
Noble gas hydrides: theoretical prediction of the first group of anionic species†
Stefano
Borocci
 ab,
Patrizio
Cecchi
a,
Felice
Grandinetti
*ab,
Nico
Sanna
ac and
Costantino
Zazza
a
ab,
Patrizio
Cecchi
a,
Felice
Grandinetti
*ab,
Nico
Sanna
ac and
Costantino
Zazza
a
aDipartimento per la Innovazione nei sistemi Biologici, Agroalimentari e Forestali (DIBAF), Università della Tuscia, L.go dell’Università, s.n.c., 01100 Viterbo, Italy. E-mail: fgrandi@unitus.it
bIstituto per i Sistemi Biologici del CNR (ISB), Sede di Roma – Meccanismi di Reazione c/o Dipartimento di Chimica, Sapienza Università di Roma, P.le A. Moro 5, Rome, Italy
cIstituto per la Scienza e Tecnologia dei Plasmi del CNR (ISTP), Via Amendola 122/D, 70126 Bari, Italy
First published on 10th February 2024
Abstract
The first group of anionic noble-gas hydrides with the general formula HNgBeO− (Ng = Ar, Kr, Xe, Rn) is predicted through MP2, Coupled-Cluster, and Density Functional Theory computations employing correlation-consistent atomic basis sets. We derive that these species are stable with respect to the loss of H, H−, BeO, and BeO−, but unstable with respect to Ng + HBeO−. The energy barriers of the latter process are, however, high enough to suggest the conceivable existence of the heaviest HNgBeO− species as metastable in nature. Their stability arises from the interaction of the H− moiety with the positively-charged Ng atoms, particularly with the σ-hole ensuing from their ligation to BeO. This actually promotes relatively tight Ng–H bonds featuring a partially-covalent character, whose degree progressively increases when going from HArBeO− to HRnBeO−. The HNgBeO− compounds are also briefly compared with other noble-gas anions observed in the gas phase or isolated in crystal lattices.
1. Introduction
Chemical compounds containing noble gas–hydrogen (Ng–H) bonds have not yet been prepared using synthetic methods. Numerous cationic and neutral noble-gas hydrides are, however, detected in the gas phase or in cold solid matrices. The first evidence of gaseous cationic species dates back to 1925,1 when Hogness and Lunn detected the simplest HeH+ and HeH2+ from ionized He/H2 mixtures. Over the years, the gas-phase chemistry of the NgmHn+ (Ng = He–Xe; m, n ≥ 1) turned out to be rich and varied,2 and the rejuvenated interest that it is currently enjoying3–13 is also stimulated by the actual detection of ArH+ and HeH+ in various galactic and extragalactic regions.14–18 Neutral compounds with Ng–H bonds emerged first in 1995,19 when Räsänen and his coworkers identified the strong infrared absorbers originating from the photodissociation of hydrogen halides in Kr and Xe matrices as the “inserted” compounds HXeI, HXeBr, HXeCl, and HKrCl. Other HNgY species (Y = electronegative group) were soon prepared using the same method, including the celebrated HArF,20 a first and still unique neutral covalent argon compound. The family currently includes more than thirty HNgY (Ng = Kr, Xe) and other related species, whose investigation is a major chapter of modern noble-gas chemistry.21,22 Anionic noble-gas hydrides are, instead, still essentially unknown. The simplest diatomic HNg− (Ng = He–Xe)23 species are weak van der Waals complexes, but more compact structures featuring Ng–H bonds are still unreported, both experimentally and theoretically. We report here – to the best of our knowledge – a first theoretically-predicted group of such species, having formula HNgBeO−. The complexes of Ng atoms with beryllium Lewis acids, including BeO and other related species, have attracted, over the years, considerable experimental and theoretical interest.24–30 The present search was inspired by the computational results that we recently obtained from the study of the complexes of ArBeO with simple ligands.31,32 When the Ng atoms, particularly the heaviest Ar, Kr, Xe, and Rn, interact with the strong electron-withdrawing BeO, the only slightly positive electrostatic potential (EP)33,34 of Ng (ca. 2 kcal mol−1 at the 0.0010 e a0−3 isodensity surface) dramatically increases up to more than 50 kcal mol−1. The EP of NgBeO features, in particular, a point of maximum on the outer prolongation of the Ng–Be bond axis, and a σ-hole35,36 at the Ng atom that allows the interaction with various ligands L. The study of numerous L-ArBeO31,32 unraveled, in particular, that the complexes with NH3 and HF (featuring a negative EP at the N and F atom, respectively) have sizable binding energies of 4–5 kcal mol−1, whose major attractive component (ca. 70%) comes from the electrostatic term. In essence, the formation of these complexes is driven by the interaction of the positive EP of Ar with the negative EP of N or F. We thus surmised that this favorable electrostatic contact could be further enhanced with anionic ligands, including the simplest H−. This picture was confirmed by the actual location of the four HNgBeO− species (Ng = Ar, Kr, Xe, Rn) as true minima on the corresponding potential energy surface (PES). These systems are actually unstable with respect to dissociation into Ng and HBeO−, but the protecting barriers are high enough to suggest their conceivable existence as metastable species, at least for the heaviest congeners. The structure, bonding situation, harmonic frequencies, and stability of these anions are discussed in this article. We also briefly compare the bonding motif occurring in the HNgBeO− species with those occurring in other observed or predicted Ng anions.2. Theoretical methods and computational details
The theoretical methods employed in this study were the Møller–Plesset37 perturbation theory truncated at the second order (MP2), the Coupled Cluster theory38 with the inclusion of single and double electronic substitutions, CCSD, and an estimate of connected triples, CCSD(T), and the B3LYP density functional.39,40 The employed basis sets, henceforth denoted as aVnZ (n = T, Q, 5), were the Dunning's correlation consistent aug-cc-pVnZ41 for H, Be, O, Ar, and Kr, and aug-cc-pVnZ-PP41 for Xe and Rn, the latter being treated with the Stuttgart/Cologne small-core, scalar-relativistic effective core potentials (ECP-28 and ECP-60, respectively).42 The MP2 and CCSD(T) calculations were performed by correlating only the outer valence electrons (i.e. frozen-core approximation).The MP2/aVTZ and B3LYP/aVTZ geometry optimization and harmonic frequency calculations were performed using Gaussian 09 (G09, Revision D1),43 and the CCSD(T)/aVTZ geometry optimizations and harmonic frequencies calculations were performed using CFOUR (V2.1).44 Electronic energies at the complete basis set (CBS) limit were obtained by extrapolating the CCSD(T)/aVQZ and CCSD(T)/aV5Z correlation energies using the cubic formula:45
 | (1) |
At each computational level, the energy change at 0 K and the energy, enthalpy and free energy changes at 298.15 K of the investigated reactions were calculated using the unscaled harmonic vibrational frequencies and by adding the translational (3/2 RT), rotational (RT or 3/2 RT for linear and non-linear species, respectively) and vibrational contributions at this temperature. The last term was calculated using standard statistical mechanics formulas.46 The total entropies were calculated using the unscaled harmonic frequencies and moments of inertia.
