
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The role of the oxime group in the excited state deactivation processes of indirubin†
Danîela C.
Nobre
a,
Estefanía
Delgado-Pinar
 ab,
Carla
Cunha
a and
J.
Sérgio Seixas de Melo
*a
ab,
Carla
Cunha
a and
J.
Sérgio Seixas de Melo
*a
aUniversity of Coimbra, CQC-ISM, Department of Chemistry, P3004-535 Coimbra, Portugal. E-mail: sseixas@ci.uc.pt
bInstituto de Ciencia Molecular, Departamento de Química Inorgánica, Universidad de Valencia, C/Catedrático José Beltrán 2, 46980, Paterna, Valencia, Spain
First published on 6th February 2024
Abstract
The introduction of an oxime group into indirubin (INR) derivatives, including INROx, MINROx, and 6-BrINROx, and its impact on the spectral and photophysical properties of INR was investigated using a combination of fast-transient absorption (fs-TA/fs-UC) and steady-state fluorescence techniques. The oxime group introduces structural modifications that promote a rapid keto–enol tautomeric equilibrium and enhance the excited-state proton transfer (ESPT) process compared to its analogue, INR. In the oxime–indirubin derivatives investigated, the ESPT process is notably more efficient than what is observed in INR and indigo, occurring extremely fast (<1 ps) in all solvents, except for the viscous solvent glycerol. The more rapid deactivation mechanism precludes the formation of an intermediate species (syn-rotamer), as observed with INR. These findings are corroborated by time-dependent density functional theory (TDDFT) calculations. The work demonstrates that introducing an oxime group to INR, whether in nature or in the laboratory, results in an enhancement of its photostability.
Introduction
While typically associated with plant products, indigo (IND) and the indirubins (INRs) have been documented in four distinct sources: (i) various plants, including Baphicacanthus cusia, Polygonum tinctorium, Isatis indigotica, Indigofera suffrutticosa, and Indigofera tinctoria; (ii) several marine mollusks, primarily within the Muricidae family of gastropods; (iii) natural or recombinant bacteria; and (iv) human urine.1–5Indirubin, a dark-red 3,2′-bisindole isomer of indigo, is also recognized as an active ingredient in Danggui Longhui Wan, a Traditional Chinese Medicine (TCM) employed in the treatment of chronic diseases like chronic myelogenous leukemia.6–10 In addition to cyclin-dependent kinases (CDKs), indirubins have been observed to interact with glycogen synthase kinase-3 (GSK-3), aurora kinases, and the aryl hydrocarbon receptor (AhR), also known as the dioxin receptor.2,3,11–13 Over the past decade, several indirubin analogs have been synthesized to enhance this promising drug scaffold.14,15 Incorporation of oxime groups enhances the kinase inhibitory activity of natural compounds.16,17 These findings have prompted a range of studies related to their physiological and pharmacological functions. Several indirubin derivatives have been synthesized and characterized for their kinase inhibition and anticancer properties.18 In this context, incorporating an oxime group into the backbone has proven to be a viable strategy for developing cytotoxic agents, and numerous oxime derivatives have been recognised for their therapeutic activity against cancer and neurodegenerative disorders.16
In the last two decades, a significant number of oximes have been documented to possess valuable pharmaceutical properties, including compounds with antibacterial, anticancer, anti-arthritis, anti-inflammatory activities, and anti-human immunodeficiency (HIV) agents capable of inhibiting HIV protease.16,19 The oxime group contains two H-bond acceptors (nitrogen and oxygen atoms) and one H-bond donor (OH group), in contrast to carbonyl groups, which have only one H-bond acceptor. This distinctive characteristic, combined with the high polarity of oxime groups, can lead to a notably different interaction mode with receptor binding sites when compared to corresponding carbonyl compounds, even in cases of slight differences in the overall size and shape of the compound.16
Considerable research has been undertaken on indirubins and oxime–indirubin derivatives, with a primary focus on biological aspects of these compounds. Nonetheless, their photophysical and photochemical properties have mostly remained unexplored.
This work involves the synthesis and comprehensive examination of various oxime–indirubin derivatives, which have shown promise for various biological applications. The study is focused on exploring the impact of substituting the oxime group on the electronic spectral, photophysical, and photochemical properties of the compounds in comparison to indirubin. The observed properties and behavior were further elucidated through TDDFT computational studies.
