface (blue); bulk (olive). The relative energies of different species optimized in the gas phase are set to zero and reported in kJ mol−1. | ||
It is also of interest to compare the results with the study from Luo et al., where the energetics of the initial decomposition were computed with an additive model. Similar to this research, this model assumes that only the central molecule is reactive in a 3-layered α-FOX-7 crystal. Restraining the neighbouring molecules to their crystal structure, Luo et al. simply replaced the central FOX-7 molecule with various stationary points (e.g., ts1, ts3, etc.) optimized in the gas phase. The energy of these hybrid systems (gas phase optimal structure plus condensed phase environment) was used as the energetics of the decomposition in the condensed phase. Luo et al.'s additive model is an attempt to balance the computational cost and the realism of the model, and their results exhibited better agreement with the experiment than the energetics computed in the gas phase. However, it is worth pointing out that the reaction occurring in a crystal structure could be directly impacted by the neighbouring molecules, and not letting them relax could undermine the validity of the results. The additive model predicts that NO2 can be observed at 337 kJ mol−1 and NO can be observed at both 225 and 337 kJ mol−1, in clear contrast to the experimental results. This discrepancy in experimental observations is overcome by considering the immediate neighbours around the reactive center during a stationary point search in our calculations.
Among all the scenarios investigated, only the decompositions on a surface and in the bulk mimic the experiments in the condensed phase. Between the two, the initial decomposition (e.g., forming NO via a nitro-to-nitrate isomerization) takes place more easily in the molecules on the surface than in the bulk. In other words, the chemical bond in the –NO2 moiety is the weakest when it is on the surface of the crystal. Given that the formation of NO is exothermic, the decomposition of α-FOX-7 is hypothesized as follows:
(1) The FOX-7 molecules on the surface of the crystal are more sensitive (i.e., less stable) than those in bulk because they are restrained by a lower amount of intermolecular hydrogen bonds. They are more likely to be involved in the initial decomposition, and create defects that destabilize molecules in the bulk.
(2) The decomposition of molecules in the bulk undermines the integrity of the crystal structure, which in turn reduces the energy barriers for the initial decomposition of more molecules.
(3) More molecules decompose, and the crystal structure is completely destroyed. Enough energy is released for the explosion.
As noted earlier, although experiments with more levels of excitation are required to further distinguish all the models employed in this manuscript, the surface and bulk models can accurately describe the decomposition of α-FOX-7. It is of interest to examine the performance of these models with other energetic materials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the support from Naval Research (A9550-21-1-0221). The authors acknowledge the Information Technology Service (ITS) from the University of Hawaii at Manoa for the computational resources.References
- N. V. Latypov, J. Bergman, A. Langlet, U. Wellmar and U. Bemm, Synthesis and Reactions of 1, 1-Diamino-2, 2-Dinitroethylene, Tetrahedron, 1998, 54(38), 11525–11536 CrossRef CAS.
- M. Anniyappan, M. B. Talawar, G. M. Gore, S. Venugopalan and B. R. Gandhe, Synthesis, Characterization and Thermolysis of 1, 1-Diamino-2, 2-Dinitroethylene (FOX-7) and Its Salts, J. Hazard. Mater., 2006, 137(2), 812–819 CrossRef CAS PubMed.
- N. V. Latypov, M. Johansson, E. Holmgren, E. V. Sizova, V. V. Sizov and A. J. Bellamy, On the Synthesis of 1, 1-Diamino-2, 2-Dinitroethene (FOX-7) by Nitration of 4, 6-Dihydroxy-2-Methylpyrimidine, Org. Process Res. Dev., 2007, 11(1), 56–59 CrossRef CAS.
- X. Zhao, D. He, X. Ma, X. Liu, Z. Xu, L. Chen and J. Wang, Preparation, Characterization of Spherical 1, 1-Diamino-2, 2-Dinitroethene (FOX-7), and Study of Its Thermal Decomposition Characteristics, RSC Adv., 2021, 11(53), 33522–33530 RSC.
- G. F. Adams and R. W. Shaw Jr, Chemical Reactions in Energetic Materials, Annu. Rev. Phys. Chem., 1992, 43(1), 311–340 CrossRef CAS.
