
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Characteristics and long-term kinetics of an azobenzene derivative and a donor–acceptor Stenhouse adduct as orthogonal photoswitches†
Tanja
Schmitt
 a,
Christian
Huck
a,
Nils
Oberhof
b,
Li-Yun
Hsu
c,
Eva
Blasco
c,
Andreas
Dreuw
b and
Petra
Tegeder
*a
a,
Christian
Huck
a,
Nils
Oberhof
b,
Li-Yun
Hsu
c,
Eva
Blasco
c,
Andreas
Dreuw
b and
Petra
Tegeder
*a
aPhysikalisch-Chemisches Institut, Ruprecht-Karls-Universität Heidelberg, 69120 Heidelberg, Germany. E-mail: tegeder@uni-heidelberg.de
bInterdisziplinäres Zentrum für Wissenschaftliches Rechnen, Ruprecht-Karls-Universität Heidelberg, 69120 Heidelberg, Germany
cInstitute for Molecular System Engineering and Advanced Materials, Ruprecht-Karls-Universität Heidelberg, 69120 Heidelberg, Germany
First published on 8th February 2024
Abstract
Light-triggered molecular switches are extensively researched for their applications in medicine, chemistry and material science and, if combined, particularly for their use in multifunctional smart materials, for which orthogonally, i.e. individually, addressable photoswitches are needed. In such a multifunctional mixture, the switching properties, efficiencies and the overall performance may be impaired by undesired mutual dependences of the photoswitches on each other. Within this study, we compare the performance of the pure photoswitches, namely an azobenzene derivative (Azo) and a donor–acceptor Stenhouse adduct (DASA), with the switching properties of their mixture using time-resolved temperature-dependent UV/VIS absorption spectroscopy, time-resolved IR absorption spectroscopy at room temperature and quantum mechanical calculations to determine effective cross sections, switching kinetics as well as activation energies of thermally induced steps. We find slightly improved effective cross sections, percentages of switched molecules and no increased activation barriers of the equimolar mixture compared to the single compounds. Thus, the studied mixture Azo + DASA is very promising for future applications in multifunctional smart materials.
Introduction
Nowadays, molecular switches are extensively applied in biology, chemistry and material science.1–7 The switching is initiated by external stimuli such as light, temperature, pH or electric fields. Light-triggered switching is of particular interest since light offers high spatiotemporal resolution while being omnipresent and sustainable.5,8 Hence, molecular photoswitches are widely studied and used in e.g. information storage, biomedicine, functionalised surfaces and nanotechnology.1,4,9–16 To date, several classes of photoswitches are available e.g. azobenzenes, diarylethenes, spiropyranes and the novel class of donor–acceptor Stenhouse adducts.17–20 Each class is characterised by distinct properties. Azobenzenes, for instance, show absorption in the UV and blue spectral region, which stimulates a reversible E/Z isomerisation,18,21–23 and hold great potential for applications in robotics, medicine and solar thermal batteries.24–26 Similar to azobenzenes, diarylethenes, spiropyranes and most other classes of photoresponsive switches are addressed by UV or blue light which induces reversible E/Z isomerisation or, as in the case of spiropyranes, reversible ring closure/opening. In contrast, the class of donor–acceptor Stenhouse adducts exhibits tuneable absorption at longer wavelengths ranging from the green to the red region of the electromagnetic spectrum.17,27 Donor–acceptor Stenhouse adducts offer high fatigue resistance and a high change in solubility upon switching.17In order to create multifunctional smart materials, components with orthogonal, i.e. independent, addressability need to be combined. Research in such materials is a growing field providing solutions e.g. for robotics, lithography and DNA nanotechnology.10,28,29 However, for orthogonal photo-addressability materials with different excitation wavelengths are required. Thus, most of the well-known classes absorbing in the UV and blue spectral region cannot be combined. Therefore, a promising way is to combine compounds of the well-known classes of photoswitches with donor–acceptor Stenhouse adducts.
Recently, Hsu et al. used a mixture of an azobenzene derivative (Azo, Fig. 1a) and a donor–acceptor Stenhouse adduct (DASA, Fig. 1b) to demonstrate the first 4D printed multifunctional smart material with wavelength-selective microactuator properties.28 Thereby 4D printing terms the 3D printing of smart materials reacting to external stimuli with the time upon response as the fourth dimension.30–33 In order to demonstrate the orthogonal addressability of the Azo + DASA mixture, which is important for the performance of such a multifunctional smart material, we recently studied the photophysical properties and especially the ultrafast excited states dynamics of the single compounds and the mixture by means of static UV/VIS spectroscopy and femtosecond time-resolved transient absorption spectroscopy.34 Therein, we investigated the reversible E/Z isomerisation of Azo (Fig. 1a) and reversible Z/E isomerisation of DASA (Fig. 1b) using different environments, namely toluene as non-polar solvent, acetonitrile as polar solvent and PMMA-blend thin films, for all possible combinations of switching one or both photoresponsive molecules. Especially important for the performance of such a multifunctional smart material is whether there are modified switching properties or undesired mutual dependencies in the mixture compared to the pure compounds. For the ultrafast dynamics, we found no relevant interactions diminishing the performance of the photoswitches.34 However, since important steps of the switching processes are thermally activated and occur on longer time scales, different experimental methods are needed to elucidate the dynamics of these steps and complete the picture upon switching the orthogonally addressable mixture of the photoswitches Azo and DASA.
 | ||
| Fig. 1 Reversible switching of the molecular switches (a) Azo and (b) DASA. The absorption spectra in toluene of (c) Azo, DASA and their equimolar mixture Azo + DASA show the orthogonal addressability of the photoswitches in the mixture. | ||
In this work, we utilise time-resolved temperature-dependent UV/VIS absorption spectroscopy and time-resolved IR absorption spectroscopy at room temperature both on longer time scales to study the dynamics of the orthogonally addressable mixture Azo + DASA and the respective pure compounds upon switching. In addition, quantum chemical calculations are utilised. By using time-resolved temperature-dependent UV/VIS absorption spectroscopy, we determined the effective cross sections of the photoinduced switching and the activation barriers of the thermally driven switching processes. Additionally, the last step of switching DASA, the proton transfer, is analysed by kinetic IR absorption spectroscopy. We find small changes resulting in optimised switching properties, e.g. higher effective cross sections, in the mixture compared to the pure compounds, while the activation energies are mainly unchanged.