Results and discussion
Synthesis of the oxime–indirubin derivatives: general synthetic procedure
The synthesis of the indirubin-3′-oxime derivatives was carried out following a modified procedure described elsewhere.20 The corresponding indirubin derivative (100 mg) was placed in a round-bottom flask, and 10 mL of pyridine was added. After 5 minutes of stirring, hydroxylamine hydrochloride (10 equiv.) was introduced, and the solution was heated under reflux for 2 h. Then, the solvent was evaporated under reduced pressure, and the residue was washed with the appropriate solvent, see Scheme 1. The molecular structures of all the indirubin derivatives and oxime–indirubin derivatives were confirmed by 1H and 13C nuclear magnetic resonance (NMR) spectroscopy and further by high resolution mass spectrometry (HRMS), please see Fig. SI1–SI16 (ESI†). The obtained data were found in accordance with the structure proposed. | ||
| Scheme 1 Synthetic pathway for the synthesis of the oximes: indirubin–3′-oxime (INROx), 6-bromoindirubin-3′-oxime (6-BrINROx) and N-methylindirubin-3′-oxime (MINROx). | ||
The influence of the oxime group on the spectral and photophysical properties of indirubin (INR)
Absorption and fluorescence spectra of oxime–indirubin derivatives (INROx, 6-BrINROx, and MINROx) were recorded in various organic solvents with different dielectric constant (ε) and viscosities (η) at room temperature (T = 293 K).Fig. 1 presents the absorption and fluorescence emission spectra of the oxime–indirubin derivatives in dioxane. For the sake of comparison, previously acquired data for indirubin (INR) in dioxane were also considered.21
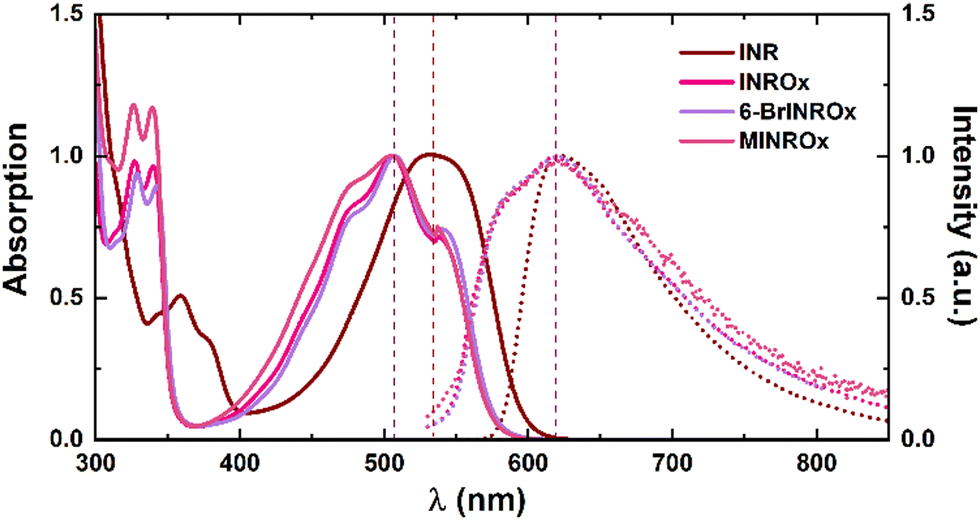 | ||
| Fig. 1 Normalized absorption (solid line) and fluorescence emission (dashed line) spectra of INROx, 6-BrINROx and MINROx in dioxane at room temperature. Indirubin (INR) spectra are also presented for comparison. The dashed vertical lines are just meant to be guidelines for the eye. | ||
Table 1 highlights important characteristics of the oxime–indirubin derivatives, including that (i) the absorption wavelength maxima of oxime–indirubin derivatives are blue-shifted by approximately 24–40 nm, when compared to indirubin (INR). This indicates that (i) the fundamental core responsible for the highest occupied molecular orbital (HOMO) – lowest unoccupied molecular orbital (LUMO) transition is affected by the oxime group substitution; (ii) in comparison with INR, it is noteworthy that oxime–indirubin derivatives exhibit a significantly larger Stokes shift value. Indeed, oxime–indirubin derivatives display a broad fluorescence spectrum, providing direct evidence that the emission spectra likely involve convolution of more than one contributing species, with a faster keto–enol equilibrium compared to indirubin; (iii) also, in the oxime–indirubin derivatives, the substituent (bromide-, methyl- or non-substituted) and the polarity of the solvents do not appear to interfere significantly with the absorption and emission maxima.
| Compound | η | ε | Solvent | λ absmax (nm) | λ emmax (nm) | Δ SS (cm−1) |
|---|---|---|---|---|---|---|
| a Data from ref. 21. | ||||||
| INRa | 1.20 | 2.209 | Dx | 530 | 640 | 3243 |
| 0.58 | 7.58 | 2-MeTHF | 531 | 635 | 3084 | |
| 0.92 | 36.7 | DMF | 546 | 638 | 2641 | |
| 1.41 | 42.5 | Glycerol | 548 | 676 | 3455 | |
| INROx | 1.20 | 2.209 | Dx | 506 | 617 | 3555 |
| 0.48 | 7.58 | THF | 507 | 616 | 3490 | |
| 0.92 | 36.7 | DMF | 506 | 616 | 3529 | |
| 0.33 | 37.5 | ACN | 502 | 620 | 3791 | |
| 1.41 | 42.5 | Glycerol | 510 | 620 | 3479 | |
| 6-BrINROx | 1.20 | 2.209 | Dx | 507 | 617 | 3516 |
| 0.48 | 7.58 | THF | 507 | 618 | 3543 | |
| 0.92 | 36.7 | DMF | 506 | 622 | 3686 | |
| 0.33 | 37.5 | ACN | 503 | 620 | 3752 | |
| MINROx | 1.20 | 2.209 | Dx | 507 | 620 | 3595 |
| 0.48 | 7.58 | THF | 507 | 616 | 3490 | |
| 0.92 | 36.7 | DMF | 507 | 624 | 3698 | |
| 0.33 | 37.5 | ACN | 502 | 620 | 3791 | |
The introduction of the oxime group in oxime–indirubin derivatives results in significant electronic spectral and photophysical changes when compared to INR. These changes arise from distinct contributions to the potential energy curves of the ground and excited states of the compounds. These shows that the deactivation mechanism varies between INR and INROx. Additional insights will be obtained from the time-resolved data presented in Table 2 and will be explained at the light of the DFT and TDDFT calculations.