The CCSD(T)/aVTZ natural bond orbital (NBO) atomic charges were calculated using the NBO program.47
The functions examined within the bonding analysis, including the electron density ρ(r),48 the electron energy density H(r),49–51 and the reduced density gradient (RDG) s(r),52,53 are defined as follows. The ρ(r) is defined by the equation:48
 | (2) |
The H(r) is the sum of the kinetic energy density G(r) and the potential energy density V(r). The presently-employed definition48,54 of the G(r) is given by the equation:
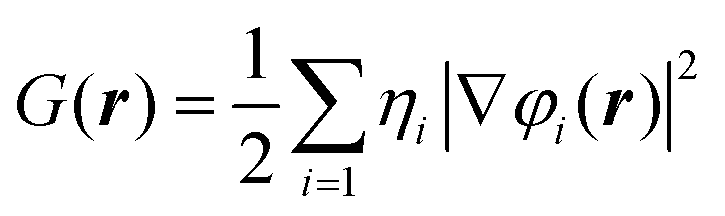 | (3) |
The potential energy density V(r) is evaluated48 from the local form of the virial theorem:
 | (4) |
At last, the RDG is defined by the equation:52,53
 | (5) |
Low-value s(r) isosurfaces (typically 0.2–0.6) appear among atoms undergoing any type of interaction. The integral of a given property P [particularly the ρ(r) and the H(r)] over the volume Ωs enclosed, in particular, by the s(r) = 0.2 isosurface (s) at around the BCP located on any bond path, P(Ωs), was calculated by producing an orthogonal grid of points that encloses the isosurface and applying the formula:
 | (6) |
The Extended Transition State – Natural Orbitals for Chemical Valence (ETS-NOCV) calculations were performed at the B3LYP/aVTZ level of theory using the CCSD(T)/aVTZ optimized geometries. In the ETS scheme,55 the interaction energy between two fragments A and B in a molecular system AB, ΔEint, is partitioned into five components:
| ΔEint = ΔEprep + ΔEelstat + ΔEPauli + ΔEdisp + ΔEorb | (7) |
| Vφk = vkφk | (8) |
The φk can be grouped into pairs of complementary orbitals (φk, φ−k) corresponding to the same eigenvalue with the opposite sign, ±vk. In particular, the NOCV orbitals decompose the deformation density Δρorb(r) (namely, the difference between the density of the complex and that of the separated fragments) into the sum of the various pair contributions:
 | (9) |
 | (10) |
The ΔEorb term of eqn (7) and the ρ(r), the H(r), and the s(r) were evaluated and analyzed with the Multiwfn package (version 3.8.dev).60 The CCSD(T)/aVTZ wave functions were stored in the molden files generated using CFOUR,44 and properly formatted using the Molden2AIM utility.61 The Multiwfn was also employed to produce the two-(2D) plots of the Δρkorb(r) and of the H(r), the latter including the contour lines corresponding to the critical points located for each structure from the topological analysis of the H(r) and the standard contour lines belonging to the patterns ± k × 10n (k = 1, 2, 4, 8; n = −5 to 6).
3. Results and discussion
3.1 Structure and bonding
All the linear HNgBeO− (Ng = He–Rn) species were located as stationary points on the MP2/aVTZ PES. However, the lightest HHeBeO− and HNeBeO− species turned out to be second-order saddle points (unstable to dissociation through the H–Ng–Be bending), and were not explored in further detail. The HArBeO−, HKrBeO−, HXeBeO−, and HRnBeO− were instead characterized as true energy minima, and as such confirmed also at the B3LYP/aVTZ and CCSD(T)/aVTZ levels of theory. The NgBeO species were also investigated for comparison.In general, as shown in Table S1 of the ESI,† compared with the two ab initio methods, the B3LYP method tends to underestimate the Ng–H and Be–O distances, while overestimating the Ng–Be distances, with mutual differences arriving up to ca. 0.05 Å. The MP2/aVTZ and CCSD(T)/aVTZ values are, instead, quite similar, with the largest difference of only 0.02 Å. The forthcoming discussion will be based on the in principle most accurate CCSD(T)/aVTZ values shown in Fig. 1. At these geometries, the CCSD/aVTZ T1 diagnostic62 of any HNgBeO− species resulted close to the accepted threshold of 0.02, thus suggesting the effectiveness of single-determinant methods to describe their electronic structure. The geometries and NBO atomic charges of the HNgBeO− species suggest their structural assignment as tight complexes between H− and NgBeO, best formulated as H−(NgBeO). As shown in Fig. 1, in any HNgBeO−, the H atom bears a negative charge lower than −0.8e, arriving at −0.943e in HArBeO−. The Ng–H distances range between 2.5267 Å (Ng = Rn) and 2.5764 Å (Ng = Kr). These values are definitely higher than the sum of the covalent radii63 of H (0.32 Å) and of the Ng atoms (Ar: 1.07 Å, Kr: 1.21 Å, Xe: 1.35 Å, Rn: 1.45 Å), but lower by ca. 0.5–1 Å than the sum of the van der Waals radii of H64 (1.06 Å) and of the Ng atoms65 (Ar: 1.97 Å, Kr: 2.12 Å, Xe: 2.32 Å, Rn: 2.43 Å). They are also appreciably lower than the distances of the diatomic HNg−, predicted23 between 3.47 Å (Ng = Rn) and 3.78 Å (Ng = Ar). This overall suggests that the Ng–H bonds of the HNgBeO−, while certainly rather far from typical covalent interactions, are, however, appreciably tighter than simple van der Waals contacts. This was confirmed by the results of the bonding analysis, performed at the CCSD(T)/aVTZ level of theory using our recently proposed method.49,66–68 It involves the study of three functions, namely the electron density ρ(r),48 the electron energy density H(r),49–51 and the reduced density gradient (RDG) s(r).52,53 Any Ng–X bond (X = binding partner) is assigned following the step-by-step procedure68 briefly recalled below. Further details are given in ref. 49 and 66–68.
 | ||
| Fig. 1 CCSD(T)/aVTZ optimized bond lengths (Å) and NBO atomic charges (e, in italics) of the HNgBeO− and NgBeO species (Ng = Ar, Kr, Xe, Rn). | ||
Step 1. The Ng–X contact is ascertained by analyzing the topology of the ρ(r) and locating the corresponding bond path (BP) and bond critical point (BCP) (the classical AIM analysis).
Step 2. The topology of the H(r) of the whole molecule is analyzed. This typically produces various critical points (HCPs) of rank 3 and signature −3, −1, +1, or +3. The contour lines these points belong to are collected as the HCP lines.
Step 3. The HCP lines are combined with a set of standard (STD) H(r) lines, typically the patterns ± k × 10n (k = 0, 1, 2, 4, 8; n = −5 to 6).
