
The regulatory function of the d-orbital structure in TM@g-t-C4N3 for bifunctional catalysis of the oxygen evolution/reduction reaction†
Zhenduo
Wang
,
Meichen
Wu
,
Yuhong
Huang
 ,
Jianmin
Zhang
* and
Xiumei
Wei
*
,
Jianmin
Zhang
* and
Xiumei
Wei
*
School of Physics and Information Technology, Shaanxi Normal University, Xi’an, 710119, China. E-mail: weixiumei@snnu.edu.cn; jmzhang@snnu.edu.cn
First published on 29th November 2023
Abstract
Highly efficient catalysts for the oxygen evolution/reduction reaction (OER/ORR) have attracted great attention in research for energy devices with high conversion efficiency. Herein, systematic first-principles investigations are performed to explore the catalytic performance of graphitic C4N3 loaded with single transition metal atoms (TM@g-t-C4N3) for the OER/ORR. The results show that Fe, Co, Ni and Rh@g-t-C4N3 exhibit fascinating bifunctional catalytic activities for both the OER and ORR. Moreover, it is observed that better activities are easily achieved when the valence d orbitals of doped TM atoms are nearly fully occupied. Further analysis reveals the volcano relationship between the OER/ORR performance and the adsorption Gibbs free energy. The adsorption free energy of intermediates in the OER/ORR process is also found to highly correlate with the electronic structures of TM@g-t-C4N3, which are mainly characterized by two quantities, one is the descriptor φ related to the electronegativity and the number of valence electrons in d orbitals, and the other is the projected d band center. The results indicate that it is possible to predict the catalytic performance of TM@g-t-C4N3 by a detailed examination of the electronic properties of the doped TM atoms to some extent. This research not only provides several highly active g-t-C4N3-based single-atom catalysts (SACs) for the OER/ORR, but also reveals some potential regularities of SAC systems.
1. Introduction
The energy issue has drawn widespread concerns because of massive consumption of fossil fuels and ever-increasing power demands.1,2 Developing energy devices with high conversion efficiency is a promising strategy to deal with the energy crisis. Among various devices, water splitting cells and fuel cells are paid close attention as water splitting cells can produce clean and high-energy-density hydrogen by using renewable energy and fuel cells are able to efficiently utilize hydrogen energy.3–5 Unfortunately, their commercial applications are hindered by the sluggish electrode reactions, i.e., the oxygen evolution reaction (OER) and oxygen reduction reaction (ORR).6–8 Therefore, catalysts with high activities are desired to accelerate the sluggish reactions. Currently, noble metal-based materials are commonly used as effective catalysts for the OER and ORR.9–14 However, the low reserves and high price restrict the large-scale application of these precious materials. As a consequence, developing alternative catalysts with high activity and low cost becomes more and more urgent in practice.15,16Single-atom catalysts (SACs) have been considered to have broad prospects in catalysis owing to the maximized utilization of loaded metal atoms and the excellent catalytic activity.17,18 Numerous studies have revealed the commendable catalytic ability of SACs for a series of reactions, such as the OER,19–21 ORR,22–24 hydrogen evolution reaction (HER),25–28 nitrogen reduction reaction (NRR),29,30 carbon dioxide reduction reaction (CO2RR),31,32etc. In order to acquire a SAC structure with sufficiently dispersed transition metal (TM) atoms, a suitable substrate is firstly required, which should restrain the aggregation of loaded TM atoms.33 Two-dimensional (2D) carbonaceous materials have shown promising prospects as substrates to anchor TM atoms due to their porous structures and large surface area.34–39 Particularly, graphene (Gr),22–24,34 graphyne (Gy)35,36 and graphitic carbon nitride (g-CxNy)37–39 have been widely reported as appropriate substrates for the anchoring of TM atoms. Among various g-CxNy species, g-C4N3 has received widespread interest due to its unique geometric and electronic structures.40–44 Many reports have already shown that the single-ring g-C4N3 (g-s-C4N3) is appropriate to provide support for SACs, which can efficiently catalyze many reaction processes, such as the NRR,45 OER/ORR46 and oxidation reaction of formaldehyde (HCHO).47 On the other side, another structure of g-C4N3 with tri-ring units ((g-t-C4N3) was reported theoretically in 2013.41 Furthermore, Sakshi Agarwal et al. found that the g-t-C4N3 doped with single TM atoms can catalyze the NRR process with high performance,48 clarifying that the g-t-C4N3 has broad prospects in catalysis. However, relevant studies for the application of g-t-C4N3 for the OER and ORR are rare and need to be further conducted. Therefore, in this work, a series of TM-doped g-t-C4N3 SACs (TM@g-t-C4N3) are designed so as to investigate the potential ability for catalyzing the OER and ORR.