Experimental and computational details
Temperature-controlled kinetic UV/VIS measurements
The temperature-controlled kinetic UV/VIS measurements were performed using an Agilent 8453 spectrometer with a USP 203 A cooling unit from Unisoku. The measurements were conducted under inert gas atmosphere using cuvettes with 10 mm optical pathway. The integration time for a single measurement was kept at 500 ms. The solutions were constantly stirred to guarantee homogeneous samples and kinetics. Before starting a kinetic measurement series, steady state conditions are ensured by subsequent measurements until no changes are observed. To induce photoswitching, LEDs (mounted LEDs M430L5, M590L4 and M505L4 with focusing unit from Thorlabs) were used. For orthogonal Azo photoswitching, the LED with maximum intensity at a wavelength of 434 nm held an output power of 2.78 mW. For orthogonal DASA photoswitching, the LED with maximum intensity at a wavelength of 594 nm held an output power of 4.85 mW. For simultaneous photoswitching of Azo and DASA, the LED with maximum intensity at a wavelength of 507 nm hold an output power of 20.04 mW.Kinetic IR absorption measurements
Kinetic IR absorption measurements at room temperature were performed using the FTIR spectrometer Tensor 27 from Bruker equipped with the flow-through measuring cell Aquaspec which contains a calcium fluoride measuring cell with an optical pathway of about 7 μm. The whole FTIR spectrometer was purged with dry air to avoid atmospheric absorptions. The integration time was kept at 6 s. The solutions were injected into the flow-through system which includes a filter prior to the transmission cell to ensure complete solvation. Before starting a kinetic measurement series, steady state conditions are ensured by subsequent measurements until no changes are observed. To induce photoswitching of DASA, an LED (mounted LED M590L4 with focusing unit from Thorlabs) with maximum intensity at a wavelength of 594 nm held an output power after reflection on a high-reflective mirror of 2.5 mW and 56.1 mW for toluene and acetonitrile solution, respectively.DFT calculations
In order to relate the experimentally observed IR absorption bands to the different DASA isomers, DFT calculations were carried out. Therefore, the B3LYP functional, the 6-311G(d,p) basis set and a scaling factor of 0.9682 was used. The literature known scaling factor35 was validated by plotting the calculated frequencies against the experimentally observed frequencies of DASA in an KBr pellet with no environmental influences and applying a linear fit curve. The calculations of the energy landscape to extract the activation energies were performed using the Qchem 5.3 software packages.36 Ground state geometry optimizations were done at the density functional theory (DFT) level, employing the range-separated hybrid exchange–correlation (xc)-functional ωB97X-D3 with the 6-311G(d,p) basis set.37,38 Solvation was considered using the conductor-like screening model (COSMO, acetonitrile ε = 37.5).39 For the approximate transition states, the freezing-string method was used as implemented and afterwards the received structures were further refined employing a P-RFO algorithm.40,41 Subsequent frequency calculations verified the geometries as local minima with no imaginary frequencies or as transition states with exactly one imaginary frequency (along the reaction coordinate). For computational efficiency, the long alkyl chains of the azobenzene derivative were substituted with short methyl chains. This is expected to have little to no influence on the general potential energy surface landscape of the system.Results and discussion
Optical properties of the molecular switches
The studied molecular photoswitch Azo undergoes reversible light-induced E-to-Z isomerisation (Fig. 1a) via excitation with blue light with maximum absorption at around 435 nm (Fig. 1c and 2). The Z-to-E backswitching can be stimulated either photochemically with light of shorter wavelengths or thermally. The shorter wavelength of the Z-to-E photoisomerisation compared to E-to-Z isomerisation is caused by the push–pull character of the azobenzene substituents.34 In contrast, photoexcitation of DASA causes a linear-to-closed switching composed of a photochemically induced Z-to-E isomerisation of the linear Z-DASA (isomer A) followed by a thermally induced C–C bond rotation and electrocyclisation (isomer C) with subsequent thermally driven proton transfer to generate the zwitterionic end product (isomer C′, see Fig. 1b).17,42 The first step, the Z-to-E isomerisation, is initiated by absorption of green or yellow light with a maximum absorption at about 570 nm (Fig. 1c). All steps of the closed-to-linear backswitching are thermally activated. Due to the different excitation wavelengths, Azo and DASA can be addressed orthogonally in a mixture containing both molecular photoswitches. Due to a small spectral overlap, simultaneous switching of both photoswitches can still be induced by choosing a wavelength of e.g. 505 nm. This way, an additional wavelength selectivity exists which broadens the range of future applications.The absorption bands of both photoswitches are sensitive to the environment with Azo and DASA exhibiting positive and negative solvatochromism, respectively (Fig. S1, ESI†). For example, by changing the nonpolar solvent toluene to the polar solvent acetonitrile, the absorption maximum of Azo shifts about 10 nm towards longer wavelengths while the absorption maximum of DASA shifts around 20 nm towards shorter wavelengths. Quantum chemical calculations reveal a higher polar character of the excited state of Azo and a less polar character of the excited state of DASA compared to the respective ground states causing the observed solvatochromism.34 Hence, the absorption wavelength can be fine-tuned by altering the environment.