| Compound | Solvent | ϕ F | τF (ps) | k F (s−1) × 107 | k NR (s−1) × 1011 | k NR/kF × 104 |
|---|---|---|---|---|---|---|
| INR | Dx | 0.0020 | 38 | 5.26 | 0.263 | 499 |
| Glycerol | 0.0043 | 50 | 8.60 | 0.199 | 232 | |
| INROx | Dx | 0.00004 | 6.8 | 0.587 | 1.47 | 0.0025 |
| THF | 0.00001 | 4.9 | 0.200 | 2.00 | 0.010 | |
| ACN | 0.00001 | 3.0 | 0.330 | 3.30 | 0.010 | |
| Glycerol | 0.00800 | 93 | 8.60 | 0.11 | 124 | |
| 6-BrINROx | Dx | 0.00005 | 7.9 | 0.637 | 1.27 | 0.002 |
| THF | 0.00002 | 5.6 | 0.357 | 1.79 | 0.005 | |
| ACN | 0.00001 | 4.1 | 0.242 | 2.42 | 0.010 | |
| MINROx | Dx | 0.00001 | 6.8 | 0.147 | 1.47 | 0.010 |
| THF | 0.00001 | 4.6 | 0.217 | 2.17 | 0.010 | |
| ACN | 0.00005 | 3.5 | 1.4 | 2.86 | 0.002 | |
Photophysical data, including fluorescence quantum yields (ϕF), fluorescence lifetimes (τF) and radiative (kF) and radiationless rate constants (kNR) for the oxime–indirubin derivatives are provided in Table 2. When comparing the oxime–indirubin derivatives to the parent compound (INR), three main observations can be highlighted: (i) oxime–indirubin derivatives exhibit fluorescence quantum yields that are at least one order of magnitude lower than INR; (ii) quantum yield values increase significantly with the increase of the viscosity of the solvent; (iii) in dioxane, the lifetimes are shorter for the oxime–indirubin derivatives, consistent with an ultrafast keto–enol tautomerization equilibrium.
Very often, molecules capable of undergoing excited-state proton transfer (ESPT) have the possibility to establish intramolecular hydrogen bonding interactions between a hydrogen-bond donor (e.g., –OH and NH2) and a hydrogen-bond acceptor (e.g., C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N– and CO).22–32 Tautomerism is a form of isomerization involving species that can easily interconvert. One of the most common type of tautomerism is prototropic tautomerism, which involves a shift in the position of a proton within a molecule.28,33–36 Even though it may seem like a subtle change, this process leads to substantial modifications in chemical bonding and polarity, ultimately determining molecular properties.
N– and CO).22–32 Tautomerism is a form of isomerization involving species that can easily interconvert. One of the most common type of tautomerism is prototropic tautomerism, which involves a shift in the position of a proton within a molecule.28,33–36 Even though it may seem like a subtle change, this process leads to substantial modifications in chemical bonding and polarity, ultimately determining molecular properties.
To confirm the presence of an efficient ESPT mechanism in the oxime–indirubin derivatives, femtosecond transient absorption (fs-TA) and femtosecond Up-Conversion (fs-UC) spectroscopy experiments were carried out in various solvents. The fs-TA experiments were carried out in the 440–800 nm range, covering various aerated solvents with different viscosities and dielectric constants. An excitation wavelength of 500 nm was employed for the oxime–indirubin derivatives, whereas for INR, excitation was performed at 530 nm.21Fig. 2 displays the fs-TA spectrum for INROx, MINROx and 6-BrINROx in dioxane. The transient spectrum for INR in dioxane has been previously described.21
 | ||
| Fig. 2 Time-resolved transient absorption data for indirubin and oxime–indirubin derivatives in dioxane obtained with λexc = 500 nm and 530 nm at T = 293 K. The vertical dashed line is just meant to be a guideline to the eye, showing the spectral shift of the ESA band for each compound. | ||
For the oxime–indirubin derivatives, the fs-TA spectra exhibit positive and broad transient absorption bands in the 530–630 nm range, with maxima at around 580 nm, attributed to an excited singlet-state absorption (ESA). This is accompanied by a negative band in the long wavelength region (640–730 nm) of the spectra, associated to stimulated emission (SE). A bleaching of the ground-state absorption (GSA) in the 440–480 nm range is observed. Comparable variations in the transient absorption bands were noted for the oxime–indirubin derivatives in the other studied solvents.
Characteristic fs-TA data is provided in Table 3. The global analysis of the time-resolved data yields the best-fit results with a bi-exponential decay law, indicating the presence of two excited species for all compounds. In the case of INR, in dioxane, two ESA bands are observed at ∼580–600 nm and 775 nm, which disappear after 38 ps. For the oxime–indirubin derivatives, the fs-TA spectra in dioxane shows one ESA band, observed at ∼545–630 nm, which now disappear after ∼7–8 ps. Both the τ1 (associated with the formation of the keto species and decaying in approximately 1 ps, i.e., – an ultrafast proton transfer) and τ2 (associated with the enol species decaying within 3–8 ps) decay time values, obtained in various organic solvents for the oxime–indirubin derivatives are remarkably fast, especially when compared to the values observed with INR (see Table 3). These results further validate that the significant blue-shift of the fluorescence, observed with the oxime–indirubin derivatives when compared to indirubin, is attributed to an efficient excited-state proton transfer (ESPT) process with high proton transfer rate constant values. In glycerol, formation of the enol species is hindered by the solvent's viscosity, leading to a more efficient radiative decay channel, as indicated by the increase in the ϕF value (and in the radiative rate constant).