Herein, 26 TMs (Fig. 1) are selected to be embedded into the g-t-C4N3 sheet to form TM@g-t-C4N3 whose stability and electronic properties are explored firstly. Then the catalytic performance of TM@g-t-C4N3 for the OER/ORR is evaluated by investigating the reaction process. The relationship between adsorption Gibbs free energy and OER/ORR activity is revealed from the volcano curve. Furthermore, the catalytic origins of TM@g-t-C4N3 are analyzed using the introduced descriptor φ and the projected d-band center, where the former is determined from Pauling's electronegativity and the number of valence electrons in d orbitals of TM atoms while the latter directly represents the electronic properties of doped-TM atoms. Meanwhile, the electronic structures of typical adsorbed systems are analyzed to gain a deeper insight into the physical image of catalytic activity.
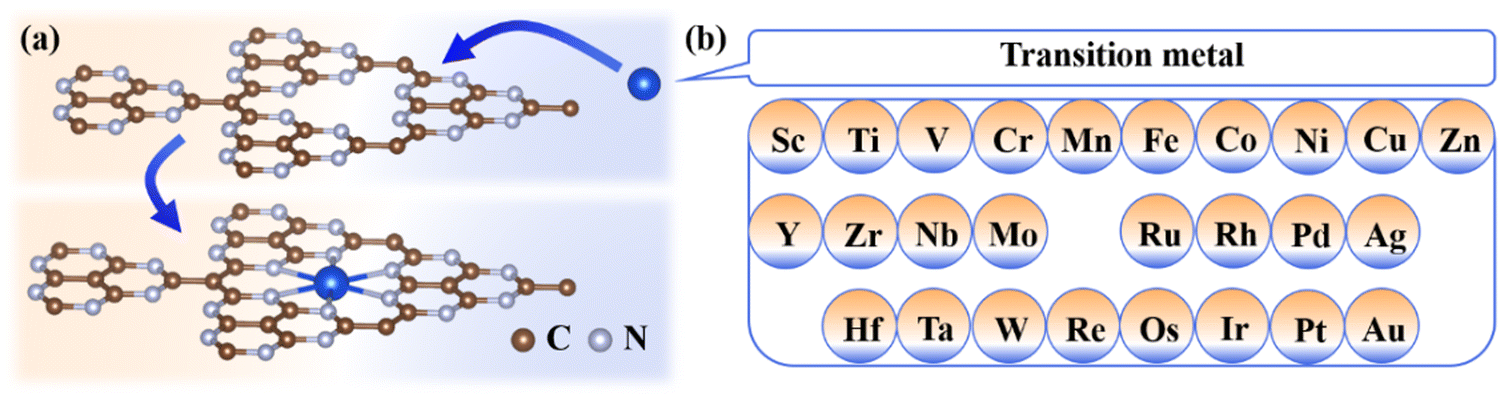 | ||
| Fig. 1 (a) Schematic diagram of g-t-C4N3 with anchored TM atoms, and (b) 26 TM atoms considered in this work. | ||
2. Computational methods
Spin-polarized density functional theory (DFT) calculations are performed using the Vienna ab initio simulation package (VASP),49,50 with the DFT-D3 van der Waals (vdW) corrections.51 The interactions between ions and electrons are treated by the project-augmented wave (PAW) method52 with a cut-off energy of 500 eV. The Perdew–Burke–Ernzerhof (PBE) functional within the generalized gradient approximation (GGA) is adopted to describe the exchange–correlation interaction.53 A 3 × 3 × 1 gamma-centered k-point grid is employed in the structural optimization, while a denser grid of 6 × 6 × 1 is used for the calculation of density of states (DOS). Besides, the thermodynamic stability of TM@g-t-C4N3 is assessed via ab initio molecular dynamics (AIMD) simulations.54 The convergence criteria of energy and force are set as 10−5 eV and 0.01 eV Å−1, respectively. A vacuum region of 20 Å is created to preclude the influence between adjacent layers.The changes of Gibbs free energy (ΔG) are calculated based on the computational hydrogen electrode (CHE) model10via the equation:
| ΔG = ΔE + ΔEZPE − TΔS | (1) |
3. Results and discussion
3.1. Construction and stability of TM@g-t-C4N3
A steady substrate is a prerequisite for the construction of SACs. Herein, a 2 × 2 × 1 supercell of g-t-C4N3 is selected to construct TM@g-t-C4N3 systems. As shown in Fig. S1 (ESI†), the optimized structure of g-t-C4N3 possesses planar properties with net pores encircled by six sp2-bonded N atoms, which could be beneficial to the anchoring of TM atoms. The AIMD simulations indicate the good thermodynamic stability of g-t-C4N3 as the total energy oscillates around the equilibrium value. In addition, the insets of Fig. S1 (ESI†) demonstrate that the structure of g-t-C4N3 is robust due to the little deformation after a 5 ps evolution.Fig. 1 shows the construction process and structural prototype of TM@g-t-C4N3. There are 26 TMs that are chosen to be embedded in the g-t-C4N3 sheet to obtain initial models of TM@g-t-C4N3. Then, all these configurations are optimized and the final structures are shown in Fig. S3 (ESI†). It can be seen that most TM atoms locate at the pore center, while Fe, Co, Ni, Cu and Zn prefer to stay at off-center positions, which may be owing to their smaller atomic radii. Furthermore, crinkles appear around the TM sites in the structures of Fe, Ni, Cu and Zn@g-t-C4N3. The asymmetric position of TM might be the primary reason for the slight deformation of the structure. The energy stability of TM@g-t-C4N3 is evaluated by calculating the binding energy (Eb) defined as:
| Eb = ETM@g-t-C4N3 − Eg-t-C4N3 − ETM | (2) |
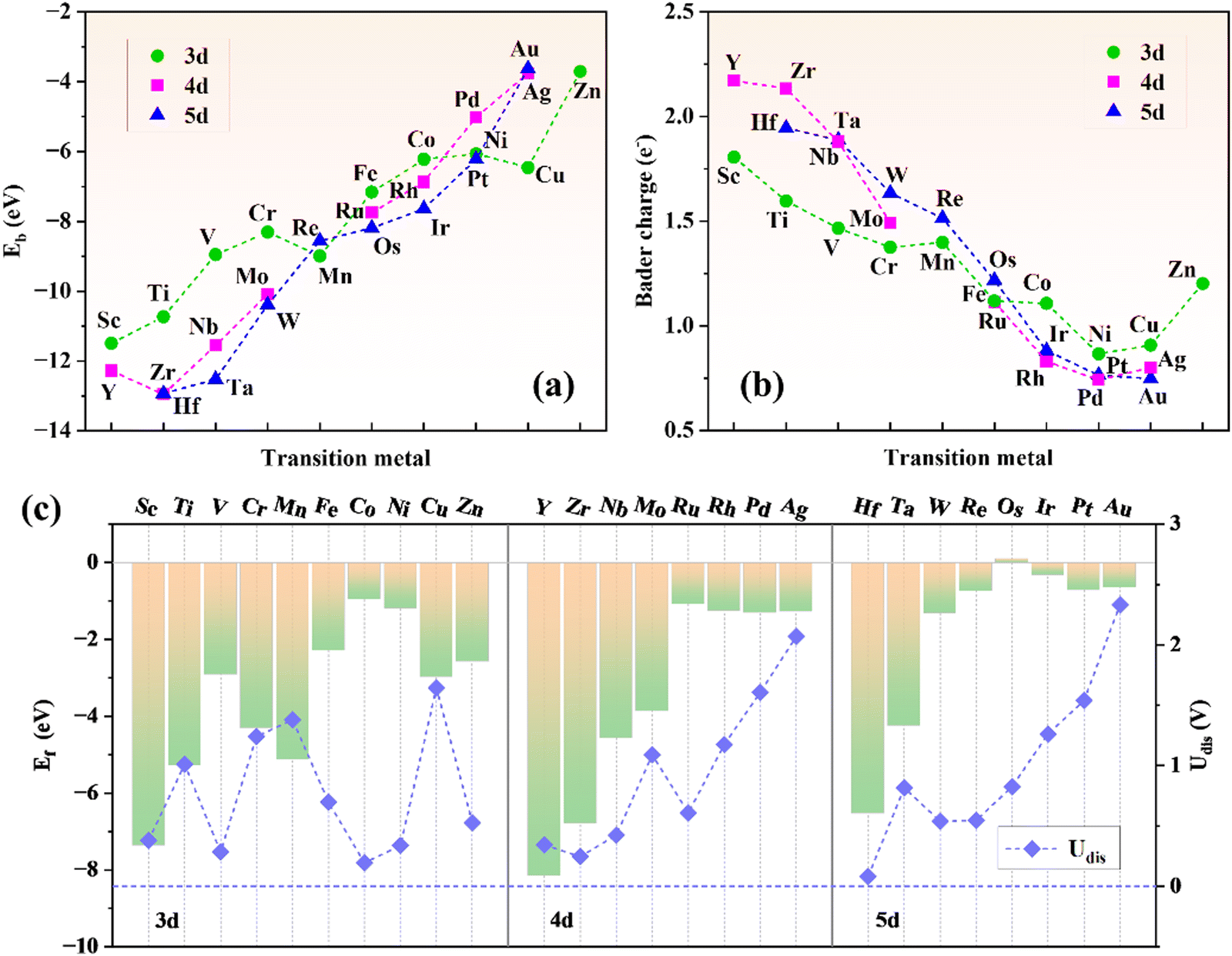 | ||
| Fig. 2 (a) The binding energy of TM@g-t-C4N3. (b) The Bader charge transferred from the metal atom to the substrate in the TM@g-t-C4N3. (c) The formation energy (left label) and dissolution potential (right label) of TM@g-t-C4N3. The dashed line marks the zero value of dissolution potential. | ||