Time-resolved UV/VIS spectroscopy: dynamics, cross sections and activation energies
Upon irradiation with blue light using an LED with maximum emission at around 434 nm, Azo undergoes E-to-Z isomerisation (Fig. 2a and b). During illumination, the intensity of the absorption band of the thermodynamically stable E-Azo isomer at about 450 nm decreases while the hypsochromically shifted absorption band of Z-Azo at around 380 nm increases (Fig. 2a). The thermally activated backswitching, i.e. the Z-to-E isomerisation, shows the reverse process in the absorption data (Fig. 2b). These switching processes of Azo can also be initiated in an equimolar mixture of Azo and DASA (Fig. 2c and d). Upon E-to-Z photoisomerisation of Azo, the Z-DASA signal stays basically constant whereas a small increase in Z-DASA absorption is observed upon thermally activated Z-to-E isomerisation of Azo with the reason being subject of current research. By changing the excitation wavelength to green light with maximum LED emission at about 507 nm, Azo and DASA are addressed simultaneously with successful E-to-Z isomerisation of Azo and the same effect of slightly increased Z-DASA absorption upon thermally activated Z-to-E isomerisation of Azo is observed (Fig. S2e and f, ESI†). In order to compare the photo- and thermally induced isomerisation processes in terms of their kinetics and efficiencies, the maximum absorption of the thermodynamically stable E-Azo isomer is studied at around 450 nm which slightly hypsochromically shifts with increasing temperature (Fig. S42a, ESI†). By monitoring the temporal evolution of the maximum absorption wavelength of E-Azo upon E-to-Z photoisomerisation and extracting the lifetimes from monoexponential fit curves (Fig. S3, ESI†), the effective cross section is determined (Table S1, ESI†). It increases in the presence of DASA and is higher for orthogonal switching with maximum LED emission at 434 nm compared to simultaneous switching with maximum LED emission at 507 nm amounting to 6.8 × 10−18 cm2. Hence, the determined effective cross sections of Azo are in the typical range of other azobenzene derivatives in solution.43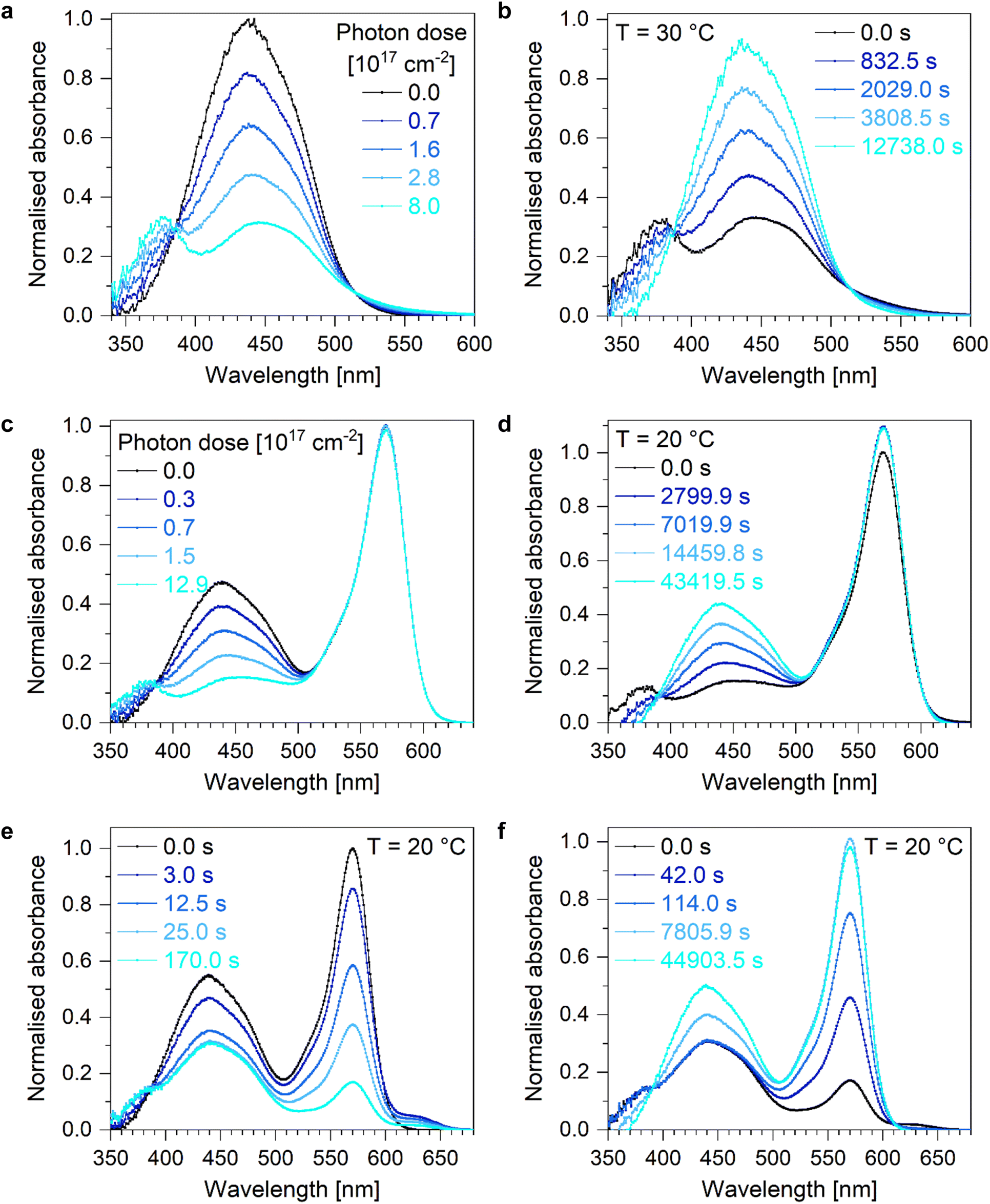 | ||
| Fig. 2 UV/VIS absorption spectra upon switching Azo. UV/VIS absorption spectra upon (a) photochemically initiated E-to-Z isomerisation of Azo in toluene using an LED with maximum emission at a wavelength of 434 nm and (b) thermally activated Z-to-E backswitching at 30 °C. The absorption maxima of E-Azo and Z-Azo are observed at around 450 and 380 nm, respectively. UV/VIS absorption spectra upon (c) photochemically initiated E-to-Z and (d) thermally activated Z-to-E isomerisation of Azo within an equimolar mixture of Azo and DASA in toluene at 20 °C. The absorption maxima of E-Azo at around 450 nm decreases in intensity while the absorption maxima of Z-Azo at about 380 nm increases in intensity upon E-to-Z isomerisation and vice versa. The absorption of Z-DASA stays basically constant upon E-to-Z isomerisation of Azo while it slightly increases upon Z-to-E isomerisation of Azo. UV/VIS absorption spectra upon (e) photochemically initiated E-to-Z isomerisation of Azo and simultaneous Z-to-E photoisomerisation of DASA and (f) thermally activated Z-to-E isomerisation of Azo and simultaneous E-to-Z isomerisation of DASA within an equimolar Azo and DASA mixture in toluene at 20 °C. | ||
Analysing the thermally initiated Z-to-E backswitching of Azo at different temperatures, the corresponding lifetimes are fitted to generate an Arrhenius plot in order to determine the activation energy (Fig. 3, lifetimes see Table S2, ESI†). The Arrhenius plot reveals an activation energy for thermally initiated Z-to-E backswitching reaction of 86.2 kJ mol−1 for a pure Azo in toluene solution (Table 1). Computing this activation barrier using density functional theory (DFT) and the ωB97X-D3 functional yields a value of 124 kJ mol−1 (Tables S13–S15, ESI†). The functional is chosen due to the presence of push-pull charge transfer and dispersion contributions although it is known to overestimate reaction barriers.44 Studying this isomerisation in the presence of Z-DASA within an equimolar Azo + DASA solution in toluene reveals a similar behaviour (Fig. S4, lifetimes see Table S2, ESI†) but a slightly smaller activation energy of 83.6 kJ mol−1 (Table 1). However, if the thermally initiated backreactions of Azo (Z-to-E) and DASA (E-to-Z) occur simultaneously (Fig. S5, lifetimes see Table S2, ESI†), the activation energy is highest at 89.0 kJ mol−1 (Table 1). Overall, the determined activation energies of Azo in different environments are similar to the activation energy of the parent azobenzene molecule which amounts to 95 kJ mol−1.18
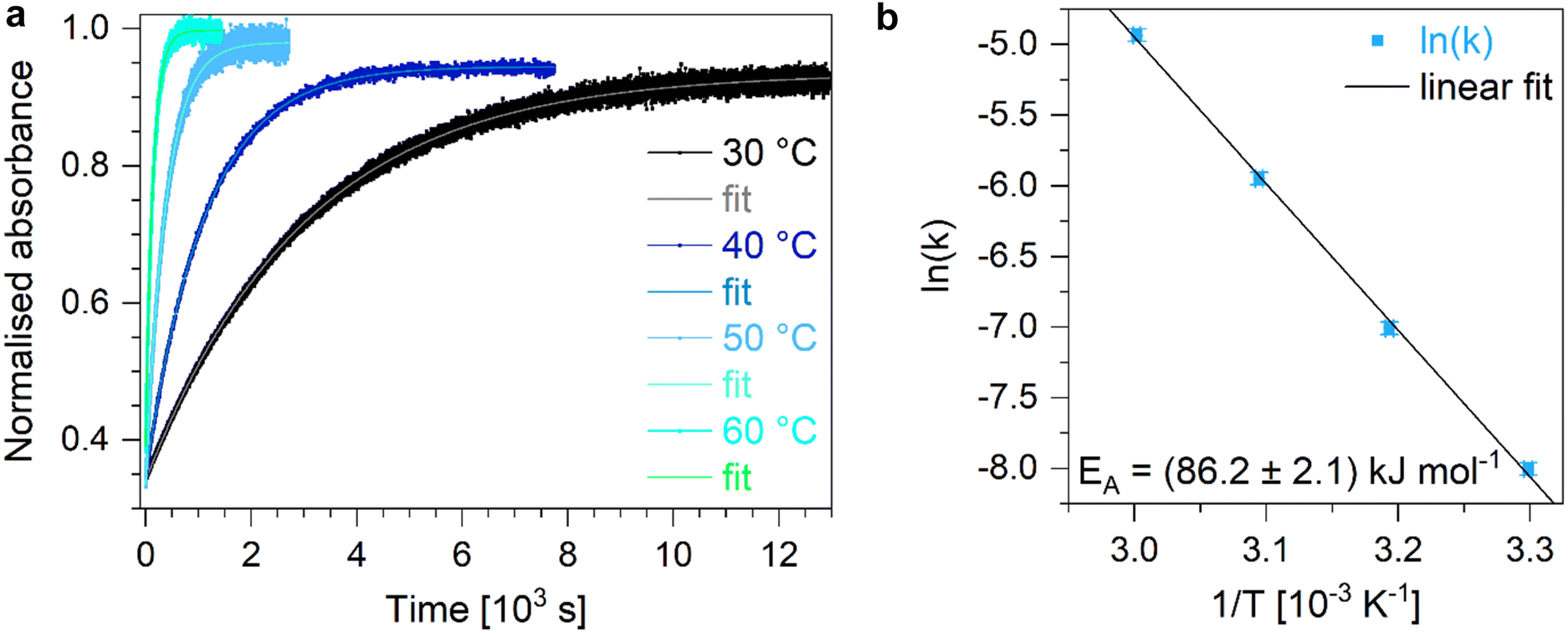 | ||
| Fig. 3 (a) Temporal evolution of the absorption of E-Azo at its maximum absorption wavelength upon thermally activated Z-to-E isomerisation of Azo in toluene at different temperatures with the respective monoexponential fit curves. Using the fitted lifetimes, the corresponding rate constants are calculated and an (b) Arrhenius plot is applied to extract the activation energy. | ||
| Activation energy of the Z-to-E isomerisation of Azo [kJ mol−1] | |
|---|---|
| Azo | 86.2 ± 2.1 |
| Azo + Z-DASA | 83.6 ± 1.4 |
| Azo + DASA upon simultaneous thermal backswitching | 89.0 ± 0.5 |