- A. K. Sikder and N. Sikder, A Review of Advanced High Performance, Insensitive and Thermally Stable Energetic Materials Emerging for Military and Space Applications, J. Hazard. Mater., 2004, 112(1–2), 1–15 CrossRef CAS PubMed.
- D. M. Badgujar, M. B. Talawar, S. N. Asthana and P. P. Mahulikar, Advances in Science and Technology of Modern Energetic Materials: An Overview, J. Hazard. Mater., 2008, 151(2–3), 289–305 CrossRef CAS PubMed.
- T. B. Brill and K. J. James, Kinetics and Mechanisms of Thermal Decomposition of Nitroaromatic Explosives, Chem. Rev., 1993, 93(8), 2667–2692 CrossRef CAS.
- G. Majano, S. Mintova, T. Bein and T. M. Klapötke, Confined Detection of High-Energy-Density Materials, J. Phys. Chem. C, 2007, 111(18), 6694–6699 CrossRef CAS.
- A. A. L. Michalchuk, S. Rudić, C. R. Pulham and C. A. Morrison, Predicting the Impact Sensitivity of a Polymorphic High Explosive: The Curious Case of FOX-7, Chem. Commun., 2021, 57(85), 11213–11216 RSC.
- G. D. Kozak, Factors Augmenting the Detonability of Energetic Materials, Propellants, Explos., Pyrotech., 2005, 30(4), 291–297 CrossRef CAS.
- R. P. Singh, R. D. Verma, D. T. Meshri and J. M. Shreeve, Energetic Nitrogen-Rich Salts and Ionic Liquids, Angew. Chem., Int. Ed., 2006, 45(22), 3584–3601 CrossRef CAS PubMed.
- U. Bemm and H. Östmark, 1,1-Diamino-2,2-Dinitroethylene: A Novel Energetic Material with Infinite Layers in Two Dimensions, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1998, 54(12), 1997–1999, DOI:10.1107/S0108270198007987.
- W. Trzciński and A. Belaada, 1, 1-Diamino-2, 2-Dinitroethene (DADNE, FOX-7)-Properties and Formulations (a Review), Cent. Eur. J. Energ. Mater., 2016, 13(2), 527–544 CrossRef PubMed.
- P. B. Kempa and M. Herrmann, Temperature Resolved X-Ray Diffraction for the Investigation of the Phase Transitions of FOX-7, Part. Part. Syst. Charact., 2005, 22(6), 418–422 CrossRef.
- H. Ostmark, H. Bergman, U. Bemm, P. Goede, E. Holmgren, M. Johansson, A. Langlet, N. Latypov, A. Pettersson and M.-L. Pettersson, 2,2-Dinitro-Ethene-1, 1-Diamine(FOX-7)-Properties, Analysis and Scale-Up, Energetic materials-Ignition, combustion and detonation, Karlsruhe, Germany, 2001, pp. 21–26.
- J. M. Winey, K. Zimmerman, Z. A. Dreger and Y. M. Gupta, Structural Transformation and Chemical Stability of a Shock-Compressed Insensitive High Explosive Single Crystal: Time-Resolved Raman Spectroscopy, J. Phys. Chem. A, 2020, 124(32), 6521–6527 CrossRef CAS PubMed.
- J. I. N. Peng-gang, C. Hai and C. Zhi-qun, Studies on Kinetics and Mechanisms of Thermal Decomposition of 1, 1-Diamino-2, 2-Dinitroethylene (FOX-7), Explos. Shock Waves, 2006, 26(6), 528–531 Search PubMed.
- M. Civis, S. Civis, K. Sovová, K. Dryahina, P. Španěl and M. Kyncl, Laser Ablation of FOX-7: Proposed Mechanism of Decomposition, Anal. Chem., 2011, 83(3), 1069–1077 CrossRef CAS PubMed.
- A. M. Turner, Y. Luo, J. H. Marks, R. Sun, J. T. Lechner, T. M. Klapötke and R. I. Kaiser, Exploring the Photochemistry of Solid 1, 1-Diamino-2, 2-Dinitroethylene (FOX-7) Spanning Simple Bond Ruptures, Nitro-to-Nitrite Isomerization, and Nonadiabatic Dynamics, J. Phys. Chem. A, 2022, 126(29), 4747–4761 CrossRef CAS PubMed.
