
What kind of interactions we may get moving from zwitter to “dritter” ions: C–O⋯Re(O4) and Re–O⋯Re(O4) anion⋯anion interactions make structural difference between L-histidinium perrhenate and pertechnetate†
Anton P.
Novikov
 *,
Alexey V.
Safonov
,
Konstantin E.
German
and
Mikhail S.
Grigoriev
*,
Alexey V.
Safonov
,
Konstantin E.
German
and
Mikhail S.
Grigoriev
Frumkin Institute of Physical Chemistry and Electrochemistry, Russian Academy of Sciences, 31 Bldg. 4, Leninsky Prosp., Moscow, 119071, Russian Federation. E-mail: tony.novickoff@yandex.ru
First published on 1st December 2023
Abstract
L-Histidinium perrhenate was synthesized by the reaction of magnesium perrhenate with the corresponding nitrogenous base in hydrochloric acid. Pertechnetate was obtained from the nitrous base by reaction with pertechnetic acid. Their crystal structure was studied using single-crystal XRD and a comparative analysis of noncovalent interactions in the obtained structures was carried out. Pertechnetate is the second example of the combination of technetium with this amino acid, and both compounds described in this work are the first examples of structures of tetrahedral transition metal anions with histidine. The histidine molecules in the present structures are in the open conformation (1)(g−). Not only are nitrogen atoms of imidazole rings and amino groups involved in the formation of N–H⋯O type hydrogen bonds, but also CH group atoms form weaker C–H⋯O type hydrogen bonds. Histidinium perrhenate contains anion⋯anion interactions, including those described for the first time: C–O⋯Re(O4) between the carboxyl group and the rhenium atom of the perrhenate anion. This type of interaction can be found (but was not discussed) in other Re and Mn compounds but is absent for Tc. Hirshfeld surface analysis showed that the most important contribution to the packing organization is made by the O⋯H/H⋯O interaction for cations and anions.
Introduction
Hydrogen bonds and various weaker valent and noncovalent intra- and intermolecular interactions are of interest because of their widespread biological occurrence.1 They are involved in the active-site substrate-binding mechanism,2 regulation of the oxygen affinity,3 DNA structures,4etc. At the same time, knowledge about the mechanisms and role of these interactions in biomolecules is constantly deepening. An analysis of noncovalent interactions in compounds of metals and bioorganic molecules can provide valuable data on the properties of compounds, in terms of recognition processes5 and receptor-information chains,6 the influence of co-factors,7 and also for the interpretation of intermolecular energy transfer processes.8 Even weak intermolecular interactions affect the properties such as luminescence,9,10 solubility in different solvents,11 phase transition temperature12 and type.One of the most interesting groups of compounds that provide ample opportunities for the analysis of various types of noncovalent interactions are the salts of tetraoxidoanions of group 7 (ref. 11 and 13–17) with multiply charged cations bearing spatially separated charges. These structures provide a variety of pertechnetate bonding options due to the redistribution of the single negative charge in equal formal fractions of −1/4 by the four oxygen atoms of tetrahedral pertechnetate, perrhenate or perchlorate anions for the weak but numerous H-bond formation. Recently the usage of the compositions of this type made it possible to isolate the evasive rhenium(VII) polyoxometalate anion in a solid structure18 – a species that has been elusive for many years. Some more examples are provided by the salts of polyfunctional dipolar cations like morpholinium19 where one may note such interactions of a σ-hole type as Re–O⋯Re(O4), discovered recently.17,20 Of extreme interest are the molecules of amino acids that a priori bear two or three charged groups in form of zwitterions (Gly, Ala, Ley) or even of dritterions (Asp, His, Glu, etc.) that could crystalize at a reasonably narrow pH region as mixed salts with supplementary anions e.g. tetraoxidoanions. In this work we elaborated the line of L-histidinium derivatives with tetraoxidometalate anions in view of their options for weak bond (H-bonds, σ-hole bonds etc.) formation.
It is important to note that the perrhenate and pertechnetate tetraoxidoanions are of great interest in nuclear medicine, both for diagnostic and therapeutic applications.21 Therefore, the synthesis of new compounds with the anions and the study of their structures are both promising fundamental and applied directions. It is generally accepted that the chemistry of technetium and rhenium is very similar, and it is possible to model the behavior of technetium using rhenium. However, a number of works have shown that rhenium and technetium (perrhenate and pertechnetate anions) can behave differently under the same conditions.13,18,22
Moreover, histidine is an important biological compound, as an essential amino acid, and it is widely involved in cell metabolism, required for synthesis of proteins. It plays a particularly important role in the active sites of enzymes.23 As a precursor of carnosine24 and other biological molecules it plays an important role in the antioxidant immune system, in human muscle, neurotransmitters, etc.25 For this reason, the synthesis and study of the structural features of new compounds based on histidine are promising for the creation of new medical drugs, toxicological studies and many other areas of medicine.26 Also, this study may bring new understanding of chemical bonding options. Thus, in the case of effective binding with ReO4− or TcO4−, they can be promising deliverers of rhenium to the cells of organs and tissues. It is also worth noting that only one structure has been described for technetium with histidine.27
Experimental
Synthesis and characterization
All reagents and solvents used in the work are high grade (ACS, Sigma Aldrich) and were not further purified. Magnesium perrhenate and ammonium pertechnetate (VO Isotop) were used in the work.
Synthesis of L-histidinium pertechnetate (2) (HHisTcO4)
From a solution of ammonium pertechnetate and L-histidine in hydrochloric acid, the initial ammonium pertechnetate or histidinium chloride usually precipitated. Therefore, it was decided to obtain histidinium pertechnetate from pertechnetic acid, as described below.In a 10 mL two-necked flask equipped with a thermometer and a reflux condenser, 20 mg of L-histidine was dissolved in 2.5 mL of 1 M HCl. After dissolving all the L-histidine, 2.5 ml of 1 M technetium acid was added. Technetium acid solution was obtained by dissolution of Tc2O7 in deionized water. In the present study, we used a facile synthetic route for Tc2O7 preparation by a direct combustion of NH4TcO4 in a stream of oxygen followed by the condensation of Tc2O7 at the cooled walls of the tubular furnace. The mixture of L-histidine and HTcO4 was stirred for 5 min at room temperature, then the temperature was raised to 70 °C and held for 15 min. The resulting solution was cooled to room temperature and left for 10 days for crystal growth. The resulting crystals were washed with two 5 mL portions of cold methanol and dried in an oil pump vacuum at room temperature. The yield of the reaction by Tc is 62%. Elemental analysis (theor.): C, 22.59; H, 3.16; N, 13.17; Tc, 31.00; (exp.): C, 22.65; H, 3.19; N, 13.20; Tc, 31.12.