Since the switching of DASA includes several isomers, multiple absorption features are observed upon photoinduced linear-to-closed switching and thermally activated closed-to-linear backreaction (Fig. 4a and b). The linear Z-DASA isomer exhibits an absorption band at about 570 nm whereas the E-DASA isomers shows a bathochromically shifted absorption feature at around 625 nm while the closed isomers C and C′ exhibit no absorption feature in the experimentally available spectral window, but at higher energies at around 270 nm.17 The assignment of the absorption bands to the corresponding isomers has been validated by quantum chemical calculations.34 The linear/closed switching processes of DASA can also be initiated in an equimolar mixture of DASA + Azo when E-Azo is present (Fig. S4c and d, ESI†) or when Azo undergoes simultaneous E/Z isomerisation (Fig. S2e and f, ESI†). Note that the photostimulated Z-to-E isomerisation of DASA as well as the thermally stimulated E-to-Z isomerisation occur on the picosecond time scale which is orders of magnitude faster than the subsequent thermally initiated ring closure/opening reaction.34,45 Hence, the ring closure/opening is the rate determining step such that the absorption of E-DASA is clearly observed upon linear-to-closed switching (Fig. 4a) whereas it is barely observed upon closed-to-linear backswitching due to the fast interconversion to Z-DASA (Fig. 4b). Consequently, the temporal evolution of the Z-DASA absorption band is pseudo monoexponential and in order to characterise the ring closure and opening, it is thus sufficient to describe the absorption feature of Z-DASA by monoexponential fit curves (Fig. S6, ESI†).
 | ||
| Fig. 4 UV/VIS absorption spectra upon switching DASA. UV/VIS absorption spectra upon (a) photochemically initiated Z-to-E isomerisation and subsequent thermally activated ring closure and (b) thermally activated ring opening with subsequent E-to-Z isomerisation of DASA in toluene at 0 °C. The absorption maxima of the linear Z-DASA and E-DASA isomers are observed at about 570 and 625 nm, respectively, while the closed species exhibit no absorption in the studied UV/VIS region. UV/VIS absorption spectra upon (c) photochemically initiated Z-to-E isomerisation and subsequent thermally activated ring closure and (d) thermally activated ring opening with subsequent E-to-Z isomerisation of DASA within an equimolar mixture of Azo and DASA in toluene at 10 °C. The absorption features of Azo stay basically constant upon switching and backswitching. | ||
To determine the activation barrier of the thermally activated electrocyclisation of DASA in toluene, the maximum absorption wavelength of Z-DASA at around 570 nm is studied as a function of time at different temperatures (Fig. 5). The exact maximum absorption wavelength shifts slightly hypsochromically with increasing temperature (Fig. S42b, ESI†). Furthermore, the relative amount of closed isomer decreases with increasing temperature (Fig. 5a). Using the fitted lifetimes (Table S3, ESI†) and the resulting Arrhenius plot (Fig. 5b), the activation energy numbers 15.4 kJ mol−1 (Table 2). The measured low activation energy corresponds most likely not to the absolute energetic barrier but to an effective barrier. Our quantum chemical calculations yield barriers for the two subsequent steps of the C–C bond rotation and the ring closure of 69 kJ mol−1 and 59 kJ mol−1, respectively, with a difference of the total energies of the transition states of 6 kJ mol−1 (Tables S6–S12 and Fig. S18, ESI†). Since the reaction coordinate of both steps is very similar, molecules passing the first rotation barrier will most likely immediately undergo ring closure thereby having higher energy. Since the first step cannot be resolved experimentally, a smaller effective barrier is observed for the second step.
 | ||
| Fig. 5 (a) Temporal evolution of the absorption of Z-DASA at its maximum absorption wavelength upon linear-to-closed switching of DASA in toluene at different temperatures with the respective monoexponential fit curves. Using the fitted lifetimes, the corresponding rate constants are calculated and an (b) Arrhenius plot is applied to extract the activation energy for the thermally activated electrocyclisation. | ||
| Activation energies of DASA [kJ mol−1] | ||
|---|---|---|
| Electrocyclisation | Ring opening | |
| DASA | 15.4 ± 1.2 | 49.7 ± 2.2 |
| DASA + Azo | 15.6 ± 2.6 | 51.1 ± 2.3 |
| DASA + Azo upon si-multaneous switching | 21.7 ± 0.6 | 51.2 ± 1.3 |
Inducing the linear-to-closed switching of DASA in an equimolar mixture of DASA + Azo in the presence of E-Azo (Fig. S7, lifetimes see Table S3, ESI†) yields a similar value of the activation energy (Table 2). Inducing the linear-to-closed reaction of DASA while Azo undergoes simultaneous E-to-Z isomerisation (Fig. S8, lifetimes see Table S3, ESI†), however, increases the activation energy to 21.7 kJ mol−1 (Table 2).
Although the ring closure is thermally activated, the photoinitiated Z-to-E isomerisation has to occur first. Thus, the effective cross section for the linear-to-closed switching can be calculated based on the fitted lifetimes. The values are temperature dependent with higher effective cross sections at higher temperatures (Table S3, ESI†). Similar to Azo, the effective cross sections of DASA are higher for orthogonal switching compared to simultaneous switching of both photoswitches and they are higher within the equimolar mixture compared to pure DASA with a maximum effective cross section of 3.8 × 10−18 cm2 (Table S4, ESI†). Thus, the values are in the same order of magnitude than the effective cross sections of Azo.