Step 4. The HCP/STD lines are plotted as 2D or 3D graphs, whose visual inspection allows assignment of the bond as type A, B, or C. As best discussed previously,49,66–68 the H(r) generally partitions the atomic space into inner regions of negative values, indicated as H−(r), and outer regions of positive values, indicated as H+(r). When two atoms form a chemical bond, their H−(r) and H+(r) regions combine in modes that signal the nature of the interaction. Particularly for the Ng–X bonds, it is possible to recognize three major situations. In interactions of type A, the atoms overlap all the contour lines of their H+(r) regions, and part of the contour lines of their inner H−(r) regions, the bond appearing as a continuous region of negative values of H(r), plunged in a zone of positive values. The bond is topologically signed by a (3, +1) HCP falling on the bond axis. Typical examples are covalent bonds, or donor–acceptor interactions with some degree of electron sharing. In interactions of type B, the H−(r) region of Ng is, again, overlapped with the H−(r) region of the binding partner, but (i) the bond is not signed by a HCP on the axis, and (ii) the Ng–X inter-nuclear region includes a (more or less wide) region of positive H(r). Typical examples are the complexes of Ng donors with strongly electropositive Lewis acceptors. In interactions of type C, Ng and X overlap only part of their H+(r) regions, their H−(r) regions remaining perfectly closed and separated by a region of positive H(r). The bond thus appears as two clearly distinguishable H−(r) regions, separated by a region of positive values of H(r). Typical examples are non-covalent contacts of variable nature.
Step 5. The assignment of the bond is refined by examining the H(r) along the Ng–X BP, particularly at around the BCP. This serves to confirm the interactions of type A, and to distinguish the interactions of type B and C as B-loose (Bl) or B-tight (Bt), and C-loose (Cl) or C-tight (Ct). The adopted criteria are given in Table 1.
| H(r) at around the BCP | ||
|---|---|---|
| a e a 0 −3. | ||
| Assignment | Ng side | X side |
| A | Negative | Negative |
| Bl or Cl | Positive | Positive |
| Bt or Ct | Positive | Negative |
| Negative | Positive | |
| Negative | Negative | |
Step 6. Once assigned as of type A, Bl/Bt, or Cl/Ct, the Ng–X bond is assayed in terms of contribution of covalency. This is accomplished by integrating the ρ(r) and the H(r) over the volume Ωs enclosed by the s(r) isosurface associated with the Ng–X BCP. The value of the s(r) is chosen by examining, particularly at around the BCP, the s(r) vs. sign(λ2) × ρ(r) 2D plot [λ2 is the second eigenvalue of the Hessian matrix of ρ(r), with λ1 < λ2 < λ3]. The selected value of s(r) is the highest one that still avoids the contribution of the tails of the atomic densities, and typically ranges between 0.2 and 0.5. Relevant quantities calculated over Ωs include the average ρ(r), ρs (ave), and the average, maximum, and minimum H(r), Hs (ave/max/min). Based on the obtained values, and on the sign of H(r) over Ωs, H(Ωs), the bond is therefore assigned as covalent (Cov), partially-covalent (pCov) or non-covalent (nCov) according to the criteria listed in Table 1.
Step 7. The bond is finally classified using the notations Cov(Type), pCov[Type/H(Ωs)], or nCov(Type), for example, Cov(A), pCov(Bt/H−/+), or nCov(Cl).
In developing the method, we also found it convenient to introduce some additional numerical indices that allow further assay of the degree of the various interactions. Of relevance in the present context is the average bond degree over Ωs, BDs (ave). Borrowing the concept of BD introduced by Espinosa et al.,69 this index is defined as the average over Ωs of the ratio −H(r)/ρ(r). Formulated in this way, the index is de facto applicable to any type of interaction (A, B, or C).
The results of the bonding analysis of the HNgBeO− are given in Fig. 2 and in Table 2. The NgBeO species were also investigated for comparison. As shown in Fig. 2(a), in HArBeO−, the H−(r) regions of the H atom and of the ArBeO moiety are separated by a zone of positive values, and the interaction is, therefore, of type C. In addition, along the BCP, the H(r) is positive at both the H and the Ar side. Over Ωs, however, although positive on the average, the H(r) is partially negative (see Table 2). The bond is, therefore, partially covalent and overall assigned as pCov(Cl/H+/−). It is, indeed, possible from Fig. 2(a) to clearly appreciate the strong deformation of the spherical H−(r) region of the H atom due to the interaction with ArBeO. When going to the heaviest congeners KrArBeO, XeArBeO, and RnArBeO, this polarization markedly increases up to promoting the overlapping of the H−(r) regions of the H atom and of the NgBeO moiety (Fig. 2(b)–(d)). Consequently, the type of the interaction changes from C to A. The H(r) is also invariably negative over Ωs, but the values of ρs (ave), ranging between 0.0202 and 0.0299 e a0−3, are definitely lower than those typical of covalent bonds. All the Ng–H contacts (Ng = Kr, Xe, Rn) are, therefore, assigned as pCov(A/H−). Not unexpectedly, the quantitative indices indicate that the interaction becomes progressively tighter when going from HArBeO− to HRnBeO−. One notes, in particular, the BDs (ave), predicted to be negative for Ng = Ar (−0.0104 hartree e−1) and increasingly positive for Ng = Kr (0.0492 hartree e−1), Ng = Xe (0.0989 hartree e−1), and Ng = Rn (0.121 hartree e−1). This suggests that the irregular trend of the Ng–H bond distances (an increase from 2.5567 Å to 2.5764 Å when going from HArBeO− to HKrBeO−, and then a decrease to 2.5968 Å and 2.5267 Å when going to HXeBeO− and HRnBeO−) is, most likely, a balance of atomic size and degree of interaction.
 | ||
| Fig. 2 2D-plots of the CCSD(T)/aVTZ H(r) in the main plane of HNgBeO− (a–d) and NgBeO (e–h) (Ng = Ar, Kr, Xe, Rn) (solid/brown and dashed/blue lines correspond, respectively, to positive and negative values). The (red) dots indicate the HCP of the interactions of type A. | ||
| Species | Bond | Type | Ω s | N(Ωs) | ρ s (ave) | H s (ave/max/min) | H(Ωs)a | BDs (ave) | Assignment |
|---|---|---|---|---|---|---|---|---|---|
| a Depending on the sign of Hs (ave/max/min), H(Ωs) = H+/−, H−/+, or H−. | |||||||||
| HArBeO− | Ar–H | Cl | 0.0585 | 0.98 | 0.0167 | 0.00017/0.00052/−0.00022 | H+/− | −0.0104 | pCov(Cl/H+/−) |
| Ar–Be | Bt | 0.0135 | 0.59 | 0.0435 | −0.0044/−0.00087/−0.0089 | H− | 0.101 | pCov(Bt/H−) | |
| ArBeO | Ar–Be | Bl | 0.0104 | 0.33 | 0.0319 | 0.0024/0.0052/−0.00024 | H+/− | −0.0754 | pCov(Bl/H+/−) |
| HKrBeO− | Kr–H | A | 0.0821 | 1.66 | 0.0202 | −0.00099/−0.00074/−0.0014 | H− | 0.0492 | pCov(A/H−) |
| Kr–Be | Bt | 0.0262 | 1.11 | 0.0423 | −0.0093/−0.0041/−0.0142 | H− | 0.221 | pCov(Bt/H−) | |
| KrBeO | Kr–Be | Bt | 0.0151 | 0.48 | 0.0321 | −0.0016/0.0011/−0.0049 | H−/+ | 0.0497 | pCov(Bt/H−/+) |
| HXeBeO− | Xe–H | A | 0.1203 | 3.14 | 0.0261 | −0.0026/−0.0022/−0.0032 | H− | 0.0989 | pCov(A/H−) |
| Xe–Be | Bt | 0.0595 | 2.49 | 0.0418 | −0.0152/−0.0092/−0.0198 | H− | 0.364 | pCov(Bt/H−) | |
| XeBeO | Xe–Be | Bt | 0.0267 | 0.84 | 0.0325 | −0.0055/−0.0011/−0.0094 | H− | 0.168 | pCov(Bt/H−) |
| HRnBeO− | Rn–H | A | 0.1373 | 4.10 | 0.0299 | −0.0036/−0.0031/−0.0043 | H− | 0.121 | pCov(A/H−) |
| Rn–Be | Bt | 0.1162 | 4.67 | 0.0402 | −0.0170/−0.0125/−0.0202 | H− | 0.422 | pCov(Bt/H−) | |
| RnBeO | Rn–Be | Bt | 0.0350 | 1.10 | 0.0314 | −0.0066/−0.0028/−0.0101 | H− | 0.210 | pCov(Bt/H−) |
Like those occurring in the NgBeO species, the Ng–Be bonds of the HNgBeO− species are invariably assigned as of type B. The bonding analysis unravels, however, that the ligation of NgBeO to H− increases the extent of the Ng–Be interaction. This is particularly evident when going from ArBeO to HArBeO−: as shown in Table 2, the character of the Ar–Be bond changes from pCov(Bl/H+/−) to pCov(Bt/H−), and the BDs (ave) changes from negative (−0.0754 hartree e−1) to positive (0.101 hartree e−1). As shown in Fig. 1, this is accompanied by a shortening of the Ar–Be distance from 2.0752 Å to 2.0173 Å. The BDs (ave) of the Ng–Be bond of the other HNgBeO− is also higher by ca. 0.2 hartree e−1 than that of the corresponding NgBeO. Only for HKrBeO−, however, is this change accompanied by a decrease of the Kr–Be bond length (from 2.1830 Å to 2.2119 Å). The Xe–Be and Rn–Be bond lengths of HXeBeO− and HRnBeO− increase by 0.0167 Å and 0.053 Å, respectively.