X-ray analysis
The crystal structures of all synthesized substances were determined by X-ray structural analysis using an automatic four-circle area-detector diffractometer (Bruker KAPPA APEX II) with MoKα radiation. The cell parameters were refined over the entire data set, together with data reduction using the SAINT-Plus software.28 Absorption corrections were introduced using the SADABS program.29 The structures were solved using the SHELXT-2018/2 program30 and refined by full-matrix least squares on F2 in the anisotropic approximation for all non-hydrogen atoms (SHELXL-2018/3 (ref. 31)). The hydrogen atoms were placed in geometrically calculated positions and refined in an idealized geometry with isotropic temperature factors equal to 1.2Ueq (C, N) for CH- and NH- and 1.5Ueq (N) for NH2-groups. Structures were refined as inversion twins. Tables and figures for the structures were generated using Olex2.32Crystal data, data collection, and structure refinement details are summarized in Table 1. All other crystallographic parameters of the structures are indicated in Tables S1–S8.† The atomic coordinates were deposited at the Cambridge Crystallographic Data Centre.33
| 1 | 2 | |
|---|---|---|
| CCDC number | 2299031 | 2299030 |
| Empirical formula | C6H10N3O6Re | C6H10N3O6Tc |
| Formula weight | 406.37 | 319.08 |
| Temperature/K | 100(2) | 100(2) |
| Crystal system | Orthorhombic | Triclinic |
| Space group | P212121 | P1 |
| a/Å | 9.2915(3) | 9.9684(11) |
| b/Å | 17.0748(5) | 10.4769(12) |
| c/Å | 19.5885(5) | 11.0045(12) |
| α/° | 90 | 89.147(4) |
| β/° | 90 | 87.966(4) |
| γ/° | 90 | 62.592(4) |
| Volume/Å3 | 3107.73(16) | 1019.6(2) |
| Z | 12 | 4 |
| d calc/g cm−3 | 2.606 | 2.073 |
| μ/mm−1 | 11.750 | 1.432 |
| F(000) | 2280.0 | 632.0 |
| Crystal size/mm3 | 0.22 × 0.18 × 0.12 | 0.06 × 0.03 × 0.03 |
| Radiation | MoKα (λ = 0.71073) | MoKα (λ = 0.71073) |
| 2θ range for data collection/° | 8.282 to 69.996 | 8.184 to 59.996 |
| Index ranges | −14 ≤ h ≤ 14, −27 ≤ k ≤ 26, −30 ≤ l ≤ 31 | −14 ≤ h ≤ 13, −14 ≤ k ≤ 14, −15 ≤ l ≤ 15 |
| Reflections collected | 53![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 445 445 |
18722 |
| Independent reflections | 13630 [Rint = 0.0625, Rsigma = 0.0632] |
9780 [Rint = 0.0662, Rsigma = 0.1272] |
| Data/restraints/parameters | 13630/0/436 |
9780/33/582 |
| Goodness-of-fit on F2 | 0.969 | 1.031 |
| Final R indices [I > = 2σ(I)] | R 1 = 0.0318, wR2 = 0.0505 | R 1 = 0.0743, wR2 = 0.1584 |
| Final R indices [all data] | R 1 = 0.0421, wR2 = 0.0535 | R 1 = 0.1041, wR2 = 0.1785 |
| Largest diff. peak/hole/e Å−3 | 1.01/−1.89 | 3.81/−2.04 |
| Flack parameter | −0.017(6) | 0.10(5) |
Elemental analysis
The chemical composition of the compounds was determined on a sample of 50 mg. Technetium in the compounds was determined by liquid scintillation on a Tri-Carb 3180 TR/SL instrument (PerkinElmer), using a HiSafe 3 scintillator; the measurement error did not exceed 5%. C, H, N were determined using an EA 3000 EuroVector analyzer; the measurement error was not more than 0.3%.Results and discussion
Structural description
The cations in both compounds are found/present in the zwitterionic form (more precisely – “dritterionic” from the German word for “3” and means a triply charged molecular ion with space-separated charges) due to the transition of the hydrogen atom from the carboxyl group to the amino group, and the nitrogen atom of the five-membered ring is additionally protonated, as in the previously obtained analogues. However, even though the ratios of cations and anions in both compounds are 1 to 1 (Scheme 1), and the structures do not include any extra molecules, 1 and 2 are not isostructural. Histidinium perrhenate crystallizes in the space group P212121, and pertechnetate in P1. The asymmetric units are shown in Fig. 1. Three crystallographically independent formula units are formed in 1; four units are present in 2.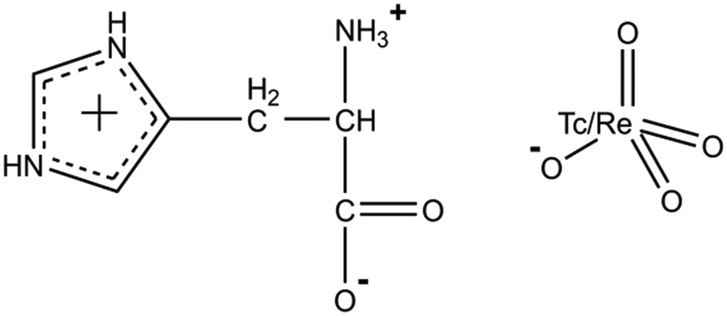 | ||