The formation energy (Ef) is used to investigate the thermodynamic feasibility for construction of TM@g-t-C4N3, which is computed using the formula:
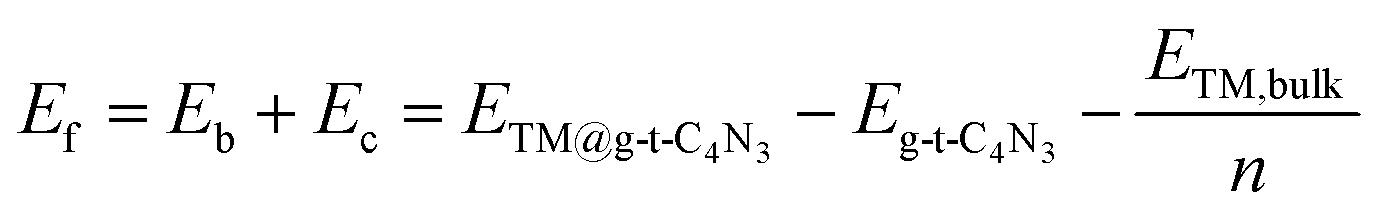 | (3) |
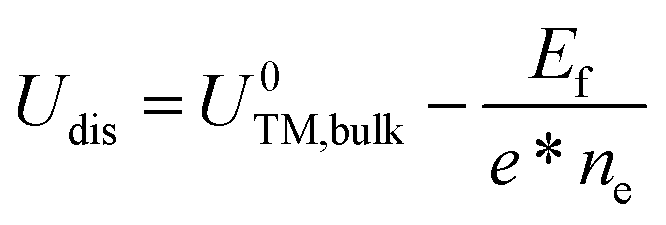 | (4) |
3.2. Catalytic performances of TM@g-t-C4N3
The adsorption properties of intermediates in the OER/ORR process (*OH, *O and *OOH) are studied to investigate the catalytic performances of TM@g-t-C4N3. According to the Sabatier principle,25 the adsorption strength of the intermediate should not be too strong or too weak, otherwise the reaction progress would be restrained somehow. Fig. 3 depicts the ΔG values of oxygen containing adsorbates with the data summarized in Table S2 (ESI†). It is seen that the ΔG of different intermediates vary in a similar trend. Particularly, the changing of ΔG*OH and ΔG*OOH is almost synchronous, which indicates that there exists a strong correlation between these two quantities and it will be further proved in Section 3.3. The Ti, V, Zr, Nb, Mo, Hf, Ta and W@g-t-C4N3 systems possess low ΔG*O with the value in the range of −2.16 to −0.56 eV, indicating too strong adsorption of *O. It could be related to the obvious d states near the Fermi level in the spin-up channel as presented in Fig. S5 (ESI†). Hence, these SAC candidates probably display poor activity for the OER/ORR.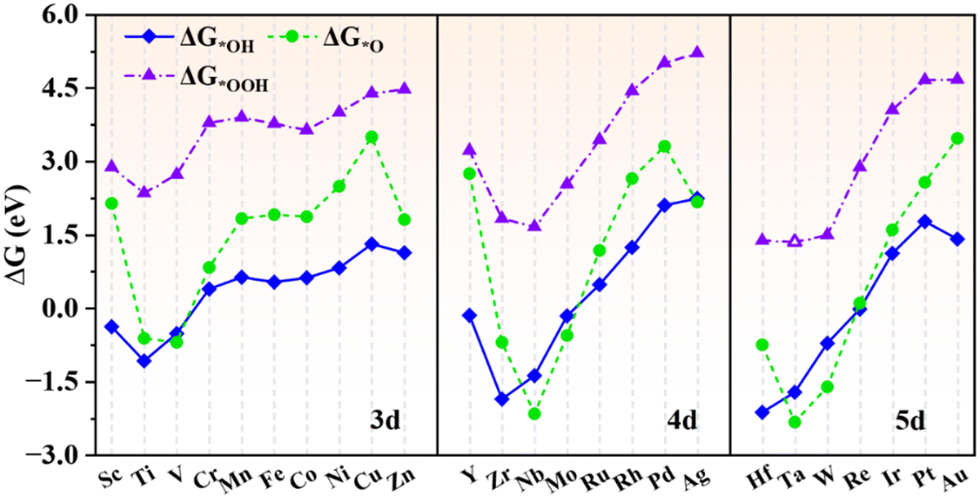 | ||
| Fig. 3 The adsorption Gibbs free energies of *OH, *O and *OOH on TM@g-t-C4N3. The ΔG*OOH of Ta@g-t-C4N3 is marked with hollow triangles because the adsorption of *OOH on Ta@g-t-C4N3 is not stable. The *OOH group will automatically separate into *O + OH after structural optimization, and the optimized structure is shown in Fig. S6 (ESI†). | ||
In order to clarify the OER and ORR performances of TM@g-t-C4N3, the reaction Gibbs free energy of each fundamental step is calculated whose value is summarized in Table S4 (ESI†). Besides, the theoretical overpotential (η) is also computed from the reaction Gibbs free energy in the potential-determining step (PDS) for the OER/ORR, which is listed in Table S5 (ESI†). For the OER, it can be seen from Fig. 4(a)–(d) that the Fe, Co, Ni and Rh@g-t-C4N3 possess low ηOER values of 0.63, 0.54, 0.44 and 0.56 V, respectively, which shows comparable catalytic ability to the previous IrO2 (ηOER = 0.56 V),10 while other TM@g-t-C4N3 have relatively poor OER activity due to their large ηOER. Specifically, for example, the Ni@g-t-C4N3 possesses lowest ηOER of 0.44 V among all candidates, indicating that when the equilibrium potential of 1.23 V is applied, the OER still proceeds non-spontaneously owing to the uphill steps of both *OH to *O and *O to *OOH. An external potential of 1.67 V is required at least to fully inverse the upward trend of the steps and to make the OER process occur spontaneously. For the ORR, Ni@g-t-C4N3 is also the best catalyst due to the lowest ηOER (0.41 V) among all the TM@g-t-C4N3, and it presents comparable catalytic activity to the Pt catalyst (ηORR = 0.45 V).60 In addition, the doped systems with Mn (0.60 V), Fe (0.70 V), Co (0.61 V), Cu (0.70 V), Zn (0.78 V), Ru (0.75 V), Rh (0.75 V) and Ir (0.74 V) also exhibit relatively good ORR activity.
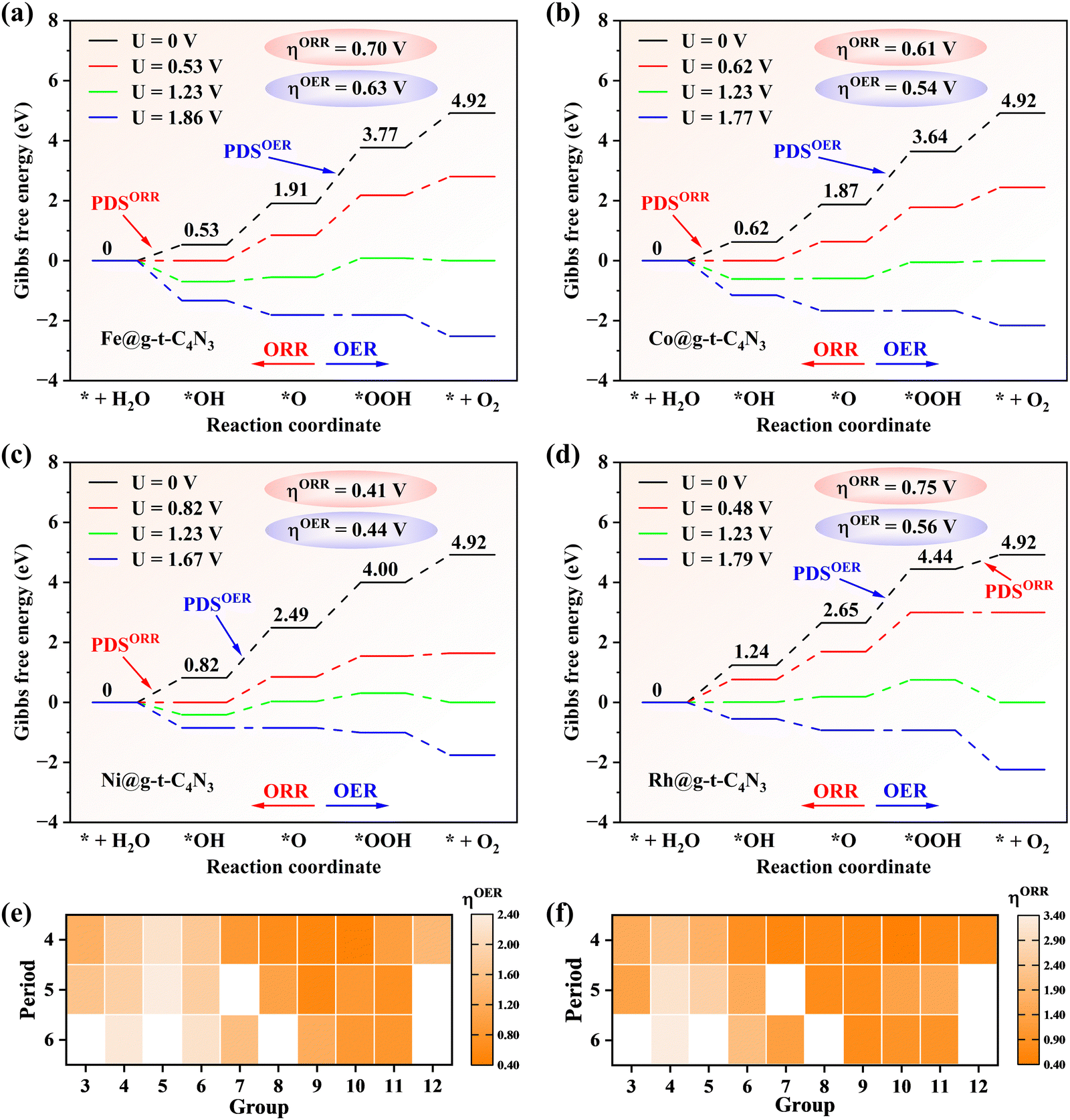 | ||
| Fig. 4 The Gibbs free energy diagrams of (a) Fe@g-t-C4N3, (b) Co@g-t-C4N3, (c) Ni@g-t-C4N3 and (d) Rh@g-t-C4N3 under different external potentials. (e) and (f) show the heat maps of overpotentials of the OER and ORR for TM@g-t-C4N3, respectively. | ||
Interestingly, it is found that the active SACs for the OER and ORR are those catalysts doped with TM atoms at the upper center-right area of the d-block in the periodic table as shown in Fig. 4(e) and (f). The preferable performances of the OER and ORR are achieved when the valence d orbitals of TM atoms are nearly full in each period, and among that, the overpotential tends to be lower with the decrease of the electron shell number (following the reduction of atomic radius). Therefore, the lowest value for both the OER and ORR in the TM@g-t-C4N3 system is found in Ni@g-t-C4N3. A similar trend has been also observed in some other SAC studies.5,16,36,61 This may partly originate from the moderate electronegativity and partly from the over half occupation of valence d orbitals of corresponding TM atoms, which enables the TMs’ middle affinity for electrons and appropriate adsorption strength of the intermediates. Quantitative analysis will be described in Section 3.4 with the introduced φ-descriptor.