Within the classical AIM analysis,48 a chemical bond is characterized in terms of ρ(BCP), of the Laplacian of the electron density at the BCP, ∇2ρ(BCP), and of H(BCP). The values of these quantities calculated for the HNgBeO− and NgBeO are given in Table S2 (ESI†). When compared with those quoted in Table 2, these data unravel some peculiar features of our employed method of bonding analysis. For example, the Ar–H bond of HArBeO−, presently assigned as pCov, would have been assigned as non-covalent, based on a positive value of both ∇2ρ(BCP) and H(BCP). In addition, even though the AIM analysis is sufficient in predicting the partially-covalent character of the Ng–H and Ng–Be bonds of any other HNgBeO− and NgBeO (∇2ρ(BCP) is positive but H(BCP) is negative), our employed indices, particularly the H(Ωs) and BDs (ave), provide further insights into their quantitative aspects and trends of intrinsic strength.
The assignment of the HNgBeO− as a tight donor–acceptor complex between H− and NgBeO is further supported by the results of the ETS-NOCV analysis. In general, the energy decomposition analysis performed according to the ETS scheme may be used to address the question about the choice of the most appropriate fragments that should be used to discuss a chemical bond. It has been, in particular, suggested70–73 that those fragments, which exhibit the smallest change in the electronic structure that is associated with bond formation, should be used for discussing the nature of the chemical interactions. Thus, the absolute value of the orbital term ΔEorb indicates which fragments should be used for the bonding analysis. The data obtained for the HNgBeO− species are given in Table 3. Irrespective of Ng, the smallest (less negative) ΔEorb is predicted for the H−/NgBeO interacting fragments, the values ranging between −9.99 kcal mol−1 (Ng = Ar) and −29.40 kcal mol−1 (Ng = Rn). If the Ng–H bond would be a covalent interaction between H and NgBeO−, the ΔEorb would be definitely more negative, ranging between −65.78 kcal mol−1 (Ng = Ar) and −77.32 kcal mol−1 (Ng = Rn). As for the Ng–Be bond, the analysis clearly unravels that there is a dative bond between HNg− and BeO rather than a covalent bond between HNg and BeO−. In fact, if one assumes the former pair of interacting fragments, the ΔEorb ranges between −29.81 kcal mol−1 (Ng = Ar) and −37.41 kcal mol−1 (Ng = Rn), but the values strongly decrease to −106.64 kcal mol−1 (Ng = Ar) and −122.31 kcal mol−1 (Ng = Xe) for the alternative interacting pair.
| Ng | H−/NgBeO | H/NgBeO− | HNg−/BeO | HNg/BeO− |
|---|---|---|---|---|
| Ar | −9.99 | −65.78 | −29.81 | −106.64 |
| Kr | −14.00 | −68.58 | −32.57 | −108.94 |
| Xe | −23.81 | −75.09 | −36.42 | −122.31 |
| Rn | −29.40 | −77.32 | −37.41 | −117.83 |
The analysis of the deformation density Δρorb(r) accompanying the formation of HNgBeO− from the two most favourable interacting pairs H−/NgBeO and HNg−/BeO unravelled that, in both cases, for any Ng, the by far prevailing contribution is the σ polarization of H− or HNg− toward NgBeO or BeO. The corresponding isosurfaces are shown in Fig. 3. The interaction of H− with NgBeO (Fig. 3(a)) produces the polarization of the electronic cloud of the anion already highlighted by the analysis of the H(r) (vide supra), and a charge accumulation particularly at the BeO moiety. The ensuing stabilization ranges between −7.71 kcal mol−1 (Ng = Ar) and −24.26 kcal mol−1 (Ng = Rn), and accounts for 77–83% of the total ΔEorb. The interaction of HNg− with BeO (Fig. 3(b)) produces the polarization of both Ng and H− toward BeO, and the ensuing stabilization, ranging between −17.55 kcal mol−1 (Ng = Ar) and −27.69 kcal mol−1 (Ng = Rn), is still the prevailing contribution to the total ΔEorb, with percentages ranging between 59% (Ng = Ar) and 74% (Ng = Rn).