| Scheme 1 Schematic representation of 1 and 2. | ||
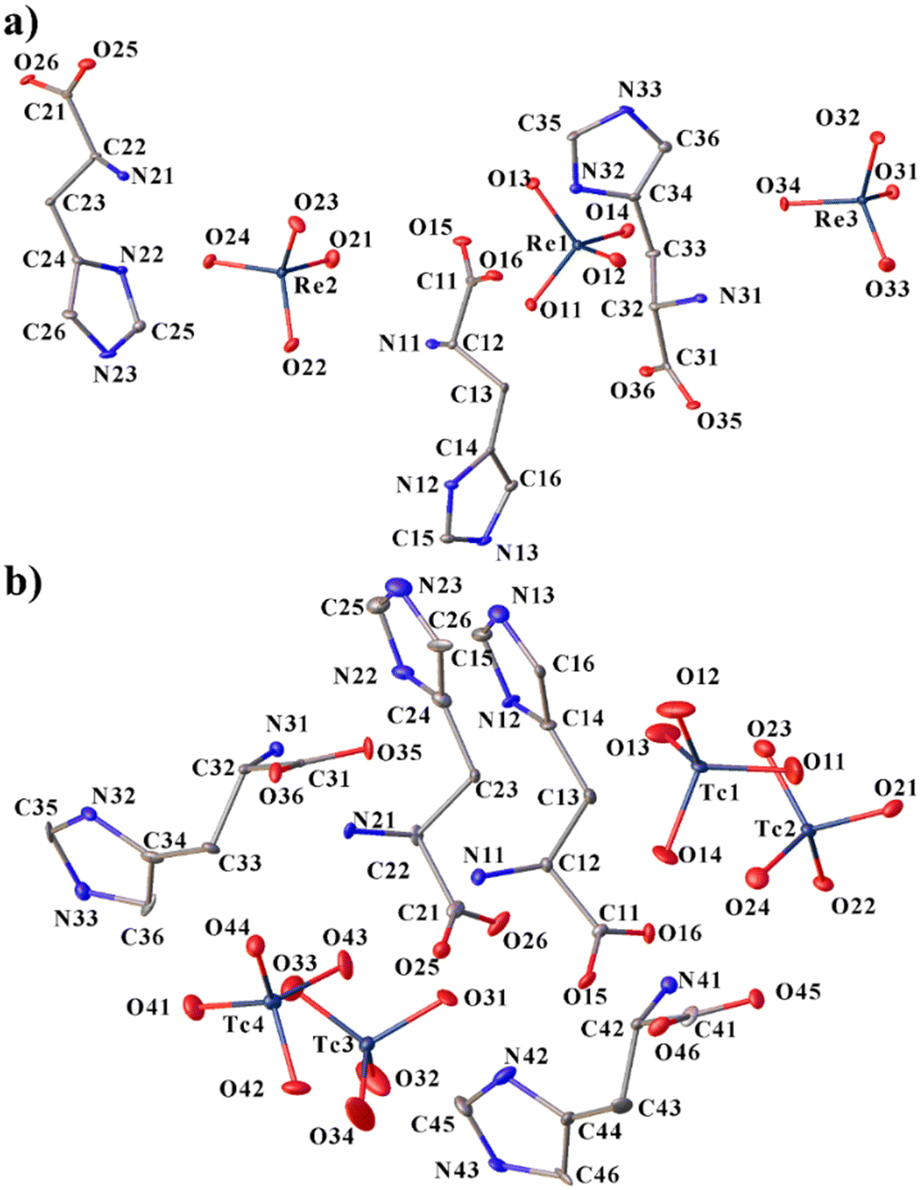 | ||
| Fig. 1 Molecular structures of 1 (a) and 2 (b) with atom labeling. H-atoms are omitted for clarity. Displacement ellipsoids are drawn at the 50% probability level. | ||
The Re–O distances in the three perrhenates of compound 1 are close and vary from 1.722(4) to 1.736(4) Å (Table S1†). In pertechnetates, the distances are shorter than in perrhenates, but vary more strongly from 1.667(16) to 1.726(14) Å in general (Table S5†). The greater variation in M–O bond lengths in pertechnetates than in perrhenates may be due to an essentially lower quality of structure 2. The Tc1–O14 bond (1.752(14) Å) lengthens the most, which may be due to the participation of the oxygen atom O14 in a N–H⋯O hydrogen bond with the imidazolium group. The M–O–M angles in anions are close to tetrahedral; the most distorted angle O33–Re3–O32 is 112.1(2)°.
Carbon atoms C_3 in the cations are in the plane of the imidazole ring (the largest deviation is 0.037(1) Å for C43 in 2). The conformation of the histidine side chain can be described by two torsion angles χ1 [C_4–C_3–C_2–N_1] (−(60.9(7)–63.3(7))° in 1 and −(55.1(2)–63.3(2))° in 2) and χ21[C_2–C_3–C_4–N_2] (−(65.5(8)–68.9(7))° in 1 and −(41(3)–56(2))° in 2) or χ22 [C_2–C_3–C_4–C_6] (110.4(15)–112.3(7)° in 1 and 121.6(19)–148.7(19)° in 2)34 (Tables S4 and S8†). The sum of the absolute values of χ21 and χ22 is 180°. χ1 can take values around −60, 60 or 180° corresponding to the open conformation (I) (g−), closed conformation (g+) and open conformation (II).35–37 It can be said that the histidinium cations in 2 have a more distorted, although the same, conformation. All histidinium cations in these structures are in the same open conformations (I) (g−). A deviation of χ21 and χ22 angles from 90° and χ1 from 60° may be due to the participation of the imidazole rings in H-bonds and is also observed in other compounds such as L-histidinium tetrafluoroborate.38 The imidazole groups of L-histidinium are in a trans conformation, and χ [C_1–C_2–C_3–C_4] values are close to 180°, which may provide higher availability of these groups to receptor interactions.
A complex system of hydrogen bonds is formed in both new compounds, a fragment of which for 1 is shown in Fig. 2. In this case, not only nitrogen atoms of imidazole rings and amino groups are involved in the formation of hydrogen bonds of the N–H⋯O type, but also carbon atoms form much weaker yet notable hydrogen bonds of the C–H⋯O type (Tables S3 and S7†). Cations and anions differ not only in their participation in hydrogen bonds in different structures, but also within the structures. For example, the first (C1_ and N1_ in Fig. 1a) and second (C2_ and N2_) cations form five H-bonds of the N–H⋯O type; the third (C3_ and N3_) cation forms six such bonds in 1. The N⋯O distances of N–H⋯O hydrogen bonds of compounds 1 and 2 are presented in Table 2. In 2, the first (C1_ and N1_) and the third (C3_ and N3_) cations each form six H-bonds of the same type, and the second (C2_ and N2_) and the fourth (C4_ and N4_) each form five. Some of these bonds bifurcate. For example, N31–H31C in 1 forms a bifurcate hydrogen bond with O22ix and O34 [symmetry code (ix) −x + 3/2, −y + 1, z + 1/2].