- A. M. Turner, J. H. Marks, Y. Luo, J. T. Lechner, T. M. Klapötke, R. Sun and R. I. Kaiser, Electron-Induced Decomposition of Solid 1, 1-Diamino-2, 2-Dinitroethylene (FOX-7) at Cryogenic Temperatures, J. Phys. Chem. A, 2023, 127(15), 3390–3401 CrossRef CAS PubMed.
- A. M. Turner, J. H. Marks, J. T. Lechner, T. M. Klapötke, R. Sun and R. I. Kaiser, Ultraviolet-Initiated Decomposition of Solid 1,1-Diamino-2,2-Dinitroethylene (FOX-7), J. Phys. Chem. A, 2023, 127(37), 7707–7717, DOI:10.1021/acs.jpca.3c03215.
- V. P. Sinditskii, A. A. Kushtaev, N. V. Yudin, A. I. Levshenkov, N. N. Kondakova and M. A. Alekseeva, 1, 1-Diamino-2, 2-Dinitroethylene: The Riddles of Thermal Decomposition and Combustion, J. Phys. Chem. Solids, 2023, 177, 111275 CrossRef CAS.
- D. Jones, M. Vachon, R. Wang and Q. Kwok, Preliminary Studies on the Thermal Properties of FOX-7, In Proc. NATAS Annu. Conf. Therm. Anal. Appl. 32nd, 2004, pp. 1–105.
- A. M. Turner, J. H. Marks, Y. Luo, J. T. Lechner, T. M. Klapötke, R. Sun and R. I. Kaiser, Electron-Induced Decomposition of Solid 1,1-Diamino-2,2-Dinitroethylene (FOX-7) at Cryogenic Temperatures, J. Chem. Phys. A, 2023, 127(15), 3390–3401, DOI:10.1021/acs.jpca.3c01035.
- P. Politzer, M. C. Concha, M. E. Grice, J. S. Murray and P. Lane, Computational Investigation of the Structures and Relative Stabilities of Amino/Nitro Derivatives of Ethylene, J. Mol. Struct., 1998, 452(1–3), 75–83 CrossRef CAS.
- A. V. Kimmel, P. V. Sushko, A. L. Shluger and M. M. Kuklja, Effect of Charged and Excited States on the Decomposition of 1,1-Diamino-2,2-Dinitroethylene Molecules, J. Chem. Phys., 2007, 126(23), 234711, DOI:10.1063/1.2741530.
- A. Gindulytė, L. Massa, L. Huang and J. Karle, Proposed Mechanism of 1, 1-Diamino-Dinitroethylene Decomposition: A Density Functional Theory Study, J. Phys. Chem. A, 1999, 103(50), 11045–11051 CrossRef.
- A. V. Kimmel, P. V. Sushko, A. L. Shluger and M. M. Kuklja, Effect of Charged and Excited States on the Decomposition of 1, 1-Diamino-2, 2-Dinitroethylene Molecules, J. Chem. Phys., 2007, 126(23), 234711 CrossRef PubMed.
- R. S. Booth and L. J. Butler, Thermal Decomposition Pathways for 1, 1-Diamino-2, 2-Dinitroethene (FOX-7), J. Chem. Phys., 2014, 141(13), 134315 CrossRef PubMed.
- B. Yuan, Z. Yu and E. R. Bernstein, Initial Decomposition Mechanism for the Energy Release from Electronically Excited Energetic Materials: FOX-7 (1, 1-Diamino-2, 2-Dinitroethene, C2H4N4O4), J. Chem. Phys., 2014, 140(7), 074708 CrossRef PubMed.
- V. G. Kiselev and N. P. Gritsan, Unexpected Primary Reactions for Thermolysis of 1,1-Diamino-2,2-Dinitroethylene (FOX-7) Revealed by Ab Initio Calculations, J. Phys. Chem. A, 2014, 118(36), 8002–8008 CrossRef CAS PubMed.