 | ||
| Fig. 3 Deformation densities Δρ(r) (plotted isosurfaces: 0.0008 e a0−3) and the corresponding orbital interaction energies ΔE associated with the two most important pairwise orbital interactions for: (a) H− interacting with NgBeO and (b) HNg− interacting with BeO. The values in parentheses are the percentage contributions to the total ΔEorb. Green and blue zones correspond, respectively, to charge accumulation and charge depletion. | ||
3.2 Harmonic frequencies
The description of the HNgBeO− species as tight complexes between H− and NgBeO emerges also from their vibrational pattern. The latter includes, in fact, the ν(Ng–Be) and ν(Be–O) stretchings, and the doubly-degenerate δ(Ng–Be–O) of the –NgBeO moiety, and the ν(Ng–H) stretching and the δ(H–Ng–Be) bending. The MP2/aVTZ, B3LYP/aVTZ and CCSD(T)/aVTZ harmonic frequencies are compared in Table S3 (ESI†). With respect to the ab initio methods, the B3LYP underestimates the ν(Ng–Be) and overestimates the ν(Be–O). The forthcoming discussion is based on the CCSD(T)/aVTZ values quoted in Table 4.| Species | ν(Ng–Be) | ν(Be–O) | ν(Ng–H) | δ(Ng–Be–O)a | δ(H–Ng–Be)a |
|---|---|---|---|---|---|
| a Doubly-degenerate bending. | |||||
| HArBeO− | 318 (0.4) | 1508 (124) | 569 (1284) | 233 (15) | 134 (713) |
| ArBeO | 272 (2) | 1536 (72) | 155 (64) | ||
| HKrBeO− | 267 (19) | 1495 (117) | 591 (1635) | 250 (8) | 153 (638) |
| KrBeO | 246 (1) | 1533 (77) | 155 (59) | ||
| HXeBeO− | 267 (69) | 1495 (107) | 591 (2273) | 250 (84) | 153 (420) |
| XeBeO | 237 (0.6) | 1527 (78) | 150 (50) | ||
| HRnBeO− | 234 (94) | 1476 (101) | 641 (2548) | 286 (90) | 161 (300) |
| RnBeO | 224 (0.5) | 1522 (81) | 145 (46) | ||
Compared with the triatomic NgBeO, the ν(Be–O) of HNgBeO−, falling between 1476 cm−1 (Ng = Rn) and 1508 cm−1 (Ng = Ar), is red-shifted by ca. 30–40 cm−1. This mirrors the elongation of the Be–O bond that occurs when going from any NgBeO to the corresponding HNgBeO−. The ν(Ng–Be), falling between 224 cm−1 (Ng = Rn) and 318 cm−1 (Ng = Ar), is, instead, blue-shifted by ca. 10–40 cm−1. This is consistent with the increase of the strength of the Ng–Be bond when going from any NgBeO to the corresponding HNgBeO− unravelled by the bonding analysis. The δ(Ng–Be–O), falling between 233 (Ng = Ar) and 286 cm−1 (Ng = Rn), is also blue-shifted with respect to NgBeO by ca. 100 cm−1. The rather tight character of the interaction occurring between H− and NgBeO is, however, best signalled by the relatively high value of the ν(Ng–H) stretching, predicted between 569 cm−1 (Ng = Ar) and 641 cm−1 (Ng = Rn). This absorption is expected to be brilliant. Thus, as shown in Table S3 (ESI†), both the MP2/aVTZ and B3LYP/aVTZ predicted intensities are higher than 1000 km mol−1, and are up to more than 2000 km mol−1 for HRnBeO−. The δ(H–Ng–Be) bending, predicted between 134 cm−1 (Ng = Ar) and 161 cm−1 (Ng = Rn), is also expected to be brilliant. The intense ν(Ng–H) of the HNgBeO− could be in principle diagnostic for their experimental detection, for example, in the gas phase or in cold matrices. In this regard, it is decisive to investigate the conceivable stability or metastability of the anions on the corresponding PES. This issue is discussed in the subsequent paragraph.
3.3 Stability
The stability or metastability of the HNgBeO− species depends on the thermodynamics and the activation barrier (E#) of their conceivable dissociation processes. These include the two-body (2B) reactions (11) and (12), and the three-body (3B) reactions (13) and (14) summarized by the equations:| HNgBeO− → H− + NgBeO | (11) |
| → Ng + HBeO− | (12) |
| → H− + Ng + BeO | (13) |
| → H + Ng + BeO− | (14) |
The values of their ΔEel, ΔE (0 K), ΔE (298.15 K), ΔH (298.15 K), and ΔG (298.15 K) computed at the B3LYP/aVTZ, MP2/aVTZ, and CCSD(T)/aVTZ levels of theory are given in Tables S4–S6 (ESI†), and the CCSD(T)/CBS data, explicitly discussed in this paragraph, are quoted in Table 5. Both the MP2/aVTZ and CCSD(T)/aVTZ values are quite similar to the benchmark CCSD(T)/CBS values, while the B3LYP/aVTZ method overestimates the energetics by ca. 1–2 kcal mol−1.
| Species | H− + NgBeO | H− + Ng + BeO | H + Ng + BeO− | Ng + HBeO− |
E
#![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)
|
|
|---|---|---|---|---|---|---|
| a Energy barrier for the reaction HNgBeO− → Ng + HBeO−. | ||||||
| HArBeO− | ΔEel | 28.4 | 40.1 | 7.6 | −83.2 | 3.0 |
| ΔE (0 K) | 26.9 | 37.7 | 5.1 | −81.5 | 2.5 | |
| ΔE (298.15 K) | 27.0 | 37.7 | 5.0 | −82.2 | 2.0 | |
| ΔH (298.15 K) | 27.6 | 38.9 | 6.2 | −81.6 | 2.0 | |
| ΔG (298.15 K) | 21.3 | 25.9 | −7.2 | −87.4 | 1.8 | |
| HKrBeO− | ΔEel | 30.4 | 43.9 | 11.4 | −79.4 | 6.0 |
| ΔE (0 K) | 28.9 | 41.5 | 8.8 | −77.7 | 5.4 | |
| ΔE (298.15 K) | 29.1 | 41.5 | 8.8 | −78.4 | 5.0 | |
| ΔH (298.15 K) | 29.7 | 42.7 | 10.0 | −77.8 | 5.0 | |
| ΔG (298.15 K) | 23.1 | 29.5 | −3.6 | −83.8 | 4.5 | |
| HXeBeO− | ΔEel | 34.6 | 50.5 | 17.9 | −72.9 | 11.1 |
| ΔE (0 K) | 33.0 | 47.9 | 15.3 | −71.3 | 10.3 | |
| ΔE (298.15 K) | 33.2 | 47.9 | 15.3 | −71.9 | 10.0 | |
| ΔH (298.15 K) | 33.8 | 49.1 | 16.5 | −71.3 | 10.0 | |
| ΔG (298.15 K) | 27.0 | 35.9 | 2.8 | −77.4 | 9.1 | |
| HRnBeO− | ΔEel | 39.4 | 56.0 | 23.4 | −67.4 | 14.4 |
| ΔE (0 K) | 37.6 | 53.3 | 20.6 | −65.9 | 13.5 | |
| ΔE (298.15 K) | 37.9 | 53.4 | 20.7 | −66.5 | 13.2 | |
| ΔH (298.15 K) | 38.5 | 54.5 | 21.9 | −65.9 | 13.2 | |
| ΔG (298.15 K) | 31.6 | 41.3 | 8.2 | −72.0 | 12.3 | |
The 2B channel (11) measures the degree of the interaction between H− and NgBeO. When going from HArBeO− to HRnBeO−, the corresponding ΔEel progressively increases from 28.4 to 39.4 kcal mol−1, and these rather high values confirm the strong character of the interaction between H− and NgBeO suggested by the geometries and bonding analysis. The ΔG (298.15 K) values are also definitely positive, ranging between 21.3 and 31.6 kcal mol−1.