Based on the aforementioned analysis, the promising bifunctional catalysts for the OER and ORR can be further examined. It is noted that the Ni@g-t-C4N3 (ηOER/ηORR = 0.44/0.41 V) exhibits best catalytic activity for both the OER and ORR among all TM@g-t-C4N3 candidates. Thus Ni@g-t-C4N3 is the most promising bifunctional catalyst for the OER and ORR, followed by Co@g-t-C4N3 (ηOER/ηORR = 0.54/0.61 V), Rh@g-t-C4N3 (ηOER/ηORR = 0.56/0.75 V) and Fe@g-t-C4N3 (ηOER/ηORR = 0.63/0.70 V). It can be inferred from the above results that the screened TM@g-t-C4N3 candidates are potential candidates for application as highly efficient catalysts for the OER and ORR.
3.3. Relationship between adsorption and activity
As intrinsic quantities in the OER/ORR process, the adsorption Gibbs free energies of intermediates (ΔG*OH, ΔG*O and ΔG*OOH) are customarily used to characterize the catalytic activity of catalysts. Hence it is important to identify the relationships between the adsorption Gibbs free energies of intermediates and the overpotentials of the OER and ORR.62 For TM@g-t-C4N3 systems, a strong linear relationship between ΔG*OH and ΔG*OOH is discovered. As depicted in Fig. 5(a), the ΔG*OOH can be expressed as a function of ΔG*OH: ΔG*OOH = 0.93ΔG*OH + 3.15 eV with a large coefficient of determination (R2 = 0.93). This result is consistent with other studies on metals and their oxides as well as SACs.5,6,35,62 Another linear equation between ΔG*O and ΔG*OOH is also revealed: ΔG*OOH = 0.60ΔG*O + 2.71 eV with an R2 of 0.77 as shown in Fig. 5(b). The difference of linearity can be attributed to the fact that *OH and *OOH require one electron to be adsorbed on active sites,5,6 while *O requires two electrons.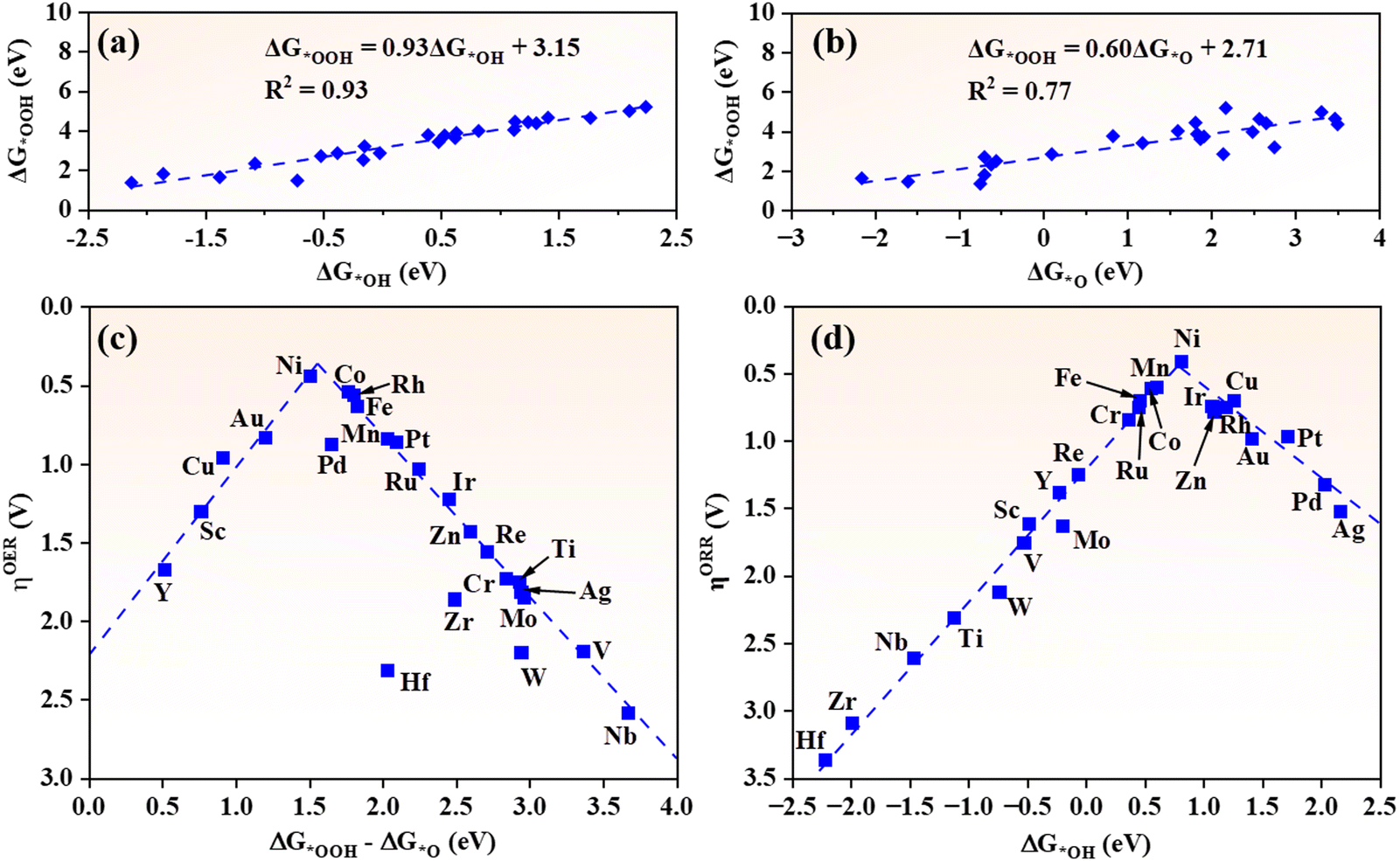 | ||
| Fig. 5 The scaling relationships of (a) ΔG*OOHversus ΔG*OH and (b) ΔG*OOHversus ΔG*O. The volcano plots of (c) ηOER against ΔG*OOH–ΔG*O and (d) ηORR against ΔG*OH, respectively. | ||
As listed in Table S4 (ESI†), the PDSs of the OER for TM@g-t-C4N3 systems appear mostly in the third step (*O to *OOH), thus ΔG*OOH–ΔG*O can be applied as a solid indicator to delineate the ηOER. As shown in Fig. 5(c), the ηOERversus ΔG*OOH–ΔG*O exhibits universal volcano characteristics. Ni@g-t-C4N3 is the closest to the peak of volcano and exhibits best catalytic activity. Besides, it is noticed that most TM@g-t-C4N3 tend to stay in the right branch of the volcano curve where the values of ΔG*OOH–ΔG*O are larger than the left ones, which implies that the relatively weak adsorption of *OOH or the relatively strong adsorption of *O are the main limiting factors to the OER activity of TM@g-t-C4N3. Table S4 (ESI†) also shows that the PDSs of the ORR for TM@g-t-C4N3 occur at the desorption step of *OH or the formation of *OOH (*OH). Considering the strong linearity between ΔG*OOH and ΔG*OH, the ΔG*OH can be employed as a sufficient descriptor for the ηORR. As shown in Fig. 5(d), a volcano curve between ηORR and ΔG*OH is obtained. Among all TM@g-t-C4N3 candidates, the Ni@g-t-C4N3 nearly locates at the peak of the volcano, indicating its best ORR activity. Moreover, the ORR activity of TM@g-t-C4N3 will increase at first and then decrease with the decreasing of ΔG*OH, and reaches its maximum when ΔG*OH equals 0.79 eV, which indicates that moderate adsorption strength of *OH will be beneficial to the ORR activity of TM@g-t-C4N3.