In contrast to the linear-to-closed switching, all steps of the DASA closed-to-linear backreaction are thermally activated. Based on the lifetimes fitted to the temporal evolution of the Z-DASA absorption feature of a pure DASA in toluene solution and the respective Arrhenius plot (Fig. S9, lifetimes see Table S5, ESI†), an activation energy of 49.7 kJ mol−1 of the ring opening is determined (Table 2). The activation barrier is supported by our quantum chemical calculations (Tables S9–S11, ESI†). Inducing this closed-to-linear backswitching of DASA within an equimolar Azo + DASA mixture in toluene with E-Azo being present (Fig. S10, lifetimes see Table S5, ESI†) does not change the activation energy (Table 2). In contrast to the closed-to-linear backswitching of DASA in a pure DASA solution, the efficiency of this process is increased in the Azo + DASA mixture since the number of backswitched molecules is near 100% for all studied temperatures (Fig. S9a and S10a, ESI†). If the closed-to-linear backswitching of DASA is induced in an equimolar mixture of DASA + Azo in toluene while Azo undergoes simultaneous Z-to-E isomerisation, the kinetics of the ring opening are described within the first 1000 s by a monoexponential rise of the Z-DASA absorption band (Fig. S141, ESI†). The corresponding Arrhenius plot provides the same activation energy of the ring opening of DASA compared to a pure DASA in toluene solution (Table 2). In comparison to other donor–acceptor Stenhouse adducts, the determined activation energies for the ring closure and opening are lower but in the same order of magnitude.42,46,47
Time-resolved IR spectroscopy: dynamics
The switching process of DASA is not completed by the electrocyclisation but a proton transfer occurs afterwards to form the zwitterionic isomer C′ (Fig. 1b). Since the protonation does not affect the electronic absorption, time-resolved IR absorption measurements at room temperature were conducted to study the formation of C′. To relate the observed IR absorption bands to the vibrational transitions of the different isomers, quantum chemical calculations at the level of DFT were carried out. Within these calculations, the linear Z-DASA isomer A, the closed isomer C and the zwitterionic closed isomer C′ were considered. The calculated spectra show distinct features with several bands in the spectral window of 1000–1550 cm−1 related to the linear species whereas less features are observed for the closed DASA isomers which are sensitive to the protonation of the tertiary amine (Fig. S12, ESI†). Comparing the calculated IR absorption bands to the experimentally observed features of Z-DASA in a KBr pellet, acetonitrile solution and toluene solution reveals only small shifts caused by the different environment (Fig. S13, ESI†).First, the changes in IR absorption of Z-DASA upon linear-to-closed switching in acetonitrile were studied (Fig. 6). In order to distinguish between absorption bands of the linear and closed isomers, the spectra were referenced to Z-DASA in acetonitrile in the dark. Thus, a zero line is observed for times up to 0 s at which an LED with maximum emission at a wavelength of 594 nm is turned on to induce the linear-to-closed switching. With increasing illumination time, the IR absorption bands related to the linear DASA isomers decrease in intensity while absorption bands of the closed DASA isomers appear. The absorption band at 1501 cm−1 was chosen as representative feature of the Z-DASA isomer. It is caused by high vibrational activity of the C–C bonds between the barbituric acid ring system and the hydroxyl group and is thus characteristic for all linear DASA isomers. In turn, the IR absorption band at 1616 cm−1 is related to the zwitterionic closed DASA species C′ and is caused by a vibration centred at the barbituric acid ring system with contribution of the vibration of the ammonium proton.
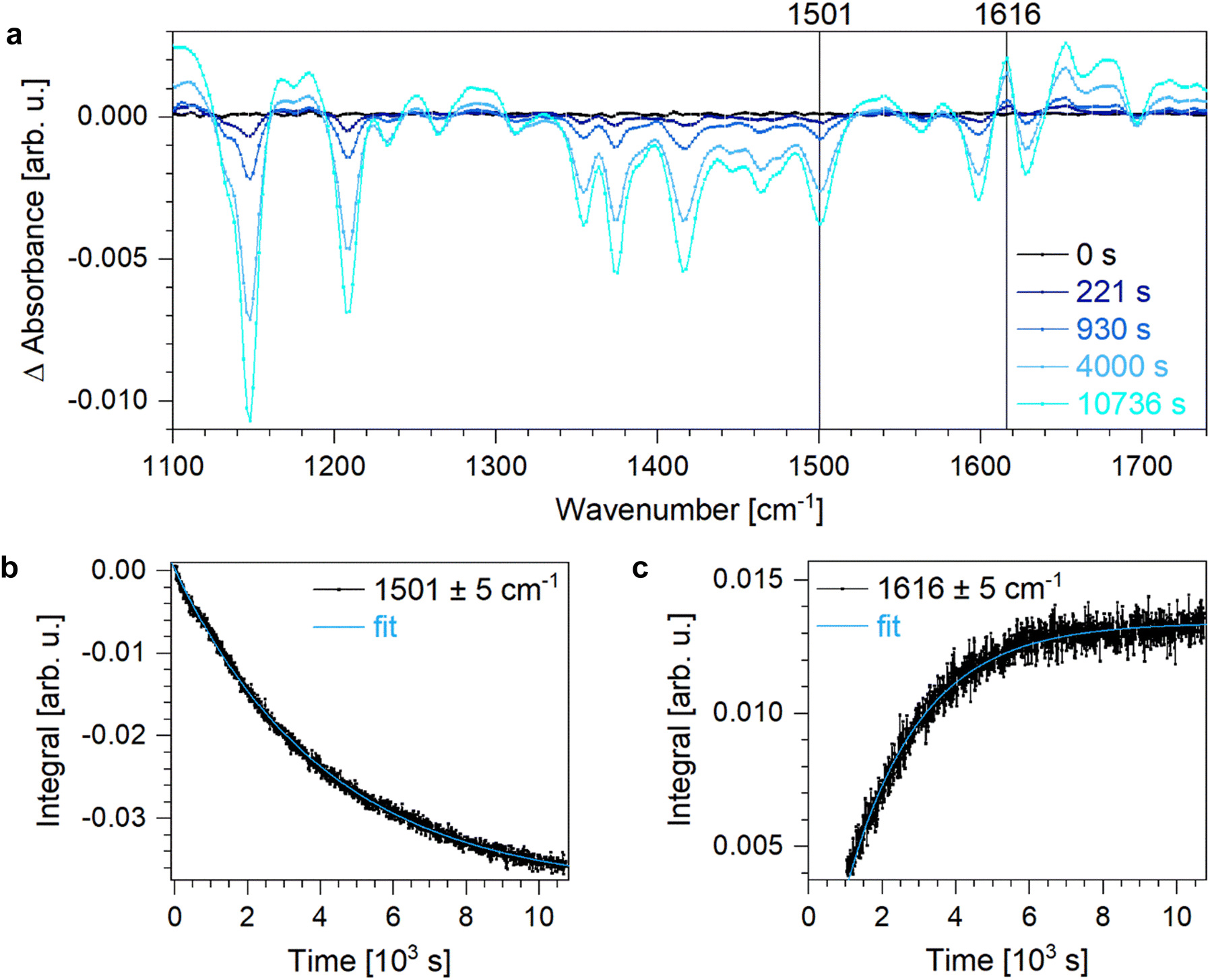 | ||
| Fig. 6 (a) Changes in IR absorption upon linear-to-closed switching of DASA in toluene-d8 at room temperature. IR absorption bands related to the linear species, e.g. at (b) 1501 cm−1, disappear while new absorption bands of the closed isomers C and C′, e.g. at (c) 1616 cm−1, emerge. | ||
Hence, in order to describe the electrocyclisation, the temporal evolution of the band located at 1501 cm−1 is fitted by a monoexponential fit curve (Fig. 6b) resulting in a lifetime of 4194 s (Table 3). The subsequent proton transfer is characterised by the monoexponential rise of the band at 1616 cm−1 (Fig. 6c) resulting in a lifetime of 1988 s (Table 3). The respective closed-to-linear backswitching reveals a lifetime of 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 384 s for the proton back-transfer to generate the non-zwitterionic closed DASA isomer C as well as a lifetime of 8332 s for the ring opening (Fig. S14, lifetimes see Table 3). Since the proton transfer occurs faster than the ring closure upon linear-to-closed switching but one order of magnitude slower than the ring opening upon closed-to-linear backswitching, the zwitterionic closed DASA isomer C′ is favoured compared to the non-zwitterionic closed DASA isomer C.