The ΔEel and ΔG (298.15 K) of the 3B channel (13) are even higher, as they include the dissociation energy of NgBeO into Ng + BeO. The ΔEel values range, in particular, between 11.7 (Ng = Ar) and 16.6 kcal mol−1 (Ng = Rn), and these estimates perfectly agree with those reported previously.24
The electron affinity of H, 17.4 kcal mol−1,74 is lower than that of BeO, 49.6 kcal mol−1,75 and, in fact, the thermodynamic values of reaction (14) are lower than those of reaction (13) by ca. 33 kcal mol−1. The ΔEel of reaction (14) thus ranges between 7.6 (Ng = Ar) and 23.4 kcal mol−1 (Ng = Rn). Once the entropy term is included, the dissociation is predicted to be exoergic for Ng = Ar (−7.2 kcal mol−1) and Ng = Kr (−3.6 kcal mol−1), but endoergic for Ng = Xe (2.8 kcal mol−1) and Ng = Rn (8.2 kcal mol−1).
The 2B channel (12) is largely exothermic, its ΔEel progressively decreasing from −83.2 (Ng = Ar) to −67.4 kcal mol−1 (Ng = Rn). This reaction occurs through the bent transition structures (TSs) whose CCSD(T)/aVTZ optimized geometries are shown in Fig. 4 (likewise the corresponding minima, their CCSD/aVTZ T1 diagnostics are close to the threshold of 0.02). The MP2/aVTZ and B3LYP/aVTZ optimized geometries are also given in Table S7 (ESI†). Compared with the linear HNgBeO− (see Fig. 1), the Ng–H bonds of these TSs (between 2.6842 Å and 2.9587 Å) are longer by ca. 0.3–0.5 Å, and the H–Ng–Be angles (between 91.6° and 114.4°) are appreciably smaller than 180°. There is also a closing of the Ng–Be–O angle by ca. 17–18°. The lengths of the Ng–Be bonds (between 2.0269 Å and 2.4681 Å) feature instead minor shortenings of 0.00015–0.0553 Å. These geometric changes suggest that the activation barriers of reaction (12) mainly arise from the shrinking of the H–Ng–Be angle and the elongation of the Ng–H bond, with an only minor contribution to the reaction coordinate of the dissociation of the Ng atom. The values of E# are, indeed, relatively high and less affected by inclusion of thermal and entropy contributions, with predicted values at 0 K ranging between 2.5 kcal mol−1 for HArBeO− and 13.5 kcal mol−1 for HRnBeO−.
 | ||
| Fig. 4 CCSD(T)/aVTZ optimized bond lengths (Å) and bond angles (°) of the HNgBeO− transition structures (Ng = Ar, Kr, Xe, Rn). | ||
The E# of reaction (12) and the ΔE of reaction (14) can be used to assay the conceivable metastability of HNgBeO−. Thus, in a benchmark study reported so far,76 it was found that for a HNgY system, in order to have a life-time of ca. 100 seconds for spectroscopic studies in the gas phase at 100 K, the 3B channel HNgY → H + Ng + Y and the 2B channel HNgY → Ng + HY must have barriers, respectively, of 13 and 8 kcal mol−1. As shown in Table 5, for HArBeO− and HKrBeO−, even the ΔE (0 K) values (5.1/8.8 kcal mol−1 and 2.5/5.4 kcal mol−1, respectively) are lower than these limits, and put into question the metastability of these species even under the conditions occurring, for example, in a cold matrix at a few kelvin. On the other hand, for HXeBeO− and HRnBeO−, even the ΔE (298.15 K) of reaction (14) (15.3 and 20.7 kcal mol−1, respectively) and the E# of reaction (12) (10.0 and 13.2 kcal mol−1, respectively) are definitely higher than the thresholds for metastability. This encourages the experimental search, particularly of HXeBeO−, overall appearing as a difficult but not impossible task.
3.4 Comparison with other noble-gas anions
The HNgBeO− species are briefly compared here with other observed or predicted noble-gas anions. Based on their geometries, one could first surmise a relationship with the FNgO− predicted by Hu and co-workers,77 and with the other “inserted” Ng anions FNgX− (X = S, Se, BN) that emerged after this first report.78–81 The compact structure of these ions, however, arises from the strong F−-induced stabilization of the singlet NgO and NgX that are intrinsically unstable or only marginally stable on their PES. This bonding motif is, indeed, substantially different from that occurring in the HNgBeO− species, instead best related to that observed in other Ng species, particularly of xenon, that were observed in the gas phase or in synthesized salts. A first one is the XeF3− species. This ion was detected in the gas phase from the collisionally-stabilized addition of F− to XeF2,82 and was subsequently established as the intermediate in the exchange between F− and XeF2 in the CH3CN solution.83 No salts of XeF3− could, however, be prepared,83 and this was explained by the predicted ground-state structure of XeF3−,83 described as a complex between F− and XeF2, stabilized by the weak covalent interaction of the fluoride anion with the positively-charged Xe atom of xenon difluoride. This situation is clearly reminiscent of the weak covalent bond between H− and the σ-hole at the Xe atom of XeBeO occurring in the HXeBeO− species. Interestingly, the hydride ion affinity of XeBeO as measured by the ΔH (298.15 K) of reaction (11), 34.6 kcal mol−1, is significantly higher than the fluoride ion affinity of XeF2, experimentally measured as 19.1 kcal mol−1.82 This suggests that species such as FNgBeO−, ClNgBeO−, and other similar anions could be even more stable than HNgBeO−, thus inviting both their theoretical and experimental investigation in the next future. A bonding motif similar to that occurring in the HNgBeO− species is also behind the stability of the [X3(XeO3)3]3− and [X4(XeO3)4]4− anions (X = Br and Cl) more recently isolated in crystal lattices by Schrobilgen and his co-workers.84 In these peculiar Xe-cage structures, the halide anions form multiple weak covalent bonds with the σ-hole at the Xe atom of XeO3, and these interactions are sufficient to ensure the overall stability of the cage.4. Conclusions
The linear HNgBeO− (Ng = Ar–Rn) species are the first theoretically-predicted examples of anionic noble-gas hydrides. Their stability is driven essentially by the favourable contact between H− and the σ-hole at the Ng atom of NgBeO, producing partially-covalent Ng–H bonds. These anions possess rather compact structures, and reside in potential wells deep enough to suggest their challenging but not impossible detection in the gas phase or in cold matrices. Other anionic species such as FNgBeO− or ClNgBeO− could be even more stable, as suggested also by the comparison of the HNgBeO− species with the gaseous XeF3−, and [X3(XeO3)3]3− and [X4(XeO3)4]4− (X = Br and Cl) isolated in crystal lattices. The experimental and theoretical search for HNgBeO− and other related species could further expand our current knowledge about the intriguing chemistry of noble-gas anions.Author contributions
Stefano Borocci: methodology, data curation – reviewing and editing. Patrizio Cecchi: reviewing and editing. Felice Grandinetti: conceptualization – methodology – writing – original draft preparation. Nico Sanna: methodology, software – reviewing and editing. Costantino Zazza: methodology, data curation – reviewing and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the “Departments of Excellence-2018” Program (Dipartimenti di Eccellenza) of the Italian Ministry of Education, University and Research, DIBAF Department of University of Tuscia, Project “Landscape 4.0 – food, wellbeing and environment”. CZ acknowledges the support from the Rome Technopole foundation within the PNRR action in the field of the NextGenerationEU – section 4: “Digital, Industry and Aerospace”.References
- T. R. Hogness and E. G. Lunn, Phys. Rev., 1925, 26, 44–55 CrossRef CAS.