3.4. Origin of the catalytic activity
In order to uncover the origin of catalytic activity of TM@g-t-C4N3, the descriptor (φ) based on intrinsic properties of TM atoms is introduced,63 which is calculated using the formula:| φ = θd × χTM | (5) |
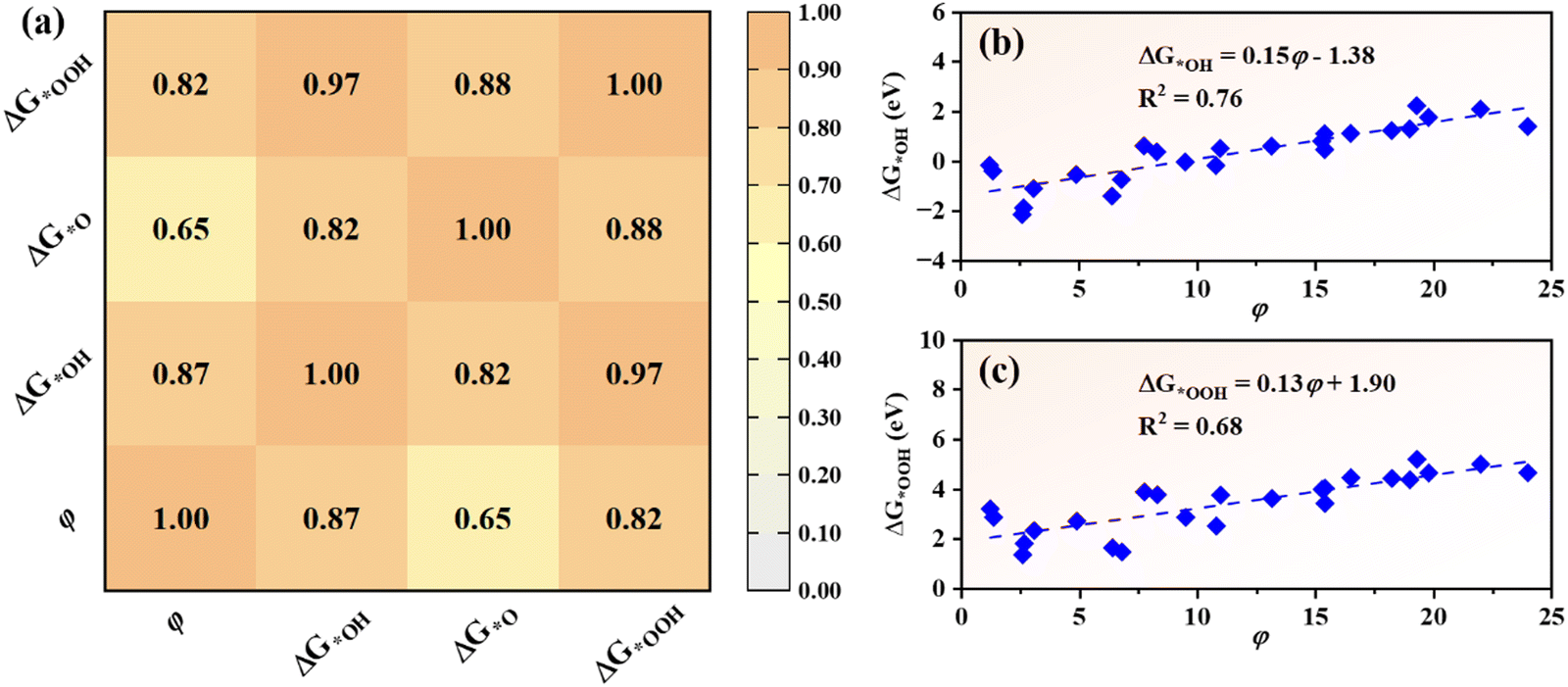 | ||
| Fig. 6 (a) Heat map of Pearson correlation matrix of φ, ΔG*OH, ΔG*O and ΔG*OOH. The color bar represents the value of Pearson correlation coefficient. (b) and (c) show the fitting lines of (b) ΔG*OH and (c) ΔG*OOHversus φ, respectively. | ||
The d-band center (εd) is then applied to gain a deeper insight into the catalytic activity of TM@g-t-C4N3, which is calculated using the formula:
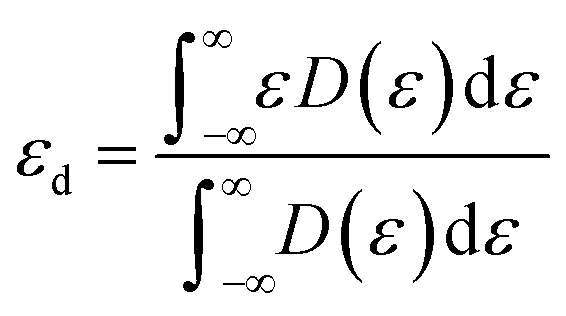 | (6) |
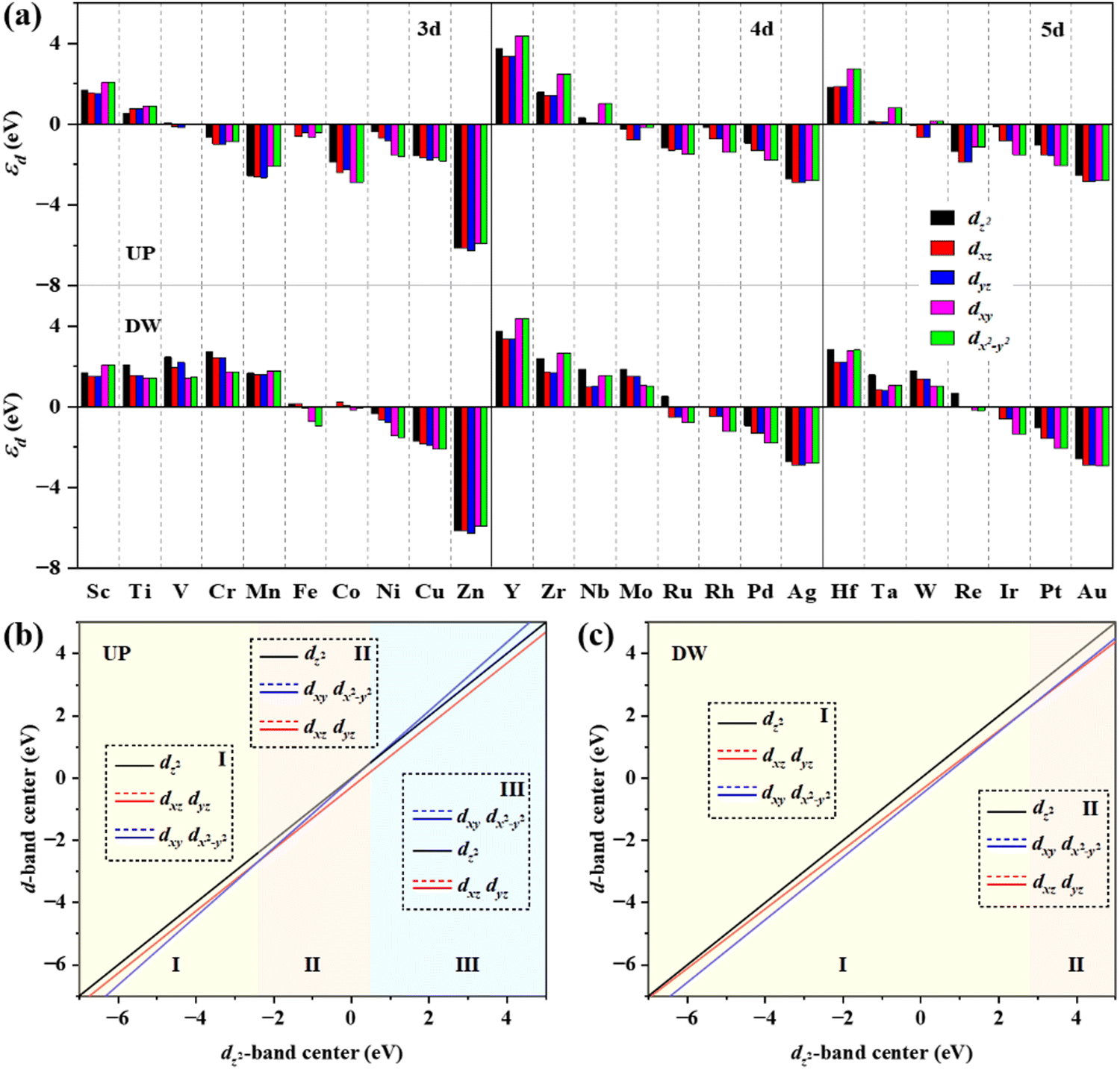 | ||
| Fig. 7 (a) The spin-up (UP) and spin-down (DW) projected d-band centers of anchored TM atoms. The fitting lines of projected d-band centers of group dxz–dyz and group dxy–dx2−y2versus dz2-band center for (b) UP and (c) DW channels, respectively. The insets show the relative order of projected d-orbitals in each region. | ||
The electronic structures of *OH adsorbed on Ni@g-t-C4N3 and Rh@g-t-C4N3 are further analyzed to reveal the specific images of activity. It is found from Fig. 8(a) and (b) that electrons are transferred from the TM to the *OH, which is also consistent with the result obtained from Bader charge analysis. Furthermore, based on the analysis of crystal orbital Hamilton population (COHP) and projected density of states (PDOS), it is indicated that when *OH is adsorbed on Ni@g-t-C4N3, the p orbitals of O and the d orbitals of Ni will strongly hybridize and form bonding and anti-bonding orbitals as shown in Fig. 8(c). The hybridization includes inert dxy and dx2−y2 orbitals. It is mainly attributed to the asymmetric and crinkling structure of the Ni@g-t-C4N3 especially near the active Ni site, which can be helpful to break out the symmetry conservation and make inert orbitals (dxy and dx2−y2) participate in orbital interactions.64 In addition, for *OH adsorbed on Rh@g-t-C4N3, strong hybridizations also occur between p orbitals of O and d orbitals of Rh as seen in Fig. 8(d). However, the participation of dxy and dx2−y2 orbitals in hybridization is negligible. It is due to the quasi-symmetric structure of Rh@g-t-C4N3 around Rh atom that keeps the restriction of orbital symmetry in orbital interactions.64 The results indicate that the concrete electronic structures of TM@g-t-C4N3 can be used to predict the adsorption behavior of intermediates in the OER and ORR, thus be effectual to investigate the origin of catalytic activity.