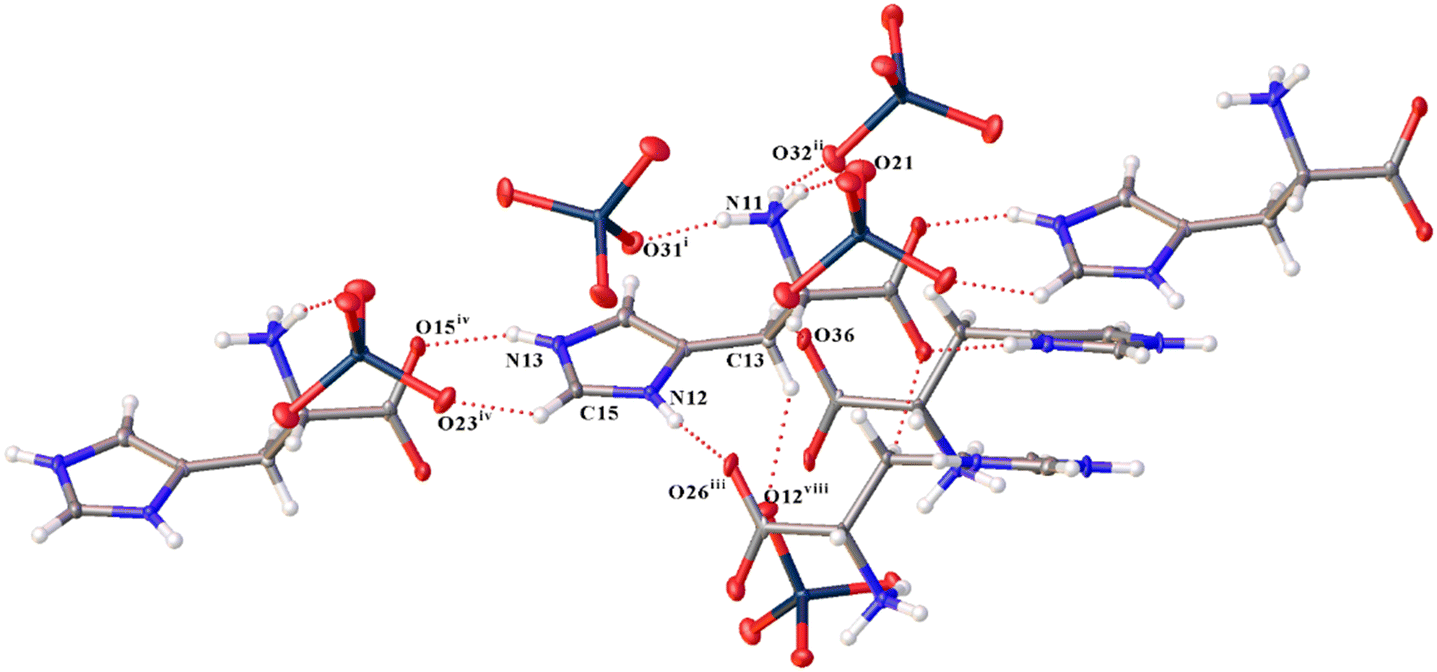 | ||
| Fig. 2 View showing the fragment of hydrogen bonds for the first cation (C1_ and N1_) in 1 [symmetry codes: (i) −x + 3/2, −y + 1, z − 1/2; (ii) −x + 1, y + 1/2, −z + 3/2; (iii) −x + 1, y − 1/2, −z + 1/2; (iv) x + 1, y, z; (viii) x + 1/2, −y + 1/2, −z + 1]. | ||
| 1 | 2 | ||||
|---|---|---|---|---|---|
| D | A | d(D–A)/Å | D | A | d(D-A)/Å |
| N11 | O21 | 2.946(7) | N33 | O421 | 2.94(2) |
| N11 | O311 | 2.801(6) | N33 | O122 | 2.81(2) |
| N11 | O322 | 2.950(7) | N32 | O153 | 2.823(17) |
| N12 | O263 | 2.651(7) | N12 | O451 | 2.947(19) |
| N13 | O154 | 2.616(7) | N31 | O251 | 2.840(17) |
| N21 | O345 | 3.264(7) | N31 | O461 | 2.743(17) |
| N21 | O24 | 2.799(6) | N31 | O223 | 2.856(19) |
| N21 | O116 | 2.830(6) | N22 | O35 | 2.815(19) |
| N22 | O367 | 2.645(6) | N21 | O44 | 2.85(2) |
| N23 | O254 | 2.653(7) | N21 | O453 | 2.997(17) |
| N31 | O138 | 2.839(6) | N21 | O164 | 2.754(18) |
| N31 | O14 | 2.787(6) | N23 | O325 | 2.69(2) |
| N31 | O229 | 3.087(7) | N11 | O31 | 2.859(18) |
| N31 | O34 | 2.844(6) | N11 | O35 | 3.063(17) |
| N32 | O16 | 2.684(7) | N11 | O26 | 2.768(19) |
| N33 | O3510 | 2.680(6) | N41 | O15 | 2.920(17) |
| N41 | O366 | 2.751(17) | |||
| N41 | O14 | 2.832(19) | |||
| N42 | O26 | 2.78(2) | |||
| N13 | O213 | 2.845(19) | |||
| N13 | O417 | 3.01(2) | |||
| N43 | O238 | 3.10(2) | |||
Though in 2 a larger number of stronger H-bonds are formed, in 1 the cations are also well linked to each other but due to supplementary CH–π interactions (Fig. 3a).39–43 Also in both structures, cations and anions are bound by noncovalent anion–π interactions (because the O⋯Cz (aromatic ring centroid) distances are shorter than 4 Å and α angles between the ring plane and the O⋯Cz vector are greater than 50° (ref. 44–49)). In this case, in 1, the anions are interconnected due to the anion–anion interaction (Re1i⋯O13 distance is 3.401(4) Å (Fig. 3a), angle O13i–Re1i⋯O13 is 164.73(9)°) thus supporting the observations and patterns found in ref. 17 and 20. In 1, there is a previously undescribed type of anion⋯anion interaction of the C–O⋯Re type with an O15ii⋯Re3 distance of 2.967(4) Å and an angle O34–Re⋯O15ii of 179.50(17)° (angle C11ii–O15ii⋯Re3 is 130.1(4)°) (Fig. 3a). The presence of directivity allows us to call it not just a short contact, but a fully-fledged σ-hole interaction or bond.
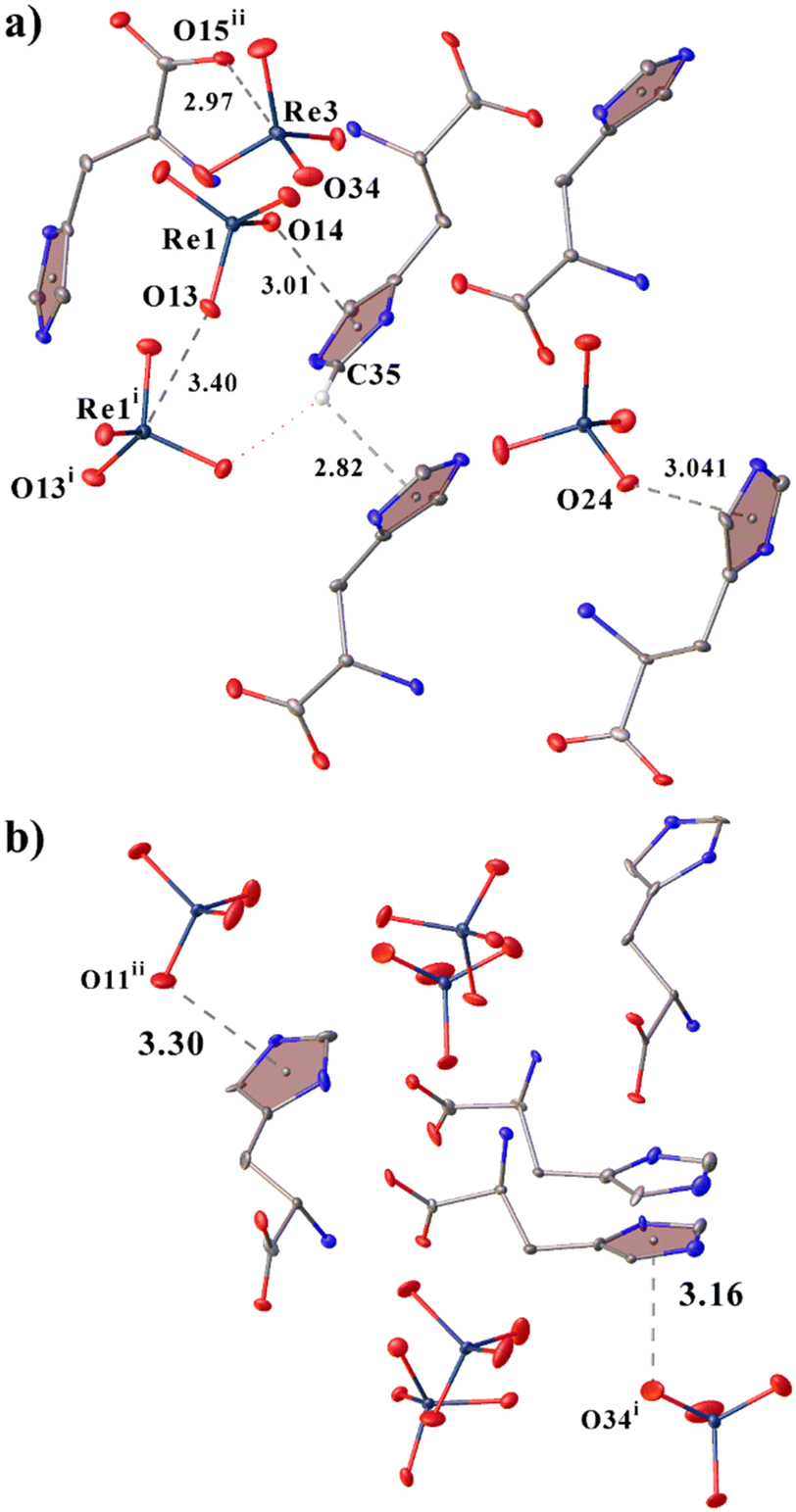 | ||
| Fig. 3 View showing noncovalent interactions in 1 (a) and 2 (b). Only hydrogen atoms involved in CH–π interactions are shown. | ||
When analysing the Cambridge Structural Database (CSD version 5.43, update of November 2022;33), only four structures were found in which there is this type of anion⋯anion interaction between the carboxyl group and the MO4 (M = 7B group) fragment (the M⋯O distance is shorter than the sum of van der Waals radii and the carboxyl group (O–C–O angle 116–130°) is deprotonated): two structures with Mn (YATVEY, YATVIC50) and two with Re (DEGXEV,51 POMKUY52). No interactions of this type have been found for technetium, which may be due to the presence of a small number of structures with it, or perhaps technetium does not form such interactions. When searching for a carboxyl group with MO4 (M = 7B Group), 15 examples of structures with a similar interaction were already found (Table S9 and Fig. S2†). By expanding the search for any transition metal with four oxygen atoms and a carboxyl group, 264 results have already been found. Of course, not all the results found are examples of C–O⋯M interactions and they require further analysis in a separate work.