- Y. Luo, C. Kang, R. Kaiser and R. Sun, The Potential Energy Profile of the Decomposition of 1, 1-Diamino-2, 2-Dinitroethylene (FOX-7) in the Gas Phase, Phys. Chem. Chem. Phys., 2022, 24(43), 26836–26847 RSC.
- Y. Guan, X. Zhu, Y. Gao, H. Ma and J. Song, Initial Thermal Decomposition Mechanism of (NH2)2C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(NO2)(ONO) Revealed by Double-Hybrid Density Functional Calculations, ACS Omega, 2021, 6(23), 15292–15299 CrossRef CAS PubMed.
C(NO2)(ONO) Revealed by Double-Hybrid Density Functional Calculations, ACS Omega, 2021, 6(23), 15292–15299 CrossRef CAS PubMed. - S. N. Rashkeev, M. M. Kuklja and F. J. Zerilli, Electronic Excitations and Decomposition of 1, 1-Diamino-2, 2-Dinitroethylene, Appl. Phys. Lett., 2003, 82(9), 1371–1373 CrossRef CAS.
- A. Hu, B. Larade, H. Abou-Rachid, L.-S. Lussier and H. Guo, A First Principles Density Functional Study of Crystalline FOX-7 Chemical Decomposition Process under External Pressure, Propellants, Explos., Pyrotech., 2006, 31(5), 355–360 CrossRef CAS.
- J.-D. Zhang and L.-L. Zhang, Theoretical Study on the Mechanism of the Reaction of FOX-7 with OH and NO2 Radicals: Bimolecular Reactions with Low Barrier during the Decomposition of FOX-7, Mol. Phys., 2017, 115(23), 2951–2960 CrossRef CAS.
- Q. Zhu, P. Zhou, J. Liu and S. Yin, Theoretical Study on the Thermal Dissociation of FOX-7 Promoted by NO2, Int. J. Quantum Chem., 2022, 122(7), e26864 CrossRef CAS.
- X. Bidault and S. Chaudhuri, How Accurate Can Crystal Structure Predictions Be for High-Energy Molecular Crystals?, Molecules, 2023, 28(11), 4471 CrossRef CAS PubMed.
- X.-H. Ju, H.-M. Xiao and Q.-Y. Xia, A Density Functional Theory Investigation of 1, 1-Diamino-2, 2-Dinitroethylene Dimers and Crystal, J. Chem. Phys., 2003, 119(19), 10247–10255 CrossRef CAS.
- J. Zhao and H. Liu, High-Pressure Behavior of Crystalline FOX-7 by Density Functional Theory Calculations, Comput. Mater. Sci., 2008, 42(4), 698–703 CrossRef CAS.
- Z. Zheng, J. Xu and J. Zhao, First-Principles Studies on the Thermal Decomposition Behavior of FOX-7, High Pressure Res., 2010, 30(2), 301–309 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu, J. Chem. Phys., 2010, 132(15), 154104 CrossRef PubMed.
- W. J. Hehre, R. Ditchfield and J. A. Pople, Self—Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian—Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules, J. Chem. Phys., 1972, 56(5), 2257–2261 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy, Phys. Chem. Chem. Phys., 2005, 7(18), 3297–3305 RSC.
- D. E. Taylor, F. Rob, B. M. Rice, R. Podeszwa and K. Szalewicz, A Molecular Dynamics Study of 1, 1-Diamino-2, 2-Dinitroethylene (FOX-7) Crystal Using a Symmetry Adapted Perturbation Theory-Based Intermolecular Force Field, Phys. Chem. Chem. Phys., 2011, 13(37), 16629–16636 RSC.
- C. J. Eckhardt and A. Gavezzotti, Computer Simulations and Analysis of Structural and Energetic Features of Some Crystalline Energetic Materials, J. Phys. Chem. B, 2007, 111(13), 3430–3437 CrossRef CAS PubMed.
- J.-Y. Fan, Z.-Y. Zheng, Y. Su and J.-J. Zhao, Assessment of Dispersion Correction Methods within Density Functional Theory for Energetic Materials, Mol. Simul., 2017, 43(7), 568–574 CrossRef CAS.