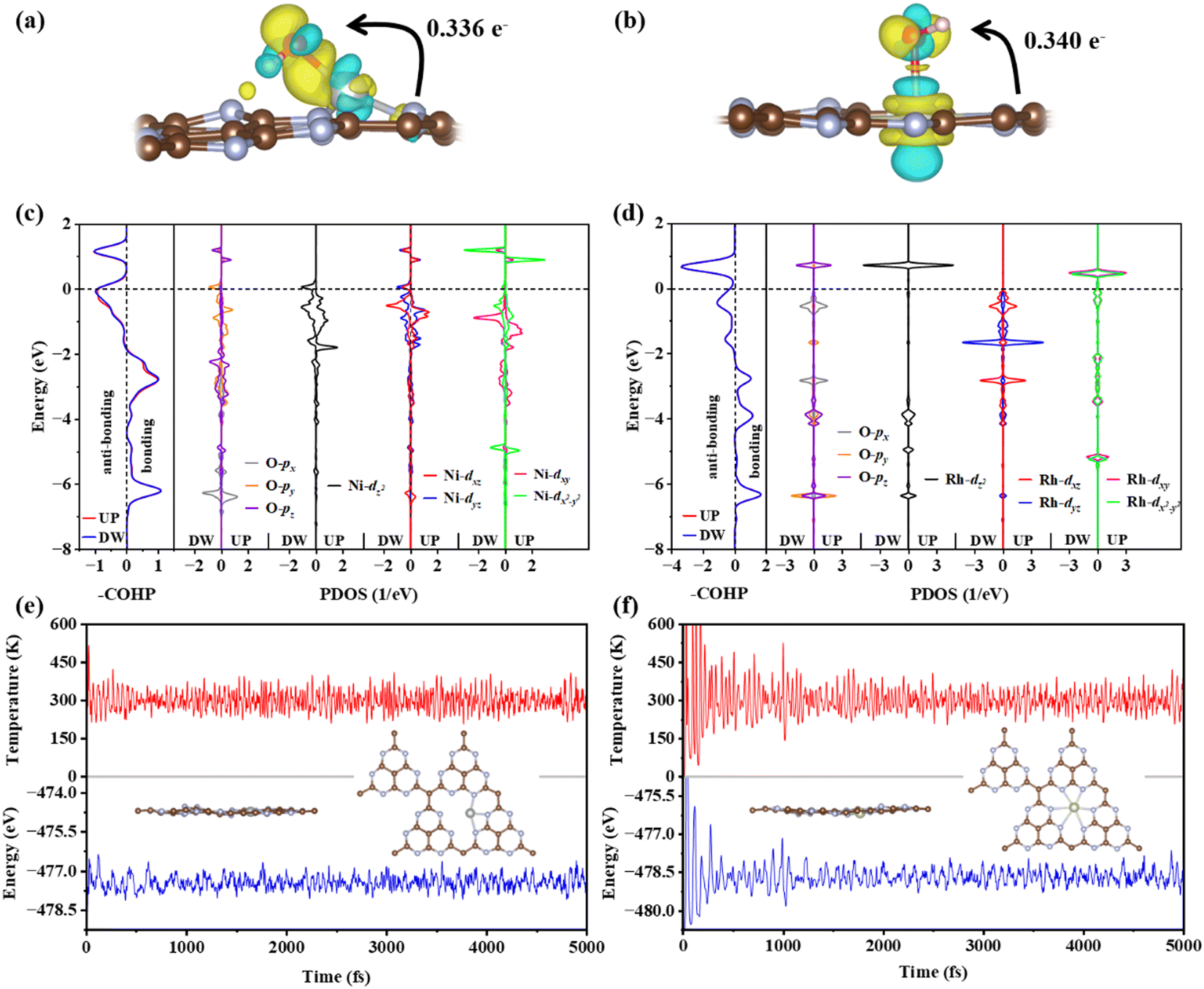 | ||
| Fig. 8 The CDDs of *OH adsorbed on (a) Ni@g-t-C4N3 and (b) Rh@g-t-C4N3, respectively. The yellow represents accumulation of electrons while the cyan represents depletion of electrons. The isovalue is set to 0.009 e Bohr−3. (c) and (d) show the COHP (PDOS) of *OH adsorbed on Ni@g-t-C4N3 and Rh@g-t-C4N3, respectively. The Fermi level is set to zero. (e) and (f) are the results of AIMD simulations (300 K) of Ni@g-t-C4N3 and Rh@g-t-C4N3, respectively. The insets show corresponding atomic structures after a 5 ps evolution. | ||
To further ensure the thermodynamic stability of Ni@g-t-C4N3 and Rh@g-t-C4N3, the AIMD simulations are carried out. As shown in Fig. 8(e) and (f), the total energies of Ni@g-t-C4N3 and Rh@g-t-C4N3 are fluctuating near the equilibrium states. For Ni@g-t-C4N3, the energy oscillation is smaller than 1.2 eV, and for Rh@g-t-C4N3, the energy oscillation is no more than 1.5 eV. Besides, the insets demonstrate that the atomic structures of Ni@g-t-C4N3 and Rh@g-t-C4N3 are robust and hardly changed after a 5 ps evolution at the temperature of 300 K, indicating their excellent thermodynamic stability.
4. Conclusions
By means of DFT calculations, the catalytic performances of TM@g-t-C4N3 (TM = Sc ∼ Au) for the OER and ORR have been studied systematically. The results show that Ni@g-t-C4N3 exhibits highest bifunctional catalytic activity for the OER and ORR (ηOER/ηORR = 0.44/0.41 V) among all TM@g-t-C4N3 systems. Meanwhile, Fe, Co and Rh@g-t-C4N3 also exhibit excellent bifunctional ability for the OER and ORR. Furthermore, it is discovered that the best OER and ORR activities are easily achieved when the doped TM atoms are located at the upper center-right area of the d-block. The volcano curves are uncovered to present the relationship between adsorption Gibbs free energies of reaction intermediates and catalytic performances.The electronic structures of TM@g-t-C4N3 are investigated to explore the origin of catalytic activity. Firstly, it is found that the ΔG*OH and ΔG*OOH can be simply assessed using the intrinsic descriptor φ. Moreover, the analysis based on the projected d-band center reveals that the relative order of different projected d orbitals will affect the adsorption strength of oxygen containing intermediates, thus promoting or inhibiting the catalytic processes of the OER and ORR. The COHP and PDOS show the orbital interactions between *OH and TM active sites (Ni and Rh), which can provide deeper insights for the excellent catalytic activity of Ni@g-t-C4N3 and Rh@g-t-C4N3. The results manifest the wide prospects of g-t-C4N3-based materials for the catalysis of the OER/ORR process. However, some key factors for the TM@g-t-C4N3 SACs need to be further studied. On the one hand, the investigation into reaction kinetics of the OER/ORR should be paid attention so as to further determine the actual barrier in the reaction process. On the other hand, the synthesis of TM@g-t-C4N3 and their stability under OER/ORR conditions ought to be studied more to evaluate the application of TM@g-t-C4N3 as efficient OER/ORR catalysts.