The crystal packing in the described histidinium perrhenate and pertechnetate can be thought of as consisting of cationic and anionic columns (Fig. S1†).
A survey of the CSD33 reveals few results for L-histidinium compounds with unicharged tetrahedral anions. L-Histidinium perchlorate was found (GASKAM,53 BOWJUT54), and this compound is very similar to the title compounds due to the similarity of the anions. The cation has the same conformation. However, in this compound, unlike 1 and 2, only one crystallographically independent unit is found. In L-histidinium dihydrogenmonophosphate (GERDOW,55 GERDOW01 (ref. 56)) crystallization water participates in H-bonds. In L-histidinium tetrafluoroborate (YEFPUV,38 YEFPUV01 (ref. 57)), the histidinium cation has a closed conformation in contrast to compounds described in this study. For rhenium with a histidine molecule, only coordination compounds, such as carbonyls, have been found.58–61 For technetium with a histidine molecule, the only structure with technetium carbonyl was found.27 It should also be noted that the described structures of histidinium perrhenate and pertechnetate are the first examples of structures of histidine with tetrahedral transition metal anions.
Overall, it can be said that despite the same conformation of the cations and similar structures of the anions, the resulting structures differ not only in the number of independent fragments, but also in the interactions in them. In both structures, a complex system of hydrogen bonds is formed and binds not only cations and anions to each other, but also cations to each other. Moreover, while more N–H⋯O type hydrogen bonds are formed in pertechnetate, in the case of perrhenate there is a greater variety of noncovalent interactions (CH–π, anion⋯anion interactions). The cation–anion interaction of the rhenium atom with the oxygen atom of the carboxyl group, described for the first time, may be of interest not only from a fundamental point of view, but also from the side of self-organization of structures and self-assembly processes.49,62 The presence of a large number of noncovalent interactions can support the stability of structures of this kind and their use for extracting technetium from solutions.14,15,22,63–65
Hirshfeld surface analysis
The CrystalExplorer 21.5 (ref. 66) program was used to analyze intermolecular contacts in all cations and anions of 1 and 2 by the Hirshfeld surface method.67,68 The donor–acceptor groups are visualized using a standard (high) surface resolution and dnorm surfaces are mapped over a fixed color scale. An example of this surface for 1 is illustrated in Fig. 4. Red spots on the surface of the dnorm plot indicate intermolecular contacts involving the hydrogen bonds. The brightest red spots correspond to the strongest hydrogen bonds of the N–H⋯O type.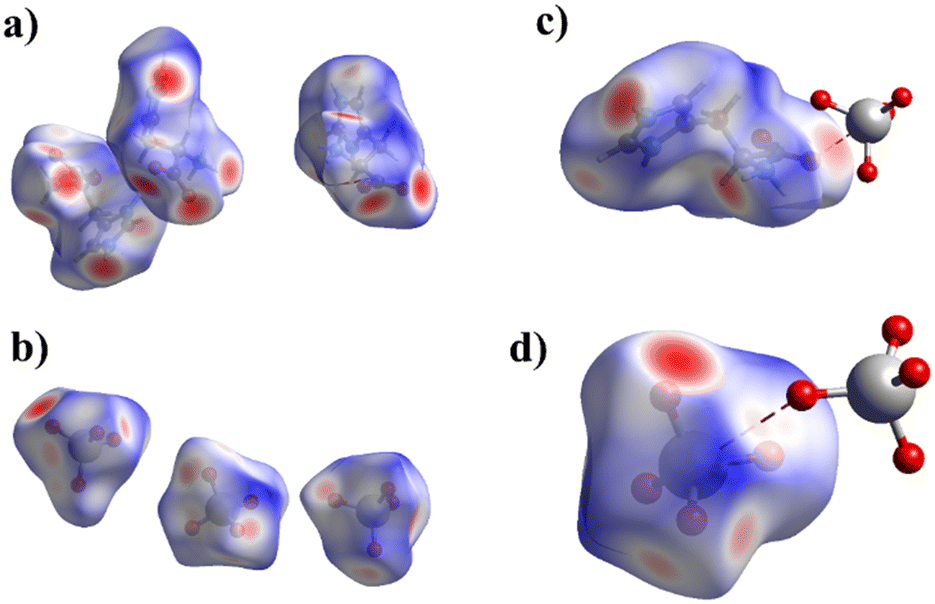 | ||
| Fig. 4 Hirshfeld surface maps over dnorm for cations (a) and anions (b) in 1 and view showing anion⋯anion interactions of the C–O⋯Re (c) and Re–O⋯Re (d) types in 1. | ||
Since the perchlorate anion has a structure similar to perrhenate and pertechnetate, Hirshfeld surfaces were constructed for the two histidinium perchlorates found in different space groups (GASKAM(P21);53 BOWJUT (Pbca)54). To elucidate the role of oxygen in tetraoxoanions, an additional Hirshfeld surface was constructed for histidinium tetrafluoroborate (YEFPUV38). For the latter, only a structure of acceptable quality was chosen.