| Comp. | Solvent | fs-TA | fs-UC | ||
|---|---|---|---|---|---|
| τ 1 (ps) | τ 2 (ps) | τ 1 (ps) | τ 2 (ps) | ||
| a Data from ref. 21. b The decay component is associated to a negative amplitude (rise-time). | |||||
| INRa | Dx | — | 38 | 3 | 37 |
| MeOH | 2 | 12 | — | — | |
| DMF | 1.8 | 14 | 1 | 8 | |
| Glycerol | 16 | 50 | — | — | |
| INROx | Dx | 0.80b | 6.81 | 0.59 | 4.03 |
| THF | 0.74b | 4.99 | 0.48 | 3.47 | |
| MeOH | 0.72b | 3.71 | — | — | |
| DMF | 0.61b | 3.28 | — | — | |
| ACN | 0.63b | 3.03 | 0.58 | 2.26 | |
| Glycerol | 4.03 | 92.9 | — | — | |
| 6-BrINROx | Dx | 1.12b | 7.85 | 0.9 | 4.69 |
| THF | 0.93b | 5.60 | 0.7 | 3.29 | |
| DMF | 0.91b | 5.28 | — | — | |
| ACN | 0.42b | 4.13 | 0.6 | 2.53 | |
| MINROx | Dx | 0.80b | 6.79 | 0.34 | 4.07 |
| THF | 0.58b | 4.61 | 0.5 | 4 | |
| DMF | 0.59b | 4.39 | — | — | |
| ACN | 0.50b | 3.50 | 0.3 | 2.23 | |
In the oxime–indirubin derivatives, the absorption occurs from the stable keto (K) form in the S0 state, while the emission arises from the favored enol (E*) isomer in the S1 state, indicating significant structural changes in the excited state (ES) and, consequently, an increase in the Stokes shift value. ESPT can be considered as a very fast phototautomerization process occurring along with a strong intramolecular hydrogen bond between two atoms presenting acidities/basicities that are significantly influenced upon electronic excitation. This implies the presence of an even more efficient ESPT process in the oxime–indirubin derivatives compared to indirubin, possibly due to a relatively lower potential energy barrier. Fig. SI17 (ESI†) depicts the energy difference between the ground state and the first keto singlet excited state (K-S1 → K-S0) for INR and INROx in dioxane.
Table 3 shows that the two decay components (τ1 and τ2), corresponding to the presence of the keto and enol forms of the studied compounds, are consistently obtained in different time-resolved experiments (fs-TA and fs-UC). The nature of the species associated with these two decay times will be further elucidated in the context of DFT calculations (as will be discussed below).
As seen in Fig. 3, the fs Up-Conversion spectra in dioxane for the oxime–indirubin derivative INROx display two bands, K* and E*. For τ < 1 ps, the K* and E* coexist, with the former being more intense. For τ > 1 ps, E* becomes dominant.
 | ||
| Fig. 3 fs time-resolved Up-Conversion, fs-UC, (solid line) data for INROx in dioxane obtained with λexc = 500 nm at T = 293 K. The dashed lines and bands in wine and purple show the bands based on the emission maximum calculated (from TDDFT) for the keto (wine line) and enol (purple line) species. See text for more details. | ||
The presence of these two species is consistent with the band maxima predicted by TDDFT, confirming that the enol form is the prevailing tautomer in solution for time values > 1 ps (as indicated in Table 3) and is dominant under steady-state conditions (Fig. 1).
TDDFT calculations
A more comprehensive set of density functional (DFT) data was acquired to obtain a deeper insight into the type of structures existent in the ground and excited state and to offer a rational explanation for the experimentally obtained data (both steady-state and time-resolved). INROx was employed to explore various conformational structures (see Fig. SI18, ESI†). The optimized geometries of the oxime–indirubin conformers can be readily obtained, and their energies calculated (see Fig. SI19 for structures, ESI†). Among the investigated structures, and after obtaining the most stable structure and conformer, the various molecular geometries were optimized using DFT calculations, and the primary transitions for both absorption and emission were analyzed.Fig. SI19 (ESI†) illustrates the most energetically stable conformer for the investigated oxime–indirubin derivatives in dioxane. Fig. SI20 (ESI†) maps the orbital contours of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) for INR and INROx in dioxane. In INROx the HOMO to LUMO transition retains the nature of the donor and acceptor groups observed in INR; however, the comparison between the HOMO and LUMO of the two compounds shows that the HOMO is found, with INROx, to be energetically lower. However, the most interesting feature in INROx is found in the hydrogen bond N–H⋯OC distance, with a value of 174 pm in S0 (compared to 185 pm for INR), whereas in S1, the distance is 162 pm (close to that found for INR of 165 pm), as shown in Fig. 4. This shows that for INROx, when compared to INR, this distance is shorter in S0 and approximately identical in S1. Moreoever, and in general, the bonding distance between the oxygen in CO and the hydrogen in N–H decreases with increasing polarity (see Table SI2 in ESI† for all oxime–indirubin derivatives, in different solvents). Indeed, and as observed with INR, the proximity of the amine hydrogen, in the half-indigo moiety, and the keto oxygen, in the isatin moiety, significantly affects the photophysical properties of this molecule. Indeed, in the excited state, the formed N–H⋯O hydrogen bond allows the proton, from the isatin moiety, to be transferred to the carbonyl group. Therefore, the E* form of oxime–indirubin derivatives, INROx–OH, can now be generated. The bond distance values show that in both S0 and S1, the N–H⋯OC hydrogen bond distance is reduced in INROx, suggesting a more favourable intramolecular excited-state proton transfer (ESPIT) than that observed with INR. The small hydrogen bond length value obtained in the keto form (K-S1) indicates that the central nitrogen has a stronger affinity for the proton in the excited state, facilitating an ultrafast ESIPT.
 | ||
| Fig. 4 Optimized structures in the S0 (A) and S1 (B) electronic states of INR and INROx, in dioxane. The bond distance between the oxygen in CO and the hydrogen in N–H decreases from 185.1 pm in S0 to 165.1 pm in S1 and 174.1 pm in S0 to 162.1 pm in S1, for INR and INROx, respectively; see text for more details. | ||
Considering all the above, and taking into account the spectral and structural characteristics of these conformers, the following aspects should be highlighted: (i) for all the oxime–indirubin derivatives, conformer C (Fig. SI18, ESI†) is found to be the most stable; (ii) the calculated absorption maxima is predicted to be in different solvents, to be in the range of 470–478 nm, which agrees reasonably well with the experimental λmax; (iii) TDDFT calculations predict an emission band at 543–567 nm for the keto form and an emission band around 614–625 nm for the enol form, in good agreement with the experimental values obtained for the oxime–indirubin derivatives in different solvents (see Tables SI3–SI5, ESI†).