384 s for the proton back-transfer to generate the non-zwitterionic closed DASA isomer C as well as a lifetime of 8332 s for the ring opening (Fig. S14, lifetimes see Table 3). Since the proton transfer occurs faster than the ring closure upon linear-to-closed switching but one order of magnitude slower than the ring opening upon closed-to-linear backswitching, the zwitterionic closed DASA isomer C′ is favoured compared to the non-zwitterionic closed DASA isomer C.
| Reaction | Lifetime in toluene [s] | Lifetime in acetonitrile [s] |
|---|---|---|
| B′ → C | 1216 ± 12 | 4194 ± 21 |
| C → C′ | 3843 ± 35 | 1988 ± 25 |
| C → B′ | — | 8332 ± 131 |
| C′ → C | — | 18384 ± 889 |
Analysing the linear-to-closed switching of DASA in toluene reveals vanishing absorption bands related to the linear DASA isomers and appearing IR absorption features related to the closed DASA species (Fig. S18, ESI†). The same vibrations representing the Z-DASA isomer A and the zwitterionic closed isomer C′ are chosen which are shifted due to the altered environment to 1487 cm−1 and 1576 cm−1, respectively. The characteristic lifetime of the ring closure amounts to 1216 s while the lifetime of the proton transfer generating the zwitterionic structure C′ numbers 3843 s (Fig. S15, lifetimes see Table 3). This determined lifetime of the electrocyclisation is orders of magnitude longer compared to the UV/VIS results, however, the concentrations used in the IR experiments are higher due to the experimental method such that the formation of the zwitterionic product C′ precipitates in the non-polar environment which affects the kinetics and need to be taken into account. This hypothesis is confirmed by the closed-to-linear backswitching of DASA in toluene which shows neither decreasing IR absorption bands, which are expected for the features related to the closed DASA species upon closed-to-linear switching, nor an exponential temporal behaviour of all features (Fig. S16, ESI†). However, the resolvation of the precipitated zwitterionic closed DASA isomer C′ upon closed-to-linear backswitching affects the kinetics. These results are in line with other IR absorption studies of donor–acceptor Stenhouse adducts.42
Summary of the single steps upon switching
Combining the results of our former transient absorption measurements34 with the results of the herein presented results on longer time scales, a complete picture of the switching processes of DASA (Fig. 7) and Azo (Fig. S17, ESI†) upon photoisomerisation and thermally driven isomerisation emerge. The photoinitiated Z-to-E isomerisation of DASA and the E/Z isomerisation of Azo happen on the ultrafast time scale with no significant changes within an mixture of these photoswitches.34 The subsequent steps of C–C bond rotation, ring closure/opening and proton transfer of DASA as well as the Z-to-E isomerisation of Azo are thermally driven and were investigated within this study using time-resolved temperature-dependent UV/VIS spectroscopy and time-resolved IR spectroscopy at room temperature. The ring opening/closure of DASA occur on the second time scale. The effective activation barrier of the ring closure amouts to around 18 kJ mol−1 while the activation energy of the ring opening is about 50 kJ mol−1. The seemingly lower activation energy of the ring closure is an effective barrier since the ring closure happens along the same reaction coordinate as the preceding C–C bond rotation such that molecules overcoming the barrier of the C–C bond rotation exhibit higher energy leading to a lower effective barrier of the subsequent ring closure as confirmed by our quantum chemical calculations. The proton transfer takes place on an hour time scale revealing that the zwitterionic closed isomer C′ is favoured compared to the non-zwitterionic closed isomer C. The thermally initiated Z-to-E isomerisation of Azo happens also on the hour time scale. The time scales of switching are not affected within the orthogonally addressable mixture of both photoswitches Azo and DASA compared to the pure compounds. | ||
| Fig. 7 Complete switching scheme of the (a) linear-to-closed and (b) closed-to-linear switching of DASA including the determined lifetimes and activation energies found for pure DASA solutions and in an equimolar mixture of DASA + Azo in different environments, namely toluene solutions, acetonitrile solutions and thin PMMA blend films. The lifetimes in the picosecond range were determined within our former study of the ultrafast kinetics34 while the longer lifetimes and activation energies are provided by this study. | ||
Conclusions
In summary, we have studied the dynamics of two orthogonally, i.e. independently, addressable photoswitches, namely an azobenzene derivative (Azo) and a donor-acceptor Stenhouse adduct (DASA). We confirmed the orthogonal addressability of the mixture of Azo and DASA which is a prerequisite for their application as multifunctional smart materials in 3D and 4D printing. We used time-resolved temperature-dependent UV/VIS experiments to determine the effective cross sections, activation energies and dynamics of the E/Z isomerisation of Azo and the linear/closed switching of DASA with particular attention to the changes upon mixing the photoswitches in an equimolar ratio. In order to study the proton transfer after electrocyclisation of DASA, further time-resolved IR absorption measurements were carried out at room temperature. The thermally initiated ring closure and opening of DASA occur on the second time scale showing improved performance in the orthogonal equimolar mixture of DASA + Azo since the effective cross section and the number of backswitched DASA molecules are increased independent of the temperature. The activation energies are not significantly increased in the mixture, beside the activation energy of the electrocyclisation of DASA while Azo undergoes simultaneous E-to-Z isomerisation. However, this activation barrier is still very low and thus does not affect the performance negatively. The time-resolved IR absorption study revealed that the proton transfer, occuring on the hour time scale, plays an important role as the zwitterionic closed DASA isomer C′ is favoured compared to the non-zwitterionic closed DASA isomer C. At very high concentrations, the zwitterionic DASA isomer C′ precipitates in non-polar toluene solution which hinders the switching process. The thermally initiated Z-to-E isomerisation of Azo happens on the hour time scale with improved performance in an orthogonally addressable equimolar mixture of Azo + DASA as the effective cross section increases. All activation barriers are validated by our quantum chemical calculations revealing that the experimentally observed activation barrier of the DASA ring closure represents an effective activation barrier since the C–C bond rotation and the subsequent ring closure happen along the same reaction coordinate.Overall, the equimolar mixture of the orthogonal photoswitches Azo + DASA shows improved performance compared to the pure compounds on the longer time scale, e.g. higher effective cross sections and higher percentage of backswitched DASA molecules. Most important, our studies show that both the photochemically and the thermally induced switching steps of Azo and DASA are orthogonal such that Azo and DASA is a well-suited combination for application in 3D and 4D printed devices.
Data availability
The ESI† contains detailed information about synthesis, sample preparation and data supporting the findings of this study.Author contributions
T. S. conceived the project, conducted the measurements, analysed the results and wrote the manuscript. C. H. supported T. S. with the IR measurements. N. O. conducted the theoretical calculations. L-Y. H. synthesised the compounds. A. D. supervised N. O., E. B. supervised L-Y. H. and P. T. supervised T. S. as well as C. H. All authors reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
T. S., C. H., L.-Y. H., E. B. and P. T. acknowledge the DFG for funding via Germany's Excellence Strategy 2082/1-390761711. T. S. and L.-Y. H. thank the Carl Zeiss Foundation for the financial support. T. S. and P. T. thank Prof. Peter Coma for the access to the time-resolved temperature-dependent UV/VIS absorption spectrometer as well as Katharina Bleher and Thomas Josephy for the experimental support.Notes and references
- Molecular Switches, ed A. Ajayaghosh, V. Balzani, F. Ciardelli, J.-P. Collin, J. Daub, B. L. Feringa, F. M. Raymo, J.-P. Sauvage, S. Shinkai, P. D. Silva, J. F. Stoddart, M. K. J. T. Wiel, R. A. V. Delden, M. R. Wasielewski, B. Willner, I. Willner and Y. Yokoyama, Wiley-VCH, Weinheim, Germany, 2001 Search PubMed.