- F. Grandinetti, Front. Chem., 2020, 8, 462 CrossRef CAS PubMed.
- R. C. Fortenberry and L. Wiesenfeld, Molecules, 2020, 25, 2183 CrossRef CAS PubMed.
- S. Adhikari, M. Baer and N. Sathyamurthy, Int. Rev. Phys. Chem., 2022, 41, 49–93 Search PubMed.
- L. Gonzalez-Sanchez, N. Sathyamurthy and F. A. Gianturco, Phys. Chem. Chem. Phys., 2023, 25, 23370–23383 RSC.
- M. J. Montes de Oca-Estévez and R. Prosmiti, Front. Chem., 2021, 9, 187 Search PubMed.
- M. J. Montes de Oca-Estévez and R. Prosmiti, J. Mol. Graph. Model., 2023, 108562 CrossRef PubMed.
- M. J. Montes de Oca-Estévez, B. Darna, B. G. Ruiz, R. Prosmiti, T. González-Lezana and D. Koner, ChemPhysChem, 2023, e202300450 CrossRef PubMed.
- O. Denis-Alpizar, L. D. Cabrera-González, D. Páez-Hernández and R. Pino-Rios, AACS Earth Space Chem., 2022, 6, 1924–1929 CrossRef CAS.
- O. Denis-Alpizar, L. D. Cabrera-González, G. Orellana-González and D. Páez-Hernández, ACS Earth Space Chem., 2023, 7, 212–217 CrossRef CAS.
- J. Bretón, J. Hernandez-Rojas, M. I. Hernández, J. Campos-Martínez and T. González-Lezana, ChemPhysChem, 2023, e202300425 CrossRef PubMed.
- J. A. Tan and J. L. Kuo, Molecules, 2022, 27, 3198 CrossRef CAS PubMed.
- J. A. Tan and J. L. Kuo, J. Chem. Phys., 2021, 154, 134302 CrossRef CAS PubMed.
- M. J. Barlow, B. M. Swinyard, P. J. Owen, J. Cernicharo, H. L. Gomez, R. J. Ivison, O. Krause, T. L. Lim, M. Matsuura, S. Miller, G. Olofsson and E. T. Polehampton, Science, 2013, 342, 1343–1345 CrossRef CAS PubMed.
- P. Schilke, D. A. Neufeld, H. S. P. Müller, C. Comito, E. A. Bergin, D. C. Lis, M. Gerin, J. H. Black, M. Wolfire, N. Indriolo, J. C. Pearson, K. M. Menten, B. Winkel, Á. Sánchez-Monge, T. Möller, B. Godard and E. Falgarone, Astron. Astrophys., 2014, 566, A29 CrossRef.
- H. S. P. Müller, S. Muller, P. Schilke, E. A. Bergin, J. H. Black, M. Gerin, D. C. Lis, D. A. Neufeld and S. Suri, Astron. Astrophys., 2015, 582, L4 CrossRef.
- R. Güsten, H. Wiesemeyer, D. Neufeld, K. M. Menten, U. U. Graf, K. Jacobs, B. Klein, O. Ricken, C. Risacher and J. Stutzki, Nature, 2019, 568, 357–362 CrossRef PubMed.
- R. C. Fortenberry, Mol. Astrophys., 2020, 18, 100062 CrossRef.
- M. Pettersson, J. Lundell and M. Räsänen, J. Chem. Phys., 1995, 102, 6423–6431 CrossRef CAS.
- L. Khriachtchev, M. Pettersson, N. Runeberg, J. Lundell and M. Räsänen, Nature, 2000, 406, 874–876 CrossRef CAS PubMed.
- L. Khriachtchev, M. Räsänen and R. B. Gerber, Acc. Chem. Res., 2009, 42, 183–191 CrossRef CAS PubMed.
- W. Grochala, L. Khriachtchev and M. Räsänen, Noble-Gas Chemistry, in Physics and Chemistry at Low Temperatures, ed. L. Khriachtchev, CRC Press, 2011, ch. 13, pp. 419–446 Search PubMed.
- J. P. Harris, D. R. Manship, W. H. Breckenridge and T. G. Wright, J. Chem. Phys., 2014, 140, 084304 CrossRef PubMed.
- Q. Zhang, M. Chen, M. Zhou, D. M. Andrada and G. Frenking, J. Phys. Chem. A, 2015, 119, 2543–2552 CrossRef CAS PubMed.
- Q. Zhang, W.-L. Li, L. Zhao, M. Chen, M. Zhou, J. Li and G. Frenking, Chem. – Eur. J., 2017, 23, 2035–2039 CrossRef CAS PubMed.
- S. Pan, S. Jalife, R. M. Kumar, V. Subramanian, G. Merino and P. K. Chattaraj, ChemPhysChem, 2013, 14, 2511–2517 CrossRef CAS PubMed.
- S. Pan, D. Moreno, J. L. Cabellos, J. Romero, A. Reyes, G. Merino and P. K. Chattaraj, J. Phys. Chem. A, 2013, 118, 487–494 CrossRef PubMed . (2013_53).
- S. Pan, D. Moreno, J. L. Cabellos, G. Merino and P. K. Chattaraj, ChemPhysChem, 2014, 15, 2618–2625 CrossRef CAS PubMed.
- R. Saha, S. Pan, G. Merino and P. K. Chattaraj, J. Phys. Chem. A, 2015, 119, 6746–6752 CrossRef CAS PubMed.
- S. Pan, M. Ghara, S. Ghoshand and P. K. Chattaraj, RSC Adv., 2016, 6, 92786–92794 RSC.
- S. Borocci, F. Grandinetti and N. Sanna, Molecules, 2021, 26, 4477 CrossRef CAS PubMed.
- S. Borocci, F. Grandinetti and N. Sanna, Chem. Phys. Lett., 2021, 768, 138402 CrossRef CAS.
- J. S. Murray and P. Politzer, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 153–163 CAS.
- P. Politzer and J. S. Murray, Theor. Chem. Acc., 2002, 108, 134–142 Search PubMed.
- T. Clark, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2013, 3, 13–20 Search PubMed.
- P. Politzer, J. S. Murray, T. Clark and G. Resnati, Phys. Chem. Chem. Phys., 2017, 19, 32166–32178 RSC.
- C. Møller and M. Plesset, Phys. Rev., 1934, 46, 618 CrossRef.
- K. Raghavachari, G. W. Trucks, J. A. Pople and M. Head-Gordon, Chem. Phys. Lett., 1989, 157, 479–483 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785 CrossRef CAS PubMed.
- B. P. Pritchard, D. Altarawy, B. Didier, T. D. Gibson and T. L. Windus, J. Chem. Inf. Model., 2019, 59, 4814–4820 CrossRef CAS PubMed.