Conflicts of interest
There are no conflicts to declare.References
- K. Villa, J. R. Galán-Mascarós, N. López and E. Palomares, Photocatalytic water splitting: advantages and challenges, Sustainable Energy Fuels, 2021, 5, 4560–4569 RSC.
- S. Wang, L. Shi, X. Bai, Q. Li, C. Ling and J. Wang, Highly efficient photo-/electrocatalytic reduction of nitrogen into ammonia by dual-metal sites, ACS Cent. Sci., 2020, 6, 1762–1771 CrossRef CAS PubMed.
- T. Zheng, X. Han, J. Wang and Z. Xia, Role of heteroatom-doping in enhancing catalytic activities and the stability of single-atom catalysts for oxygen reduction and oxygen evolution reactions, Nanoscale, 2022, 14, 16286–16294 RSC.
- J. J. Yang, Y. Zhang, X. Y. Xie, W. H. Fang and G. Cui, Photocatalytic reduction of carbon dioxide to methane at the Pd-supported TiO2 interface: mechanistic insights from theoretical studies, ACS Catal., 2022, 12, 8558–8571 CrossRef CAS.
- Y. Wang, M. Wang, Z. Lu, D. Ma and Y. Jia, Enabling multifunctional electrocatalysts by modifying the basal plane of unifunctional 1T'-MoS2 with anchored transition metal single atoms, Nanoscale, 2021, 13, 13390–13400 RSC.
- J. Xu, Y. Wang, N. Song, S. Luo, B. Xu, J. Zhang and F. Wang, Doping of the Mn vacancy of Mn2B2 with a single different transition metal atom as the dual-function electrocatalyst, Phys. Chem. Chem. Phys., 2022, 24, 20988–20997 RSC.
- S. Lu, H. L. Huynh, F. Lou, K. Guo and Z. Yu, Single transition metal atom embedded antimonene monolayers as efficient trifunctional electrocatalysts for the HER, OER and ORR: a density functional theory study, Nanoscale, 2021, 13, 12885–12895 RSC.
- C. Zhang, Y. Dai, Q. Sun, C. Ye, R. Lu, Y. Zhou and Y. Zhao, Strategy to weaken the oxygen adsorption on single-atom catalysts towards oxygen-involved reactions, Mater. Today Adv., 2022, 16, 100280 CrossRef CAS.
- Y. J. Wang, N. Zhao, B. Fang, H. Li, X. T. Bi and H. Wang, Carbon-supported Pt-based alloy electrocatalysts for the oxygen reduction reaction in polymer electrolyte membrane fuel cells: particle size, shape, and composition manipulation and their impact to activity, Chem. Rev., 2015, 115, 3433–3467 CrossRef CAS PubMed.
- J. Rossmeisl, Z. W. Qu, H. Zhu, G. J. Kroes and J. K. Nørskov, Electrolysis of water on oxide surfaces, J. Electroanal. Chem., 2007, 607, 83–89 CrossRef CAS.
- N. T. Suen, S. F. Hung, Q. Quan, N. Zhang, Y. J. Xu and H. M. Chen, Electrocatalysis for the oxygen evolution reaction: recent development and future perspectives, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- A. Mahata, A. S. Nair and B. Pathak, Recent advancements in Pt-nanostructure-based electrocatalysts for the oxygen reduction reaction, Catal. Sci. Technol., 2019, 9, 4835–4863 RSC.
- N. Bhuvanendran, S. Ravichandran, S. S. Jayaseelan, Q. Xu, L. Khotseng and H. Su, Improved bi-functional oxygen electrocatalytic performance of Pt–Ir alloy nanoparticles embedded on MWCNT with Pt-enriched surfaces, Energy, 2020, 211, 118695 CrossRef CAS.
- R. Wang, X. Cao, S. Sui, B. Li and Q. Li, Study on the growth of platinum nanowires as cathode catalysts in proton exchange membrane fuel cells, Front. Chem. Sci. Eng., 2021, 16, 364–375 CrossRef.
- S. Qi, J. Wang, X. Song, Y. Fan, W. Li, A. Du and M. Zhao, Synergistic trifunctional electrocatalysis of pyridinic nitrogen and single transition-metal atoms anchored on pyrazine-modified graphdiyne, Sci. Bull., 2020, 65, 995–1002 CrossRef CAS PubMed.
- Z. Qin, Z. Wang and J. Zhao, Computational screening of single-atom catalysts supported by VS2 monolayers for electrocatalytic oxygen reduction/evolution reactions, Nanoscale, 2022, 14, 6902–6911 RSC.
- X. F. Yang, A. Q. Wang, B. T. Qiao, J. Li, J. Y. Liu and T. Zhang, Single-atom catalysts: a new frontier in heterogeneous catalysis, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed.
- H. Xu, D. Cheng, D. Cao and X. C. Zeng, A universal principle for a rational design of single-atom electrocatalysts, Nat. Catal., 2018, 1, 339–348 CrossRef CAS.
- Q. Wang, E. Yang, R. Liu, M. Lv, W. Zhang, G. Yu and W. Chen, Embedding group VIII elements into a 2D rigid pc-C3N2 monolayer to achieve single-atom catalysts with excellent OER activity: a DFT theoretical study, Molecules, 2022, 28, 254 CrossRef PubMed.
- Z. H. Zhou, W. H. Li, Z. Zhang, Q. S. Huang, X. C. Zhao and W. Cao, Ni-O4 as active sites for efficient oxygen evolution reaction with electronic metal-support interactions, ACS Appl. Mater. Interfaces, 2022, 14, 47542–47548 CrossRef CAS PubMed.
- A. Allangawi, T. Mahmood, K. Ayub and M. A. Gilani, Anchoring the late first row transition metals with B12P12 nanocage to act as single atom catalysts toward oxygen evolution reaction (OER), Mater. Sci. Semicond. Process., 2023, 153, 107164 CrossRef CAS.
- F. Ge, Q. Qiao, X. Chen and Y. Wu, Probing the catalytic activity of M-N4−xOx embedded graphene for the oxygen reduction reaction by density functional theory, Front. Chem. Sci. Eng., 2021, 15, 1206–1216 CrossRef CAS.
- X. Hu, S. Chen, L. Chen, Y. Tian, S. Yao, Z. Lu, X. Zhang and Z. Zhou, What is the real origin of the activity of Fe-N-C electrocatalysts in the O2 reduction reaction? Critical roles of coordinating pyrrolic N and axially adsorbing species, J. Am. Chem. Soc., 2022, 144, 18144–18152 CrossRef CAS PubMed.
- W. Zhang, J. K. Pan, Y. F. Yu, X. J. Zhang, J. H. Wang, W. X. Chen and G. L. Zhuang, Correlation of the spin state and catalytic property of M-N4 single-atom catalysts in oxygen reduction reactions, Phys. Chem. Chem. Phys., 2023, 25, 11673–11683 RSC.
- G. Di Liberto, L. A. Cipriano and G. Pacchioni, Universal principles for the rational design of single atom electrocatalysts? Handle with care, ACS Catal., 2022, 12, 5846–5856 CrossRef CAS.
- D. Chodvadiya, P. K. Jha and B. Chakraborty, Theoretical inspection of Ni/α-SiX (X = N, P, As, Sb, Bi) single-atom catalyst: ultra-high performance for hydrogen evolution reaction, Int. J. Hydrogen Energy, 2022, 47, 41733–41747 CrossRef CAS.
- T. Liu, X. Zhao, X. Liu, W. Xiao, Z. Luo, W. Wang, Y. Zhang and J. C. Liu, Understanding the hydrogen evolution reaction activity of doped single-atom catalysts on two-dimensional GaPS4 by DFT and machine learning, J. Energy Chem., 2023, 81, 93–100 CrossRef CAS.