The main contribution to noncovalent interactions in cations and anions is made by contacts of the O(F)⋯H/H⋯O(F) type, which are usually responsible for hydrogen bonds (Fig. 5 and 6). The proportion of these contacts in compounds and crystallographically independent fragments is slightly different. In 1, in the cations, the proportion of these interactions is on average somewhat lower (61.5%) than in 2 (66.2%), however, in 2 there is a greater spread of this type of interaction. The proportion of the H⋯H type contacts for cations in 1, in contrast, is slightly higher on average (19.0%) than that in 2 (17.5%). It can be noted that in 1, in the cations, the proportion of H⋯C/C⋯H and H⋯N/N⋯H interactions is greater, which can be explained by the presence of CH–π and NH–π interactions. In 2, in contrast, the proportion of O⋯C/C⋯O and N⋯O/O⋯N interactions increases; here, anion–π interactions are present. Fig. 5 shows that the proportion of different types of interactions in two different perchlorates, despite different space groups, is practically the same. The fraction of contributions from short contacts in histidinium perchlorate cations is rather close to that of cations in pertechnetates. At first glance, histidinium tetrafluoroborate stands out the most. However, the share of contributions of O⋯H/H⋯O and H⋯F/F⋯H contacts in total gives 64.3%, as well as the share of O⋯H/H⋯O contacts in other cations. In tetrafluoroborate, in the cations, the proportion of other types of interactions decreases, which is not shown in Fig. 5, because they are less than 2%.
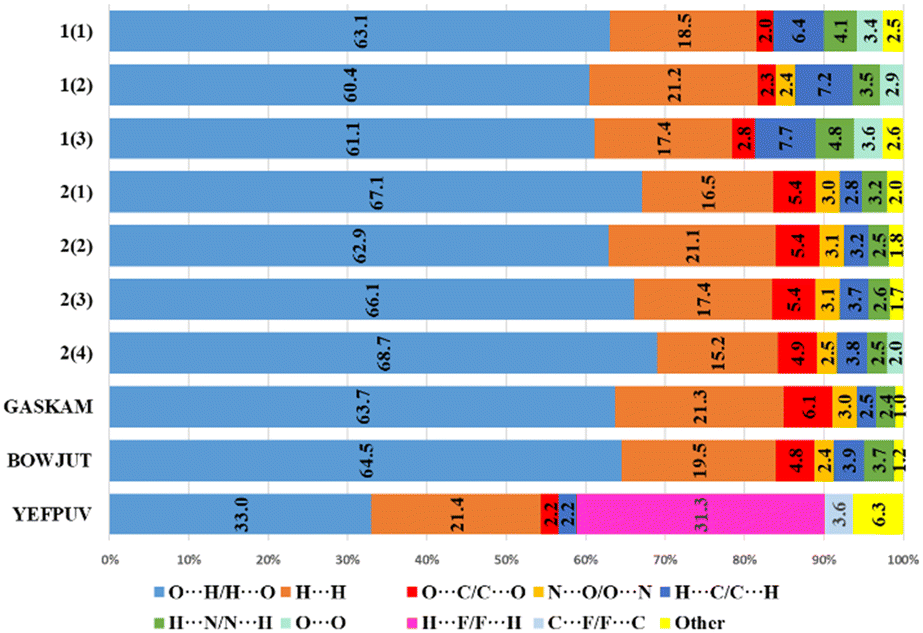 | ||
| Fig. 5 Percentage contributions of contacts to the Hirshfeld surface in cations of structures 1, 2 and taken from the CSD. The index for each crystallographically independent cation is given in parentheses. | ||
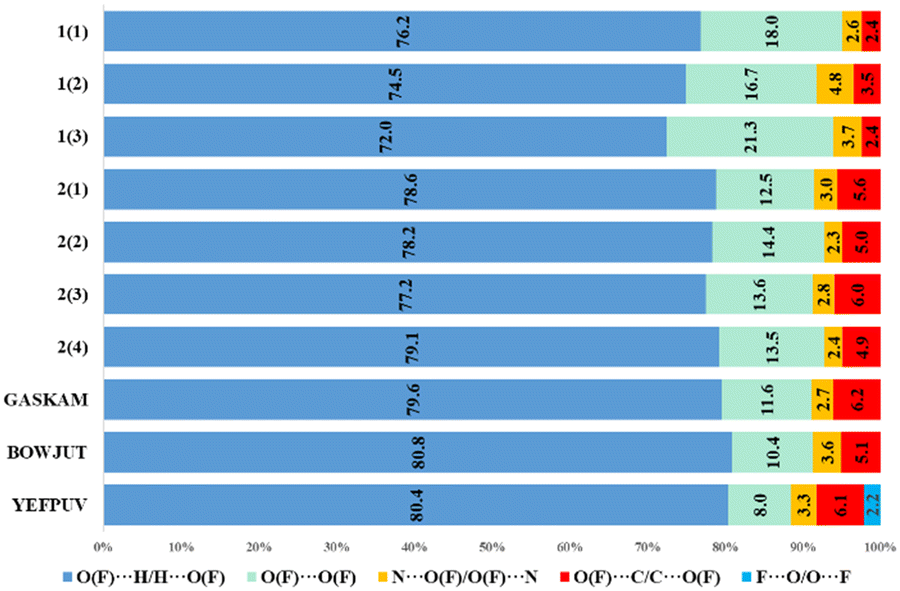 | ||
| Fig. 6 Percentage contributions of contacts to the Hirshfeld surface in anions of structures 1, 2 and taken from the CSD. The index for each crystallographically independent anion is given in parentheses. | ||
In the anions of the calculated structures, the proportion of O⋯H/H⋯O or F⋯H/H⋯F interactions decreases when passing from histidinium tetrafluoroborate to histidinium perrhenate. However, the proportion of O(F)⋯O(F) interactions increases in perrhenates, which may be due to anionic interactions or interactions of anions with the histidinium carboxyl group. Almost no differences are observed for histidinium perchlorates obtained in different space groups. In general, as for cations and for anions, histidinium tetrafluoroborate stands out the most due to the replacement of oxygen atoms by fluorine ones and the small size of anions.