- S. Grimme, A. Hansen, J. G. Brandenburg and C. Bannwarth, Dispersion-Corrected Mean-Field Electronic Structure Methods, Chem. Rev., 2016, 116(9), 5105–5154, DOI:10.1021/acs.chemrev.5b00533.
- K. Fukui, Formulation of the Reaction Coordinate, J. Phys. Chem., 1970, 74(23), 4161–4163 CrossRef CAS.
- J.-D. Chai and M. Head-Gordon, Long-Range Corrected Hybrid Density Functionals with Damped Atom–Atom Dispersion Corrections, Phys. Chem. Chem. Phys., 2008, 10(44), 6615–6620 RSC.
- I. M. Alecu and D. G. Truhlar, Computational Study of the Reactions of Methanol with the Hydroperoxyl and Methyl Radicals. 1. Accurate Thermochemistry and Barrier Heights, J. Phys. Chem. A, 2011, 115(13), 2811–2829 CrossRef CAS PubMed.
- G. L. C. de Souza and K. A. Peterson, Benchmarking Antioxidant-Related Properties for Gallic Acid through the Use of DFT, MP2, CCSD, and CCSD(T) Approaches, J. Phys. Chem. A, 2021, 125(1), 198–208 CrossRef CAS PubMed.
- E. Blokker, W.-J. van Zeist, X. Sun, J. Poater, J. M. van der Schuur, T. A. Hamlin and F. M. Bickelhaupt, Methyl Substitution Destabilizes Alkyl Radicals, Angew. Chem., Int. Ed., 2022, 61(36), e202207477 CrossRef CAS PubMed.
- D. Dwinandha, B. Zhang and M. Fujii, Prediction of Reaction Mechanism for OH Radical-Mediated Phenol Oxidation Using Quantum Chemical Calculation, Chemosphere, 2022, 291, 132763 CrossRef CAS PubMed.
- B. Long, Z.-W. Long, Y.-B. Wang, W.-J. Zhang, C.-Y. Long and S.-J. Qin, Theoretical Study on HO2-Initiated Atmospheric Oxidation of Halogenated Carbonyls, Int. J. Quantum Chem., 2012, 112(8), 1926–1935 CrossRef CAS.
- S. Nabizadeh and M. Hamzehloueian, Understanding the Mechanism of [3+2] Cycloaddition Reaction of Benzoisothiazole-2,2-Dioxide-3-Ylidene with Nitrones, Theor. Chem. Acc., 2020, 139(4), 72 Search PubMed.
- A. Yadav and P. C. Mishra, Formation of Spiroiminodihydantoin Due to the Reaction between 8-Oxoguanine and Carbonate Radical Anion: A Quantum Computational Study, Chem. Phys. Lett., 2014, 592, 232–237 CrossRef CAS.
- C. Sobhi, A. Khorief Nacereddine, A. Djerourou, M. Ríos- Gutiérrez and L. R. Domingo, A DFT Study of the Mechanism and Selectivities of the [3+2] Cycloaddition Reaction between 3-(Benzylideneamino)Oxindole and Trans-β-Nitrostyrene, J. Phys. Org. Chem., 2017, 30(6), e3637 CrossRef.
- Y. Zhao and D. G. Truhlar, How Well Can New-Generation Density Functionals Describe the Energetics of Bond-Dissociation Reactions Producing Radicals?, J. Phys. Chem. A, 2008, 112(6), 1095–1099, DOI:10.1021/jp7109127.
- M. J. Frisch, et al., Gaussian 16 Revision C.01, 2016 Search PubMed.
- U. Bemm and H. Östmark, 1, 1-Diamino-2, 2-Dinitroethylene: A Novel Energetic Material with Infinite Layers in Two Dimensions, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1998, 54(12), 1997–1999 CrossRef.
- D. C. Sorescu, J. A. Boatz and D. L. Thompson, Classical and Quantum-Mechanical Studies of Crystalline FOX-7 (1, 1-Diamino-2, 2-Dinitroethylene), J. Phys. Chem. A, 2001, 105(20), 5010–5021 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp00001c |
| This journal is © the Owner Societies 2024 |