- T. L. Wan, J. Liu, X. Tan, T. Liao, Y. Gu, A. Du, S. Smith and L. Kou, Rational design of 2D ferroelectric heterogeneous catalysts for controllable hydrogen evolution reaction, J. Mater. Chem. A, 2022, 10, 22228–22235 RSC.
- T. Bo, S. Cao, N. Mu, R. Xu, Y. Liu and W. Zhou, First principles screening of transition metal single-atom catalysts for nitrogen reduction reaction, Appl. Surf. Sci., 2023, 612, 155916 CrossRef CAS.
- S. Han, X. Wei, Y. Huang, J. Zhang, J. Yang and Z. Wang, Tuning the activity and selectivity of nitrogen reduction reaction on double-atom catalysts by B doping: a density functional theory study, Nano Energy, 2022, 99, 107363 CrossRef CAS.
- J. Min, L. Liu, F. Chen, X. Jin, T. Yuan and X. Yao, Theoretical exploration on the activity of copper single-atom catalysts for electrocatalytic reduction of CO2, J. Mater. Chem. A, 2023, 11, 7735–7745 RSC.
- P. Hou, Y. Huang, F. Ma, X. Wei, R. Du, G. Zhu, J. Zhang and M. Wang, S and N coordinated single-atom catalysts for electrochemical CO2 reduction with superior activity and selectivity, Appl. Surf. Sci., 2023, 619, 156747 CrossRef CAS.
- X. Liu, G. Li, J. Liu and J. Zhao, Transition metal atoms anchored on square graphyne as multifunctional electrocatalysts: a computational investigation, Mol. Catal., 2022, 531, 112706 CrossRef CAS.
- X. Bai, X. Zhao, Y. Zhang, C. Ling, Y. Zhou, J. Wang and Y. Liu, Dynamic stability of copper single-atom catalysts under working conditions, J. Am. Chem. Soc., 2022, 144, 17140–17148 CrossRef CAS PubMed.
- M. Guo, M. Ji and W. Cui, Theoretical investigation of HER/OER/ORR catalytic activity of single atom-decorated graphyne by DFT and comparative DOS analyses, Appl. Surf. Sci., 2022, 592, 153237 CrossRef CAS.
- Z. Zhang, S. Qi, X. Song, J. Wang, W. Zhang and M. Zhao, Stable multifunctional single-atom catalysts adsorbed on pyrazine-modified graphyne, Appl. Surf. Sci., 2021, 553, 149464 CrossRef CAS.
- H. Niu, Z. Zhang, X. Wang, X. Wan, C. Shao and Y. Guo, Theoretical insights into the mechanism of selective nitrate-to-ammonia electroreduction on single-atom catalysts, Adv. Funct. Mater., 2020, 31, 2008533 CrossRef.
- Z. Yang, Y. Li, C. Qi and C. Wang, Single atom catalysts supported on metallic C5N monolayers for oxygen reduction/evolution reactions with more active sites than loaded metal atoms, Appl. Surf. Sci., 2023, 614, 156048 CrossRef CAS.
- N. Sathishkumar, S.-Y. Wu and H.-T. Chen, Mechanistic exploring the catalytic activity of single-atom catalysts anchored in graphitic carbon nitride toward electroreduction of nitrate-to-ammonia, Appl. Surf. Sci., 2022, 598, 153829 CrossRef CAS.
- A. Du, S. Sanvito and S. C. Smith, First-principles prediction of metal-free magnetism and intrinsic half-metallicity in graphitic carbon nitride, Phys. Rev. Lett., 2012, 108, 197207 CrossRef PubMed.
- X. Li, S. Zhang and Q. Wang, Stability and physical properties of a tri-ring based porous g-C4N3 sheet, Phys. Chem. Chem. Phys., 2013, 15, 7142–7146 RSC.
- J. Cui, S. Liang and J. Zhang, A multifunctional material of two-dimensional g-C4N3/graphene bilayer, Phys. Chem. Chem. Phys., 2016, 18, 25388–25393 RSC.
- M. Mehrdad and A. Moosavi, Novel adjustable monolayer carbon nitride membranes for high-performance saline water desalination, Nanotechnology, 2021, 32, 045706 CrossRef CAS PubMed.
- R. B. Gurung, N. Sharma and H.-F. Wu, White light emission and superior photoelctrochemical response from semi-metallic graphitic carbon nitride analogue: g-C4N3, Mater. Sci. Eng. B, 2022, 280, 115663 CrossRef CAS.
- X. Wang and L.-M. Yang, Unveiling the underlying mechanism of nitrogen fixation by a new class of electrocatalysts two-dimensional TM@g-C4N3 monosheets, Appl. Surf. Sci., 2022, 576, 151839 CrossRef CAS.
- X. Wan, W. Yu, H. Niu, X. Wang, Z. Zhang and Y. Guo, Revealing the oxygen reduction/evolution reaction activity origin of carbon-nitride-related single-atom catalysts: quantum chemistry in artificial intelligence, Chem. Eng. J., 2022, 440, 135946 CrossRef CAS.
- D. Li, X. Chen, Y. Huang, G. Zhang, D. Zhou and B. Xiao, Selective catalytic oxidation of formaldehyde on single V- and Cr-atom decorated magnetic C4N3 substrate: a first principles study, J. Hazard. Mater., 2022, 439, 129608 CrossRef CAS PubMed.
- S. Agarwal, R. Kumar, R. Arya and A. K. Singh, Rational design of single-atom catalysts for enhanced electrocatalytic nitrogen reduction reaction, J. Phys. Chem. C, 2021, 125, 12585–12593 CrossRef CAS.
- G. Kresse and J. Furthmüller, Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- G. J. Martyna, M. L. Klein and M. Tuckerman, Nosé–Hoover chains: the canonical ensemble via continuous dynamics, J. Chem. Phys., 1992, 97, 2635–2643 CrossRef.
- W. Tang, E. Sanville and G. Henkelman, A grid-based Bader analysis algorithm without lattice bias, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS PubMed.
- P. Janthon, S. M. Kozlov, F. Vines, J. Limtrakul and F. Illas, Establishing the accuracy of broadly used density functionals in describing bulk properties of transition metals, J. Chem. Theory Comput., 2013, 9, 1631–1640 CrossRef CAS PubMed.
- P. Hou, Y. Huang, F. Ma, G. Zhu, J. Zhang, X. Wei, P. Du and J. Liu, Computational screening and catalytic origin of transition metal supported on g-t-C3N4 as single-atom catalysts for nitrogen reduction reaction, Appl. Surf. Sci., 2022, 599, 153880 CrossRef CAS.
- X. Guo, J. Gu, S. Lin, S. Zhang, Z. Chen and S. Huang, Tackling the Activity and Selectivity Challenges of Electro-catalysts toward the Nitrogen Reduction Reaction via Atomically Dispersed Biatom Catalysts, J. Am. Chem. Soc., 2020, 142, 5709–5721 CrossRef CAS PubMed.
- CRC Handbook of Chemistry and Physics, Internet Version 2005, ed. David R. Lide, CRC Press, Boca Raton, FL, https://www.hbcpnetbase.com, 2005 Search PubMed.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir and L. Lindqvist, Origin of the overpotential for oxygen reduction at a fuel-cell cathode, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- S. L. Li, Q. Li, Y. Chen, Y. Zhao and L. Y. Gan, Transition metal embedded graphynes as advanced bifunctional single atom catalysts for oxygen reduction and evolution reactions, Appl. Surf. Sci., 2022, 605, 154828 CrossRef CAS.
- M. Bajdich, M. Garcia-Mota, A. Vojvodic, J. K. Norskov and A. T. Bell, Theoretical investigation of the activity of cobalt oxides for the electrochemical oxidation of water, J. Am. Chem. Soc., 2013, 135, 13521–13530 CrossRef CAS PubMed.
- X. Chen, Q. Liu, H. Zhang and X. Zhao, Exploring high-efficiency electrocatalysts of metal-doped two-dimensional C4N for oxygen reduction, oxygen evolution, and hydrogen evolution reactions by first-principles screening, Phys. Chem. Chem. Phys., 2022, 24, 26061–26069 RSC.
- Y. Sun, S. Sun, H. Yang, S. Xi, J. Gracia and Z. J. Xu, Spin-related electron transfer and orbital interactions in oxygen electrocatalysis, Adv. Mater., 2020, 32, 2003297 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp04249a |
| This journal is © the Owner Societies 2024 |