Of special but complicated interest is the influence of acidity on the formation of the target compounds (both perrhenate and pertechnetate). The effect of acidity on the yield of target products is associated with a parallel change in the thermodynamic stability of the phases due to the formation of more (case of Tc) and less (case of Re) soluble phases and final products.11,63,69 According to our observations, when histidinium perrhenate is deposited from rhenium acid and a nitrogenous base (histidine), a fine-crystalline sample of HHisReO4 film is formed, which does not allow X-ray diffraction analysis of the single crystal. In the case when the precipitation of histidinium perrhenate was carried out from the salt Mg(ReO4)2 and a nitrogenous base in a slightly acidic medium supported by a small amount of HCl, a fairly complete formation of HisHReO4 crystals of good quality occurred. When attempting to synthesize HisHTcO4 crystals using the second method, the target product was not formed in the reaction mixture. When replacing the magnesium derivative (Mg(TcO4)2) with highly soluble ammonium pertechnetate, only histidinium chloride crystals precipitated, while highly soluble NH4TcO4 remained in solution. It was possible to obtain HHisTcO4 crystals only from freshly prepared HTcO4 when it was neutralized with a nitrogenous base (histidine). Due to the superposition of two effects (changes in the solubility of Tc and Re compounds with the same cations and a change in the concentration of an organic cation with a change in acidity), it is not possible to isolate the net effect of pH on the yield of the target products. At the same time, the crystallization of perrhenate and pertechnetate with the same cation in different space groups according to our observations is not related to the acidity of the solution, but to the greater diffusivity of the 5d orbitals of rhenium in comparison with the 4d orbitals of technetium, leading, as discovered in this article, to the possibility of the formation of additional weak carboxylate–perrhenate bonds, leading to a change in the space group. This observation well indicates the importance of such weak bond formation to the structure of the resulting compounds, even those with identical empirical formula, and opens the possibility of close element (like Tc and Re) separation.
Conclusions
In this work, two new L-histidinium compounds were synthesized: perrhenate and pertechnetate; for the first, anion⋯anion interactions C–O⋯Re(O4) and Re–O⋯Re(O4) were discovered. The crystal structures were studied using single-crystal XRD and a comparative analysis of noncovalent interactions in the obtained structures and their analogues was carried out. The histidine molecules in the present structures are in the open conformation (1) (g−). In L-histidinium perrhenate, a new type of anion⋯anion interaction of C–O⋯Re with the carboxylate group was firstly described with an O⋯Re distance of 2.97 Å. Anion⋯anion interactions (ReO4−⋯ReO4−) are present in the perrhenate and anion–π in both structures. Not only are NH groups of imidazole rings and amino groups involved in the formation of N–H⋯O type hydrogen bonds, but also CH groups form weaker C–H⋯O type hydrogen bonds. Hirshfeld surface analysis of noncovalent interactions in perrhenates, pertechnetates and their analogues with histidinium showed that the most important contribution to the packing organization is made by the O⋯H/H⋯O interaction for cations and anions. The size of the anion and the nature of the terminal atoms have the greatest influence in the system of noncovalent interactions. The pertechnetate is the second example of a compound of technetium with this amino acid, and both compounds described in this work are the first examples of structures of tetrahedral transition metal anions with histidine. A rare case of non-isostructurality of Tc(VII) and Re(VII) has one more example (L-histidinium MO4) and this observation demonstrates the importance of weak noncovalent bonds to the structure of the resulting compounds, even those with identical empirical formula, and opens the possibility of close element (like Tc and Re) separation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The study was supported by the Russian Science Foundation Grant No. 23-73-01068, https://rscf.ru/project/23-73-01068/. X-ray diffraction experiments were performed at the Center for Shared Use of Physical Methods of Investigation at the Frumkin Institute of Physical Chemistry and Electrochemistry, RAS. The authors express their gratitude to Volkov M. A. (employee of the technetium chemistry laboratory at Frumkin Institute of Physical Chemistry and Electrochemistry, RAS) for assistance in obtaining pertechnetic acid.Notes and references
- J. Černý and P. Hobza, Phys. Chem. Chem. Phys., 2007, 9, 5291–5303 RSC.
- R. H. Blessing, Acta Crystallogr., Sect. B: Struct. Sci., 1986, 42, 613–621 CrossRef.
- A. Subha Mahadevi and G. Narahari Sastry, Chem. Rev., 2016, 116, 2775–2825 CrossRef PubMed.
- K. E. Riley and P. Hobza, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 3–17 CAS.
- C. A. Hunter and C. A. Hunter, Angew. Chem., Int. Ed., 2004, 43, 5310–5324 CrossRef CAS PubMed.
- A. Bender and R. C. Glen, Org. Biomol. Chem., 2004, 2, 3204–3218 RSC.
- L. M. Salonen, M. Ellermann and F. Diederich, Angew. Chem., Int. Ed., 2011, 50, 4808–4842 CrossRef CAS.
- J. N. Bull, C. S. Anstöter and J. R. R. Verlet, Nat. Commun., 2019, 10, 1–9 CrossRef.
- B. F. Minaev, G. V. Baryshnikov and V. A. Minaeva, Dyes Pigm., 2012, 92, 531–536 CrossRef CAS.
- D. Recatalá, R. Llusar, F. Galindo, K. A. Brylev and A. L. Gushchin, Eur. J. Inorg. Chem., 2015, 2015, 1877–1885 CrossRef.
- M. A. Volkov, A. P. Novikov, M. S. Grigoriev, V. V. Kuznetsov, A. V. Sitanskaia, E. V. Belova, A. V. Afanasiev, I. M. Nevolin and K. E. German, Int. J. Mol. Sci., 2023, 24, 2015 CrossRef CAS PubMed.
- S. Varughese, J. Mater. Chem. C, 2014, 2, 3499–3516 RSC.
- K. E. German, A. M. Fedoseev, M. S. Grigoriev, G. A. Kirakosyan, T. Dumas, C. Den Auwer, P. Moisy, K. V. Lawler, P. M. Forster and F. Poineau, Chem. – Eur. J., 2021, 27, 13624–13631 CrossRef CAS PubMed.
- A. Ravi, A. S. Oshchepkov, K. E. German, G. A. Kirakosyan, A. V. Safonov, V. N. Khrustalev and E. A. Kataev, Chem. Commun., 2018, 54, 4826–4829 RSC.
- D. Sheng, L. Zhu, X. Dai, C. Xu, P. Li, C. I. Pearce, C. Xiao, J. Chen, R. Zhou, O. K. Farha, Z. Chai and S. Wang, Angew. Chem., Int. Ed., 2019, 58, 4968–4972 CrossRef CAS PubMed.
- K. E. German, M. S. Grigoriev, C. Den Auwer, A. Y. Maruk and Y. A. Obruchnikova, Russ. J. Inorg. Chem., 2013, 58, 691–694 CrossRef CAS.
- A. P. Novikov, K. E. German, A. V. Safonov and M. S. Grigoriev, ChemistrySelect, 2022, 7, e202202814 CrossRef CAS.
- M. A. Volkov, A. P. Novikov, N. E. Borisova, M. S. Grigoriev and K. E. German, Inorg. Chem., 2023, 62, 13485–13494 CrossRef CAS PubMed.
- M. S. Grigoriev, K. E. German and A. Y. Maruk, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, m2355 CrossRef CAS.