- K. A. Peterson, D. Figgen, E. Goll, H. Stoll and M. Dolg, J. Chem. Phys., 2003, 119, 11113–11123 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- CFOUR, a quantum chemical program package written by J. F. Stanton, J. Gauss, M. E. Harding and P. G. Szalay, with contributions from A. A. Auer, R. J. Bartlett, U. Benedikt, C. Berger, D. E. Bernholdt, Y. J. Bomble, O. Christiansen, M. Heckert, O. Heun, C. Huber, T.-C. Jagau, D. Jonsson, J. Jusélius, K. Klein, W. J. Lauderdale, D. A. Matthews, T. Metzroth, D. P. O’Neill, D. R. Price, E. Prochnow, K. Ruud, F. Schiffmann, S. Stopkowicz, A. Tajti, J. Vázquez, F. Wang and J. D. Watts, and the integral packages MOLECULE (J. Almlöf and P. R. Taylor), PROPS (P. R. Taylor), ABACUS (T. Helgaker, H. J. Aa. Jensen, P. Jørgensen and J. Olsen), and ECP routines by A. V. Mitin and C. van Wüllen, https://www.cfour.de.
- A. Halkier, T. Helgaker, P. Jørgensen, W. Klopper, H. Koch, J. Olsen and A. K. Wilson, Chem. Phys. Lett., 1998, 286, 243–252 CrossRef CAS.
- D. A. McQuarry, Statistical Mechanics, Harper & Row, New York, 1976 Search PubMed.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, C. R. Landis and F. Weinhold, NBO 6.0, Theoretical Chemistry Institute, University of Wisconsin, Madison, 2013 Search PubMed.
- R. F. W. Bader, Atoms in Molecules: a Quantum Theory, Oxford University Press, Oxford, 1990 Search PubMed.
- S. Borocci, M. Giordani and F. Grandinetti, J. Phys. Chem. A, 2015, 119, 6528–6541 CrossRef CAS PubMed.
- D. Cremer and E. Kraka, Angew. Chem., Int. Ed. Engl., 1984, 23, 627–628 CrossRef.
- D. Cremer and E. A. Kraka, Croat. Chem. Acta, 1984, 57, 1259–1281 Search PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sanchez, J. Contreras-Garcia, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- C. Narth, Z. Maroun, R. A. Boto, R. Chaudret, M. L. Bonnet, J.-P. Piquemal and J. A. Contreras-García, Applications of Topological Methods in Molecular Chemistry, Springer, Cham, 2016, pp. 491–527 Search PubMed.
- G. Saleh, C. Gatti and L. Lo Presti, Comput. Theor. Chem., 2015, 1053, 53–59 CrossRef CAS.
- T. Ziegler and A. Rauk, Theor. Chim. Acta, 1977, 46, 1–10 CrossRef CAS.
- M. Mitoraj and A. Michalak, J. Mol. Model., 2007, 13, 347–355 CrossRef CAS PubMed.
- A. Michalak, M. Mitoraj and T. Ziegler, J. Phys. Chem. A, 2008, 112, 1933–1939 CrossRef CAS PubMed.
- M. Mitoraj, A. Michalak and T. Ziegler, J. Chem. Theory Comput., 2009, 5, 962–975 CrossRef CAS PubMed.
- R. F. Nalewajski and J. Mrozek, Int. J. Quantum Chem., 1994, 51, 187–200 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- W. Zou, Molden2AIM, https://github.com/zorkzou/Molden2AIM.
- T. J. Lee and P. R. Taylor, Int. J. Quantum Chem., 1989, 36(S23), 199–207 CrossRef.
- P. Pyykkö, J. Phys. Chem. A, 2015, 119, 2326–2337 CrossRef PubMed.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- M. Rahm, R. Hoffmann and N. W. Ashcroft, Chem. – Eur. J., 2016, 22, 14625–14632 CrossRef CAS PubMed.
- S. Borocci, F. Grandinetti, N. Sanna, P. Antoniotti and F. Nunzi, J. Comput. Chem., 2019, 40, 2318–2328 CrossRef CAS PubMed.
- S. Borocci, F. Grandinetti, N. Sanna and F. Nunzi, New J. Chem., 2020, 44, 14536–14550 RSC.
- S. Borocci, F. Grandinetti and N. Sanna, J. Chem. Phys., 2022, 156, 014104 CrossRef CAS PubMed.
- E. Espinosa, I. Alkorta, J. Elguero and E. Molins, J. Chem. Phys., 2002, 117, 5529–5542 CrossRef CAS.
- R. Tonner and G. Frenking, Chem. – Eur. J., 2008, 14, 3260–3272 CrossRef CAS PubMed.
- Q. Zhang, W.-L. Li, C.-Q. Xu, M. Chen, M. Zhou, J. Li, D. M. Andrada and G. Frenking, Angew. Chem., Int. Ed., 2015, 54, 11078–11083 CrossRef CAS PubMed.
- D. M. Andrada and G. Frenking, Angew. Chem., Int. Ed., 2015, 54, 12319–12324 CrossRef CAS PubMed.
- S. J. Grabowski, J. M. Ugalde, D. M. Andrada and G. Frenking, Chem. – Eur. J., 2016, 22, 11317–11328 CrossRef CAS PubMed.
- R. C. Shiell, X. K. Hu, Q. C. J. Hu and J. W. Hepburn, Faraday Disc. Chem. Soc., 2000, 115, 331 RSC.
- K. J. Mascaritolo, A. R. Dermer, M. L. Green, A. M. Gardner and M. C. Heaven, J. Chem. Phys., 2017, 146, 054301 CrossRef PubMed.
- T.-H. Li, Y.-L. Liu, R.-J. Lin, T.-Y. Yeh and W.-P. Hu, Chem. Phys. Lett., 2007, 434, 38–41 CrossRef CAS.
- T.-H. Li, C.-H. Mou, H.-R. Chen and W.-P. Hu, J. Am. Chem. Soc., 2005, 127, 9241–9245 CrossRef PubMed.
- Y.-L. Liu, Y.-H. Chang, T.-H. Li, H.-R. Chen and W.-P. Hu, Chem. Phys. Lett., 2007, 439, 14–17 CrossRef CAS.
- P. Antoniotti, N. Bronzolino, S. Borocci, P. Cecchi and F. Grandinetti, J. Phys. Chem. A, 2007, 111, 10144–10151 CrossRef CAS PubMed.
- S. Borocci, N. Bronzolino and F. Grandinetti, Chem. Phys. Lett., 2008, 458, 48–53 CrossRef CAS.
- N. Bronzolino, S. Borocci and F. Grandinetti, Chem. Phys. Lett., 2009, 470, 49–53 CrossRef.
- I. H. Krouse, C. Hao, C. E. Check, K. C. Lobring, L. S. Sunderlin and P. G. Wenthold, J. Am. Chem. Soc., 2007, 129, 846–852 CrossRef CAS PubMed.
- N. Vasdev, M. D. Moran, H. M. Tuononen, R. Chirakal, R. J. Suontamo, A. D. Bain and G. J. Schrobilgen, Inorg. Chem., 2010, 49, 8997–9004 CrossRef CAS PubMed.
- J. T. Goettel, V. Haensch and G. J. Schrobilgen, J. Am. Chem. Soc., 2017, 139, 8725–8733 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: MP2/aVTZ and B3LYP/aVTZ optimized geometries, harmonic vibrational frequencies and dissociation energies, and CCSD(T)/aVTZ AIM indices of HNgBeO−. See DOI: https://doi.org/10.1039/d3cp05623f |
| This journal is © the Owner Societies 2024 |