The global picture of the decay mechanism, both in non-polar and polar solvents, for INROx, MINROx and 6-BrINROx, can be detailed by combining steady-state and time-resolved data with TDDFT calculations. Upon photoexcitation, electronic charge redistribution results in a higher acidity for the hydrogen bond donor group and an increased basicity for the hydrogen bond acceptor in the keto (K) form. The keto is the thermodynamically stable form with a tautomerization barrier associated with the formation of an imine-enol tautomer transition state (shown in the dashed frame of Scheme 2). As a result, an extremely fast keto-to-enol phototautomerization event occurs, taking less than 1 ps, with the excited-state keto form (K*) rapidly converting to its excited enol form (E*), which is predicted to have a higher contribution in S1. After decaying back to its electronic ground state, a reverse proton transfer takes place to produce the original K form – ground state intramolecular proton transfer (GSIPT), see Scheme 2. This process is observed with all the studied oxime–indirubin derivatives. However, in glycerol, which has a high viscosity value, formation of the enol is slowed down, as reflected in the longer decay time values associated with this species, in this solvent (Table 2). The relative energy levels of the enol and keto forms of the compounds can be schematically illustrated in Scheme 2, here exemplified for INROx. The DFT optimized geometries of the ground (S0) and first singlet excited (S1) states of both the keto (K) and enol (E) forms of INROx, are depicted in Scheme 2. The efficient ESIPT process, along with an increased Stokes shift, leads to a blue-shift in the emission while maintaining high radiative decay rates values (Table 2).
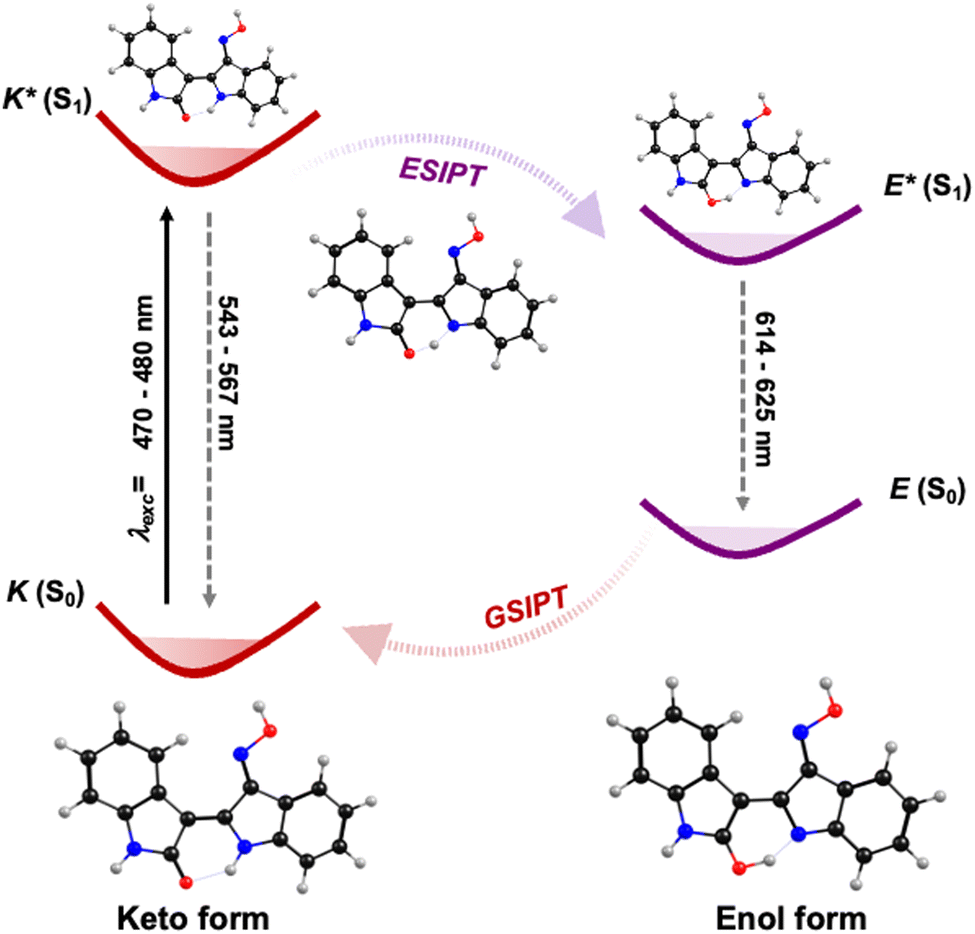 | ||
| Scheme 2 Schematic diagram of the ESPT process in oxime–indirubin derivatives (in dioxane) based on single-molecule tautomerization, here illustrated for INROx. | ||
Conclusions
In this work, we have elucidated the excited state deactivation mechanisms in oxime–indirubin derivatives, which was made by comparison with indirubin. The oxime group has introduced several distinctive features in these compounds. Like indirubin, oxime–indirubin derivatives undergo ESPT. However, a noteworthy distinction is that ESPT occurs extremely rapidly (in less than 1 ps) with all oxime–indirubin derivatives and in all solvents, with the exception of glycerol. The excited state deactivation process involves ESPT in S1, followed by a subsequent tautomerization of the formed excited enol, converting it back to its initial keto form. Importantly, this occurs without the formation of the syn-rotamer, as previously observed with indirubin. Understanding the fast excited state deactivation processes in oxime–indirubin derivatives provides valuable insights into their photophysical behavior that may be correlated with their biological activity, thereby contributing to the development of potential cytotoxic agents for therapeutic applications.Experimental
Material and methods
Chemicals were obtained from commercial sources and used as received. All the solvents (spectroscopic or equivalent grade) were used without further purification. Microwave-assisted synthesis was performed using a CEM Discover S-Class single-mode microwave reactor, featuring continuous temperature, pressure and microwave power monitoring. 1H NMR and 13C NMR spectra were recorded on a Bruker Avance III spectrometer with operating frequencies of 400.13 and 101 MHz, respectively. High-resolution mass spectrometry (HRMS) was performed on a Bruker microTOF-Focus mass spectrometer equipped with an electrospray ionization time-of-flight (ESI-TOF) source.Synthesis