- S. Inder, S. O'Rourke, N. McDermott, R. Manecksha, S. Finn, T. Lynch and L. Marignol, The Notch-3 receptor: A molecular switch to tumorigenesis?, Cancer Treat. Rev., 2017, 60, 69–76 CrossRef CAS PubMed.
- M. Natali and S. Giordani, Molecular switches as photocontrollable “smart” receptors, Chem. Soc. Rev., 2012, 41, 4010–4029 RSC.
- A. R. Buskirk and D. R. Liu, Creating Small-Molecule-Dependent Switches to Modulate Biological Functions, Chem. Biol., 2005, 12, 151–161 CrossRef CAS PubMed.
- M.-M. Russew and S. Hecht, Photoswitches: From Molecules to Materials, Adv. Mater., 2010, 22, 3348–3360 CrossRef CAS PubMed.
- M. D. Manrique-Juarez, S. Rat, L. Salmon, G. Molnar, C. M. Quintero, L. Nicu, H. J. Shepherd and A. Bousseksou, Switchable molecule-based materials for micro- and nanoscale actuating applications: Achievements and prospects, Coord. Chem. Rev., 2016, 308, 395–408 CrossRef CAS.
- N. Fuentes, A. Martin-Lasanta, L. Alvarez de Cienfuegos, M. Ribagorda, A. Parra and J. M. Cuerva, Organic-based molecular switches for molecular electronics, Nanoscale, 2011, 3, 4003–4014 RSC.
- Z. L. Pianowski, Recent Implementations of Molecular Photoswitches into Smart Materials and Biological Systems, Chem. – Eur. J., 2019, 25, 5128–5144 CrossRef CAS PubMed.
- F. M. Raymo, Digital Processing and Communication with Molecular Switches, Adv. Mater., 2002, 14, 401–414 CrossRef CAS.
- M. W. Haydell, M. Centola, V. Adam, J. Valero and M. Famulok, Temporal and Reversible Control of a DNAzyme by Orthogonal Photoswitching, J. Am. Chem. Soc., 2018, 140, 16868–16872 CrossRef CAS PubMed.
- P. Tegeder, Optically and thermally induced molecular switching processes at metal surfaces, J. Phys.: Condens. Matter, 2012, 24, 394001 CrossRef PubMed.
- C. Gahl, D. Brete, F. Leyssner, M. Koch, E. R. McNellis, J. Mielke, R. Carley, L. Grill, K. Reuter, P. Tegeder and M. Weinelt, Coverage- and Temperature-Controlled Isomerization of an Imine Derivative on Au(111), J. Am. Chem. Soc., 2013, 135, 4273–4281 CrossRef CAS PubMed.
- J. Mielke, F. Leyssner, M. Koch, S. Meyer, Y. Luo, S. Selvanathan, R. Haag, P. Tegeder and L. Grill, Imine Derivatives on Au(111): Evidence for “Inverted” Thermal Isomerization, ACS Nano, 2011, 5, 2090–2097 CrossRef CAS PubMed.
- S. Wagner, F. Leyssner, C. Kördel, S. Zarwell, R. Schmidt, M. Weinelt, K. Rück-Braun, M. Wolf and P. Tegeder, Reversible photoisomerization of an azobenzene-functionalized self-assembled monolayer probed by sum-frequency generation vibrational spectroscopy, Phys. Chem. Chem. Phys., 2009, 11, 6242–6248 RSC.
- M. Schulze, M. Utecht, A. Hebert, K. Rück-Braun, P. Saalfrank and P. Tegeder, Reversible Photoswitching of the Interfacial Nonlinear Optical Response, J. Phys. Chem. Lett., 2015, 6, 505–509 CrossRef CAS PubMed.
- M. Hänsel, C. Barta, C. Rietze, M. Utecht, K. Rück-Braun, P. Saalfrank and P. Tegeder, Two-Dimensional Nonlinear Optical Switching Materials: Molecular Engineering toward High Nonlinear Optical Contrasts, J. Phys. Chem. C, 2018, 122, 25555–25564 CrossRef.
- S. Helmy, F. A. Leibfarth, S. Oh, J. E. Poelma, C. J. Hawker and J. Read de Alaniz, Photoswitching Using Visible Light: A New Class of Organic Photochromic Molecules, J. Am. Chem. Soc., 2014, 136, 8169–8172 CrossRef CAS PubMed.
- H. M. D. Bandara and S. C. Burdette, Photoisomerization in different classes of azobenzene, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Photochromism of Diarylethene Molecules and Crystals: Memories, Switches, and Actuators, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS PubMed.
- R. Klajn, Spiropyran-based dynamic materials, Chem. Soc. Rev., 2014, 43, 148–184 RSC.
- J. Wachtveitl, T. Nägele, B. Puell, W. Zinth, M. Krüger, S. Rudolph-Böhner, D. Oesterhelt and L. Moroder, Ultrafast photoisomerization of azobenzene compounds, J. Photochem. Photobiol., A, 1997, 105, 283–288 CrossRef CAS.
- C. Slavov, C. Yang, L. Schweighauser, C. Boumrifak, A. Dreuw, H. A. Wegner and J. Wachtveitl, Connectivity matters – ultrafast isomerization dynamics of bisazobenzene photoswitches, Phys. Chem. Chem. Phys., 2016, 18, 14795–14804 RSC.
- B. Schmidt, C. Sobotta, S. Malkmus, S. Laimgruber, M. Braun, W. Zinth and P. Gilch, Femtosecond Fluorescence and Absorption Dynamics of an Azobenzene with a Strong Push-Pull Substitution, J. Phys. Chem. A, 2004, 108, 4399–4404 CrossRef CAS.
- Z. Mahimwalla, K. G. Yager, J.-i Mamiya, A. Shishido, A. Priimagi and C. J. Barrett, Azobenzene photomechanics: prospects and potential applications, Polym. Bull., 2012, 69, 967–1006 CrossRef CAS.
- J. Broichhagen, J. A. Frank and D. Trauner, A Roadmap to Success in Photopharmacology, Acc. Chem. Res., 2015, 48, 1947–1960 CrossRef CAS PubMed.
- C.-L. Sun, C. Wang and R. Boulatov, Applications of Photoswitches in the Storage of Solar Energy, ChemPhotoChem, 2019, 3, 268–283 CrossRef CAS.
- M. M. Sroda, F. Stricker, J. A. Peterson, A. Bernal and J. Read de Alaniz, Donor-Acceptor Stenhouse Adducts: Exploring the Effects of Ionic Character, Chem. – Eur. J., 2021, 27, 4183–4190 CrossRef CAS PubMed.
- L.-Y. Hsu, P. Mainik, A. Münchinger, S. Lindenthal, T. Spratte, A. Welle, J. Zaumseil, C. Selhuber-Unkel, M. Wegener and E. Blasco, A Facile Approach for 4D Microprinting of Multi-Photoresponsive Actuators, Adv. Mater. Technol., 2022, 8, 2200801 CrossRef.
- L. Garcia-Fernandez, C. Herbivo, V. S. M. Arranz, D. Warther, L. Donato, A. Specht and A. del Campo, Dual Photosensitive Polymers with Wavelength-Selective Photoresponse, Adv. Mater., 2014, 26, 5012–5017 CrossRef CAS PubMed.
- C. A. Spiegel, M. Hippler, A. Münchinger, M. Bastmeyer, C. Barner-Kowollik, M. Wegener and E. Blasco, 4D Printing at the Microscale, Adv. Funct. Mater., 2020, 30, 1907615 CrossRef CAS.
- H. Y. Jeong, S.-C. An and Y. C. Jun, Light activation of 3D-printed structures: from millimeter to sub-micrometer scale, Nanophotonics, 2022, 11, 461–486 CrossRef CAS.