Indirubin-3′-oxime (INROx), (3′E)-indirubin-3′-oxime. Indirubin (INR) (100 mg) was used and the obtained red-orange solid was washed with methanol and dichloromethane (90 mg, 85%).1H NMR (400 MHz, DMSO-d6) δ (ppm): 13.46 (s, 1H, NOH), 11.72 (s, 1H, N′H), 10.69 (s, 1H, NH), 8.63 (d, J = 7.8 Hz, 1H, H4), 8.22 (d, J = 7.6 Hz, 1H, H4′), 7.43–7.38 (m, 2H, H6′-H7′), 7.13 (dd, J = 7.6, 1.28 Hz, 1H, H6), 7.04–7.00 (m, 1H, H5′), 6.95 (dd, J = 7.7, 1.3 Hz, 1H, H5), 6.89 (d, J = 7.8 Hz, 1H, H7). 13C NMR (101 MHz, DMSO-d6) δ (ppm): 170.9 (C2), 151.3 (C3′), 145.2 (C2′), 144.8 (C7′a), 138.3 (C7a), 132.0 (C6′), 127.9 (C4′), 125.9 (C6), 122.9 (C4), 122.6 (C3a), 121.4 (C5′), 120.3 (C5), 116.5 (C3′a), 111.5 (C7′), 108.8 (C7), 98.9 (C3). ESI-MS (m/z): calculated for C16H11N3O2: 277.28; found [L + H]+: 278.0919.
N-Methylindirubin-3′-oxime (MINROx), (2′Z,3′E)-1-methylindirubin-3′-oxime. N-Methylindirubin (MINR) (100 mg) was used as reagent for obtaining the corresponding red oxime. The solid obtained was washed with dichlorometane (80 mg, 76%).
1H NMR (400 MHz, DMSO-d6) δ (ppm): 13.52 (s, 1H, NOH), 11.73 (s, 1H, N′H), 8.68 (d, J = 7.4 Hz, 1H, H4), 8.23 (d, J = 7.5 Hz, 1H, H4′), 7.41–7.40 (m, 2H, H6′-H7′), 7.21 (t, J = 7.9 Hz, 1H, H6), 7.05–7.00 (m, 3H, H5′-H5-H7), 3.30 (s, 1H, N-CH3). 13C NMR (101 MHz, DMSO-d6) δ (ppm): δ = 169.0 (C2), 151.2 (C3′), 145.4 (C2′), 144.8 (C7′a), 139.5 (C7a), 132.0 (C6′), 127.9 (C4′), 125.9 (C6), 122.7 (C4), 121.8 (C3a), 121.6 (C5′), 120.9 (C5), 116.5 (C3′a), 111.6 (C7′), 107.6 (C7), 98.0 (C3), 25.7 (N-CH3). ESI-MS (m/z): calculated for C17H13N3O2: 291.31; found [L + H]+: 292.1075.
6-Bromoindirubin-3′-oxime (6-BrINROx), (2′Z,3′E)-6-bromoindirubin-3′-oxime. 6-Bromoindirubin (6-BrINR) (100 mg) was used as a starting material and the removal of the solvent after the completion of the reaction gave a reddish solid that was washed with methanol and cold dichloromethane (80 mg, 77%).
1H NMR (400 MHz, DMSO-d6) δ (ppm): 13.61 (s, 1H, NOH), 11.75 (s, 1H, N′H), 10.83 (s, 1H, NH), 8.55 (d, J = 8.5 Hz, 1H, H4), 8.22 (d, J = 7.5 Hz, 1H, H4′), 7.43–7.39 (m, 2H, H6′-H7′) 7.11–7.04 (m, 3H, H5′-H5-H7). 13C NMR (101 MHz, DMSO-d6) δ (ppm): 170.6 (C2), 151.3 (C3′), 145.9 (C2′), 144.7 (C7′a), 139.6 (C7a), 132.1 (C6′), 127.9 (C4′), 124.2 (C4), 122.7 (C3a), 122.8 (C5′), 122.0 (C5), 117.8 (C6), 116.4 (C3′a), 111.7 (C7′), 111.5 (C7), 97.7 (C3). ESI-MS (m/z): calculated for C16H10BrN3O2: 355.00; found [L + H]+: 356.0025.
Photophysical measurements
Absorption spectra were recorded on a Shimadzu UV-2600. Fluorescence spectra were recorded in a Horiba-Jobin-Yvon Spex Fluorolog 3-2.2. spectrophotometer and corrected for the instrumental response of the system. The fluorescence quantum yields of the compounds were determined using indirubin (ϕF = 0.0003 in DMF).21Femtosecond Transient Absorption Spectroscopy (fs-TA) experiments were performed with a Helios spectrometer (Ultrafast Systems) with an instrumental response function of ∼250 fs. The instrumental response function of the system was assumed to be equal to that of the pump–probe cross correlation determined from the measurement of the instantaneous stimulated Raman signal from the pure solvent (in a 2 mm cuvette). To avoid photodegradation, the solutions were stirred during the experiments or in movement using a motorized translating sample holder. The spectral chirp of the data was corrected using the Surface Xplorer PRO program from Ultrafast Systems.
Fluorescence decays were measured using a broad band femtosecond fluorescence Up-Conversion, fs-UC, Halcyone Fire spectrometer from Ultrafast Systems (thermoelectrically cooled, −40 °C, CCD detectors with spectral range from the UV, ∼270–400 nm, Vis, 400–800 nm, to the NIR, ∼800–1600 nm), pumped by a 1 kHz Spectra Physics Solstice-Ace laser (7 W, 800 nm and 120 fs IRF) coupled to an TOPAS Prime optical parametric amplifier (OPA) with 235–2600 nm automatic tuning range. The fs-UC spectrometer comprises a delay stage and Ultrafast Systems OPA to generate the gate pulse at 800 nm or 1300 nm with a time resolution of 100 femtoseconds and time window for acquisition up to 8 nanoseconds. The time-resolved fluorescence spectra were obtained with excitation at 450 nm and 530 nm by sum frequency of the fluorescence emission with an 800 nm gate pulse in a BBO crystal. Global analysis of the data was performed using principal component Analysis via single value decomposition (SVD) implemented in the Surface Xplorer Pro program package from Ultrafast Systems.
Quantum electronic calculations
All theoretical calculations were of the DFT type, carried out using GAMESS-US37 version R3, using as functional the implemented version of LC-BPBE (ω = 0.20 au−1). In TDDFT calculations of FC (Franck–Condon) excitations the dielectric constant of the solvent was split into a “bulk” component and a fast component, which is essentially the square of the refractive index. In “adiabatic” conditions only the static dielectric constant is used. A 6-31G** basis set was used in either DFT or TDDFT calculations.The results obtained with the LC-BPBE(20) functional are essentially unscaled raw data from calculations; for the S0 → Sn transitions, a small correction, which result in the subtraction of 0.05 eV, to account for the difference between zero point and the first vibronic level, was considered. For the resulting optimized geometries time dependent DFT calculations (using the same functional and basis set as those in the previously calculations) were performed to predict the vertical electronic excitation energies. Molecular orbital contours were plotted using ChemCraft 1.7 program. Frequency analysis for each compound were also computed and did not yield any imaginary frequencies, indicating that the structure of each molecule corresponds to at least a local minimum on the potential energy surface.
Author contributions
Danîela C. Nobre: investigation, writing – review & editing. Estefanía Delgado-Pinar: methodology, writing – review & editing. Carla Cunha: formal analysis, writing – review & editing. J. Sérgio Seixas de Melo: funding acquisition, conceptualization, project administration, supervision, writing – original & final draft.Conflicts of interest
The authors declare that there are no conflicts to declare.Acknowledgements
We acknowledge funding by FEDER (Fundo Europeu de Desenvolvimento Regional) through COMPETE (Programa Operacional Factores de Competitividade). The Coimbra Chemistry Centre – Institute of Molecular Sciences (CQC-IMS) is supported by the Fundação para a Ciência e a Tecnologia (FCT), Portuguese Agency for Scientific Research, through Projects UIDB/00313/2020 and UIDP/00313/2020. E. D.-P. thanks M. Zambrano contract UP2021-044 from the Spanish MICINN financed by the European Union, Next Generation EU. FCT is also acknowledged for PhD Grants: D. C. N. (ref. SFRH/BD/140890/2018) and C. C. (ref. 2020.09661.BD). NMR data collected at the UC-NMR facility are supported in part by the EDRF through the COMPETE Program and by national funds from the FCT through grants RECI/QEQ-QFI/0168/2012 and CENTRO-07-CT62-FEDER-002012, and also through support to Rede Nacional de Ressonância Magnética Nuclear (RNRMN) and to Coimbra Chemistry Centre through grant UID/QUI/00313/2019. Dr João Pina is acknowledged for the technical help in the fs-TA and fs-UC measurements.References
- D. Olivier, M.-A. Poincelot, S. Douillard, C. Lefevre, J. Moureau, Y. Ferandin, K. Bettayeb, Z. Xiao, P. Magiatis and L. Skaltsounis, Photochem. Photobiol. Sci., 2008, 7, 328–336 CrossRef CAS PubMed.
- T. Blažević, E. H. Heiss, A. G. Atanasov, J. M. Breuss, V. M. Dirsch and P. Uhrin, J. Evidence-Based Complementary Altern. Med., 2015, 2015, 654098 Search PubMed.
- N. Gaboriaud-Kolar, K. Vougogiannopoulou and A.-L. Skaltsounis, Expert Opin. Ther. Pat., 2015, 25, 583–593 CrossRef CAS PubMed.
- E. A. Perpète and D. Jacquemin, THEOCHEM, 2009, 914, 100–105 CrossRef.
- L. Meijer, J. Shearer, K. Bettayeb and Y. Ferandin, Int. Congr. Ser., 2007, 1304, 60–74 CrossRef CAS.
- G. M. Cragg and D. J. Newman, J. Nat. Prod., 2012, 75(3), 311–335 CrossRef PubMed.
- F.-F. Yang, M.-S. Shuai, X. Guan, M. Zhang, Q.-Q. Zhang, X.-Z. Fu, Z.-Q. Li, D.-P. Wang, M. Zhou and Y.-Y. Yang, RSC Adv., 2022, 12, 25068–25080 RSC.
- W. Wang, Y. Wu, H. Xu, Y. Shang, Y. Chen, M. Yan, Z. Li and D. R. Walt, Ind. Crops Prod., 2019, 141, 111783 CrossRef CAS.
- D. T. Nguyen, G. N. Truong, T. Van Vuong, T. N. Van, C. N. Manh, C. T. Dao, T. D. T. Thuy, C. L. Van and V. T. Khac, Chem. Pap., 2019, 73, 1083–1092 CrossRef CAS.
- R. Jautelat, T. Brumby, M. Schäfer, H. Briem, G. Eisenbrand, S. Schwahn, M. Krüger, U. Lücking, O. Prien and G. Siemeister, ChemBioChem, 2005, 6, 531–540 CrossRef CAS PubMed.