- K. Jung, N. Corrigan, M. Ciftci, J. Xu, S. E. Seo, C. J. Hawker and C. Boyer, Designing with Light: Advanced 2D, 3D, and 4D Materials, Adv. Mater., 2020, 32, 1903850 CrossRef CAS PubMed.
- Smart Materials in Additive Manufacturing, ed. L.-Y. Hsu, C. A. Spiegel and E. Blasco, Elsevier, Amsterdam, Netherlands, 2022 Search PubMed.
- T. Schmitt, L.-Y. Hsu, N. Oberhof, D. Rana, A. Dreuw, E. Blasco and P. Tegeder, Ultrafast Excited States Dynamics of Orthogonal Photoswitches and The Influence of the Environment, Adv. Funct. Mater., 2023, 2300863 CrossRef.
- J. P. Merrick, D. Moran and L. Radom, An Evaluation of Harmonic Vibrational Frequency Scale Factors, J. Phys. Chem. A, 2007, 111, 11683–11700 CrossRef CAS PubMed.
- Y. Shao, Z. Gan, E. Epifanovsky, A. T. B. Gilbert, M. Wormit, J. Kussmann, A. W. Lange, A. Behn, J. Deng, X. Feng, D. Ghosh, M. Goldey, P. R. Horn, L. D. Jacobson, I. Kaliman, R. Z. Khaliullin, T. Kuś, A. Landau, J. Liu, E. I. Proynov, Y. M. Rhee, R. M. Richard, M. A. Rohrdanz, R. P. Steele, E. J. Sundstrom, H. L. Woodcock, P. M. Zimmerman, D. Zuev, B. Albrecht, E. Alguire, B. Austin, G. J. O. Beran, Y. A. Bernard, E. Berquist, K. Brandhorst, K. B. Bravaya, S. T. Brown, D. Casanova, C.-M. Chang, Y. Chen, S. H. Chien, K. D. Closser, D. L. Crittenden, M. Diedenhofen, R. A. DiStasio, H. Do, A. D. Dutoi, R. G. Edgar, S. Fatehi, L. Fusti-Molnar, A. Ghysels, A. Golubeva-Zadorozhnaya, J. Gomes, M. W. D. Hanson-Heine, P. H. P. Harbach, A. W. Hauser, E. G. Hohenstein, Z. C. Holden, T.-C. Jagau, H. Ji, B. Kaduk, K. Khistyaev, J. Kim, J. Kim, R. A. King, P. Klunzinger, D. Kosenkov, T. Kowalczyk, C. M. Krauter, K. U. Lao, A. D. Laurent, K. V. Lawler, S. V. Levchenko, C. Y. Lin, F. Liu, E. Livshits, R. C. Lochan, A. Luenser, P. Manohar, S. F. Manzer, S.-P. Mao, N. Mardirossian, A. V. Marenich, S. A. Maurer, N. J. Mayhall, E. Neuscamman, C. M. Oana, R. Olivares-Amaya, D. P. O’Neill, J. A. Parkhill, T. M. Perrine, R. Peverati, A. Prociuk, D. R. Rehn, E. Rosta, N. J. Russ, S. M. Sharada, S. Sharma, D. W. Small, A. Sodt, T. Stein, D. Stück, Y.-C. Su, A. J. W. Thom, T. Tsuchimochi, V. Vanovschi, L. Vogt, O. Vydrov, T. Wang, M. A. Watson, J. Wenzel, A. White, C. F. Williams, J. Yang, S. Yeganeh, S. R. Yost, Z.-Q. You, I. Y. Zhang, X. Zhang, Y. Zhao, B. R. Brooks, G. K. L. Chan, D. M. Chipman, C. J. Cramer, W. A. Goddard, M. S. Gordon, W. J. Hehre, A. Klamt, H. F. Schaefer, M. W. Schmidt, C. D. Sherrill, D. G. Truhlar, A. Warshel, X. Xu, A. Aspuru-Guzik, R. Baer, A. T. Bell, N. A. Besley, J.-D. Chai, A. Dreuw, B. D. Dunietz, T. R. Furlani, S. R. Gwaltney, C.-P. Hsu, Y. Jung, J. Kong, D. S. Lambrecht, W. Liang, C. Ochsenfeld, V. A. Rassolov, L. V. Slipchenko, J. E. Subotnik, T. Van Voorhis, J. M. Herbert, A. I. Krylov, P. M. W. Gill and M. Head-Gordon, Advances in molecular quantum chemistry contained in the Q-Chem 4 program package, Mol. Phys., 2015, 113, 184–215 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- Y.-S. Lin, G.-D. Li, S.-P. Mao and J.-D. Chai, Long-Range Corrected Hybrid Density Functionals with Improved Dispersion Corrections, J. Chem. Theory Comput., 2013, 9, 263–272 CrossRef CAS PubMed.
- A. Klamt and G. Schüürmann, COSMO: a new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805, 10.1039/P29930000799.
- A. Behn, P. M. Zimmerman, A. T. Bell and M. Head-Gordon, Efficient exploration of reaction paths via a freezing string method, J. Chem. Phys., 2011, 135, 224108 CrossRef PubMed.
- J. Baker, An algorithm for the location of transition states, J. Comput. Chem., 1986, 7, 385–395 CrossRef CAS.
- H. Zulfikri, M. A. J. Koenis, M. M. Lerch, M. Di Donato, W. Szymański, C. Filippi, B. L. Feringa and W. J. Buma, Taming the Complexity of Donor-Acceptor Stenhouse Adducts: Infrared Motion Pictures of the Complete Switching Pathway, J. Am. Chem. Soc., 2019, 141, 7376–7384 CrossRef CAS PubMed.
- T. Moldt, D. Przyrembel, M. Schulze, W. Bronsch, L. Boie, D. Brete, C. Gahl, R. Klajn, P. Tegeder and M. Weinelt, Differing Isomerization Kinetics of Azobenzene-Functionalized Self-Assembled Monolayers in Ambient Air and in Vacuum, Langmuir, 2016, 32, 10795–10801 CrossRef CAS PubMed.
- D. M. Adrion, D. S. Kaliakin, P. Neal and S. A. Lopez, Benchmarking of Density Functionals for Z-Azoarene Half-Lives via Automated Transition State Search, J. Phys. Chem. A, 2021, 125, 6474–6485 CrossRef CAS PubMed.
- M. Di Donato, M. M. Lerch, A. Lapini, A. D. Laurent, A. Iagatti, L. Bussotti, S. P. Ihrig, M. Medved’, D. Jacquemin, W. Szymański, W. J. Buma, P. Foggi and B. L. Feringa, Shedding Light on the Photoisomerization Pathway of Donor–Acceptor Stenhouse Adducts, J. Am. Chem. Soc., 2017, 139, 15596–15599 CrossRef CAS PubMed.
- J. R. Hemmer, S. O. Poelma, N. Treat, Z. A. Page, N. D. Dolinski, Y. J. Diaz, W. Tomlinson, K. D. Clark, J. P. Hooper, C. Hawker and J. Read de Alaniz, Tunable Visible and Near Infrared Photoswitches, J. Am. Chem. Soc., 2016, 138, 13960–13966 CrossRef CAS PubMed.
- M. M. Lerch, S. J. Wezenberg, W. Szymanski and B. L. Feringa, Unraveling the Photoswitching Mechanism in Donor–Acceptor Stenhouse Adducts, J. Am. Chem. Soc., 2016, 138, 6344–6347 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp05786k |
| This journal is © the Owner Societies 2024 |