- S. Hu, W. Cui, Z. Zhang, S. Mak, D. Xu, G. Li, Y. Hu, Y. Wang, M. Lee and K. W. Tsim, J. Mol. Neurosci., 2015, 57, 561–570 CrossRef CAS PubMed.
- L. Meijer, A. L. Skaltsounis, P. Magiatis, P. Polychronopoulos, M. Knockaert, M. Leost, X. Z. P. Ryan, C. A. Vonica, A. Brivanlou, R. Dajani, C. Crovace, C. Tarricone, A. Musacchio, S. M. Roe, L. Pearl and P. Greengard, Chem. Biol., 2003, 10, 1255–1266 CrossRef CAS PubMed.
- G. Eisenbrand, F. Hippe, S. Jakobs and S. Muehlbeyer, J. Cancer Res. Clin. Oncol., 2004, 130, 627–635 CrossRef CAS PubMed.
- J. Ribas, K. Bettayeb, Y. Ferandin, M. Knockaert, X. Garrofe-Ochoa, F. Totzke, C. Schächtele, J. Mester, P. Polychronopoulos and P. Magiatis, Oncogene, 2006, 25, 6304–6318 CrossRef CAS PubMed.
- O. Choi, Y. Cho, S. Choi, S. Lee, S. Seo, H. Kim, G. Han, D. Min, T. Park and K. Choi, Int. J. Obes., 2014, 38, 1044–1052 CrossRef CAS PubMed.
- I. A. Schepetkin, M. B. Plotnikov, A. I. Khlebnikov, T. M. Plotnikova and M. T. Quinn, Biomolecules, 2021, 11, 777 CrossRef CAS PubMed.
- S.-J. Choi, J.-E. Lee, S.-Y. Jeong, I. Im, S.-D. Lee, E.-J. Lee, S. K. Lee, S.-M. Kwon, S.-G. Ahn and J.-H. Yoon, J. Med. Chem., 2010, 53, 3696–3706 CrossRef CAS PubMed.
- Y. Ichimaru, H. Saito, T. Uchiyama, K. Metori, K. Tabata, T. Suzuki and S. Miyairi, Bioorg. Med. Chem. Lett., 2015, 25, 1403–1406 CrossRef CAS PubMed.
- S. M. M. Lopes, M. Pineiro and E. M. T. Pinho, Molecules, 2020, 25, 3450 CrossRef CAS PubMed.
- P. Polychronopoulos, P. Magiatis, A.-L. Skaltsounis, V. Myrianthopoulos, E. Mikros, A. Tarricone, A. Musacchio, S. M. Roe, L. Pearl, M. Leost, P. Greengard and L. Meijer, J. Med. Chem., 2004, 47, 935–946 CrossRef CAS PubMed.
- D. C. Nobre, E. Delgado-Pinar, C. Cunha, A. M. Galvão and J. S. Seixas de Melo, Dyes Pigm., 2023, 212, 111116 CrossRef CAS.
- G.-J. Zhao and K.-L. Han, Acc. Chem. Res., 2012, 45, 404–413 CrossRef CAS PubMed.
- J. S. Seixas de Melo, in Photochemistry, ed S. Crespi and S. Protti, The Royal Society of Chemistry, 2022, vol. 49, pp. 31–52 Search PubMed.
- D. Pinheiro, M. Pineiro, A. M. Galvão and J. S. Seixas de Melo, Chem. Sci., 2021, 12, 303–313 RSC.
- J. Pina, D. Sarmento, M. Accoto, P. L. Gentili, L. Vaccaro, A. Galvão and J. S. Seixas de Melo, J. Phys. Chem. B, 2017, 121, 2308–2318 CrossRef CAS PubMed.
- J. S. Seixas de Melo and A. L. Maçanita, J. Phys. Chem. B, 2015, 119, 2604–2610 CrossRef CAS PubMed.
- J. S. Seixas de Melo, J. Pina, F. B. Dias and A. L. Maçanita, in Applied Photochemistry, ed. R. C. Evans, P. Douglas and H. D. Burrow, Springer, Netherlands, Dordrecht, 2013, pp. 533–585.
- J. Waluk, in Conformational Analysis of Molecules in Excited States, ed J. Waluk, Wiley-VCH, New York, 1st edn, 2000 Search PubMed.
- C. A. Rumble, J. Breffke and M. Maroncelli, J. Phys. Chem. B, 2017, 121, 630–637 CrossRef CAS PubMed.
- T. Kumpulainen, B. Lang, A. Rosspeintner and E. Vauthey, Chem. Rev., 2017, 117, 10826–10939 CrossRef CAS PubMed.
- C. Sun, X. Su, Q. Zhou and Y. Shi, Org. Chem. Front., 2019, 6, 3093–3100 RSC.
- P. P. Roy, J. Shee, E. A. Arsenault, Y. Yoneda, K. Feuling, M. Head-Gordon and G. R. Fleming, J. Phys. Chem. Lett., 2020, 11, 4156–4162 CrossRef CAS PubMed.
- Y. Shiraishi, C. Yamada and T. Hirai, RSC Adv., 2019, 9, 28636–28641 RSC.
- J. S. Seixas de Melo, C. Cabral, J. C. Lima and A. L. Maçanita, J. Phys. Chem. A, 2011, 115, 8392–8398 CrossRef CAS PubMed.
- J. Seixas de Melo and A. L. Macanita, Chem. Phys. Lett., 1993, 204, 556–562 CrossRef CAS.
- W. R. Laws and L. Brand, J. Phys. Chem., 1979, 83, 795–802 CrossRef CAS.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Marsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis and J. A. Mongomery Jr., J. Comput. Chem., 1993, 14, 1347–1363 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp05260e |
| This journal is © the Owner Societies 2024 |