- A. Daolio, A. Pizzi, G. Terraneo, A. Frontera and G. Resnati, ChemPhysChem, 2021, 22, 2281–2285 CrossRef CAS.
- W. A. Volkert and T. J. Huffman, Chem. Rev., 1999, 99, 2269–2292 CrossRef CAS.
- A. A. Artemjev, A. P. Novikov, G. M. Burkin, A. A. Sapronov, A. S. Kubasov, V. G. Nenajdenko, V. N. Khrustalev, A. V. Borisov, A. A. Kirichuk, A. S. Kritchenkov, R. M. Gomila, A. Frontera and A. G. Tskhovrebov, Int. J. Mol. Sci., 2022, 23, 6372 CrossRef CAS.
- J. Rebek, Struct. Chem., 1990, 1, 129–131 CrossRef CAS.
- M. E. Brosnan and J. T. Brosnan, J. Nutr., 2020, 150, 2570S–2575S CrossRef.
- M. Holeček, Nutrients, 2020, 12, 848 CrossRef.
- M. Ramek, J. Pejić and J. Sabolović, J. Inorg. Biochem., 2021, 223, 111536 CrossRef CAS.
- D. R. Van Staveren, S. Mundwiler, U. Hoffmanns, J. K. Pak, B. Spingler, N. Metzler-Nolte and R. Alberto, Org. Biomol. Chem., 2004, 2, 2593–2603 RSC.
- SAINT, V8.40B, Bruker AXS Inc., Madison, WI, USA, 2020 Search PubMed.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3–10 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS.
- J. Mol, Biologia, 1970, 52, 1–17 Search PubMed.
- J. V. Pratap, R. Ravishankar and M. Vijayan, Acta Crystallogr., Sect. B: Struct. Sci., 2000, 56, 690–696 CrossRef CAS.
- N. Mohamedi, S. Elleuch, S. Boufas, M. Legouira and F. Djazi, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2019, 75, 823–825 CrossRef CAS.
- J. A. Krause, P. W. Baures and D. S. Eggleston, Acta Crystallogr., Sect. B: Struct. Sci., 1991, 47, 506–511 CrossRef PubMed.
- S. G. Raj, G. R. Kumar, T. Raghavalu, R. Mohan and R. Jayavel, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, o1178–o1180 CrossRef CAS.
- V. Spiwok, Molecules, 2017, 22, 1038 CrossRef.
- L. Wang, J. Hao, L. X. Zhai, Y. Zhang and W. K. Dong, Crystals, 2017, 7, 277 CrossRef.
- M. Nishio, Phys. Chem. Chem. Phys., 2011, 13, 13873–13900 RSC.
- M. Nishio, Tetrahedron, 2005, 61, 6923–6950 CrossRef CAS.
- M. A. Volkov, A. P. Novikov, M. S. Grigoriev, A. M. Fedoseev and K. E. German, Int. J. Mol. Sci., 2022, 23, 9461 CrossRef CAS.
- H. Zhuo, Q. Li, W. Li and J. Cheng, Phys. Chem. Chem. Phys., 2013, 16, 159–165 RSC.
- M. B. Shah, J. Liu, Q. Zhang, C. D. Stout and J. R. Halpert, ACS Chem. Biol., 2017, 12, 1210 Search PubMed.
- D. Mitra, N. Bankoti, D. Michael, K. Sekar and T. N. G. Row, J. Chem. Sci., 2020, 132, 93 CrossRef CAS.
- K. E. Riley and K. A. Tran, Crystals, 2017, 7, 273 CrossRef.
- D. Dumitrescu, S. Shova, I. C. Man, M. R. Caira, M. M. Popa and F. Dumitrascu, Crystals, 2020, 10, 1149 CrossRef CAS.
- A. P. Novikov, M. A. Volkov, A. V. Safonov, M. S. Grigoriev and E. V. Abkhalimov, Crystals, 2021, 11, 1417 CrossRef CAS.
- Z. Peng, S. Li, A. Li, J. Liao, Y. Wang, X. Li, W. Meng and J. Zhang, Dalton Trans., 2022, 51, 2652–2655 RSC.
- N. Budantseva, G. Andreev and A. Fedoseev, Inorg. Chem., 2017, 56, 12199–12205 CrossRef CAS.
- V. H. Rodrigues, M. M. R. R. Costa, T. Dekola and E. De Matos Gomes, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 65, m19 CrossRef CAS PubMed.
- P. Román, J. M. Gutiérrez-Zorrilla, A. Luque and A. Vegas, J. Crystallogr. Spectrosc. Res., 1987, 17, 585–595 CrossRef.
- S. A. Bahadur and S. Athimoolam, X-Ray Struct. Anal. Online, 2009, 25, 15–16 CrossRef CAS.
- M. T. Averbuch-Pouchot, A. Durif and J. C. Guitel, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1988, 44, 888–890 CrossRef.
- E. Espinosa, S. Veintemillas, C. Miravitlles and E. Molins, Z. fur Krist. - New Cryst. Struct., 1995, 210, 195–198 CAS.
- S. G. Raj, G. R. Kumar, R. Mohan, R. Jayavel and B. Varghese, Phys. Status Solidi B, 2007, 244, 558–568 CrossRef CAS.
- P. Grimminger and P. Klüfers, Dalton Trans., 2009, 715–719 Search PubMed.
- C. Tessier, F. D. Rochon and A. L. Beauchamp, Inorg. Chem., 2002, 41, 6527–6536 CrossRef CAS PubMed.
- J. K. Pak, P. Benny, B. Spingler, K. Ortner and R. Alberto, Chem. – Eur. J., 2003, 9, 2053–2061 CrossRef CAS.
- R. Alberto, R. Schibli, R. Waibel, U. Abram and A. P. Schubiger, Coord. Chem. Rev., 1999, 190–192, 901–919 CrossRef CAS.
- N. Chongboriboon, K. Samakun, W. Dungkaew, F. Kielar, M. Sukwattanasinitt and K. Chainok, Crystals, 2021, 11, 198 CrossRef CAS.
- R. Xie, N. Shen, X. Chen, J. Li, Y. Wang, C. Zhang, C. Xiao, Z. Chai and S. Wang, Inorg. Chem., 2021, 60, 6463–6471 CrossRef CAS PubMed.
- L. Zhu, D. Sheng, C. Xu, X. Dai, M. A. Silver, J. Li, P. Li, Y. Wang, Y. Wang, L. Chen, C. Xiao, J. Chen, R. Zhou, C. Zhang, O. K. Farha, Z. Chai, T. E. Albrecht-Schmitt and S. Wang, J. Am. Chem. Soc., 2017, 139, 14873–14876 CrossRef CAS PubMed.
- L. Zhu, C. Xiao, X. Dai, J. Li, D. Gui, D. Sheng, L. Chen, R. Zhou, Z. Chai, T. E. Albrecht-Schmitt and W. Shuao, Environ. Sci. Technol. Lett., 2017, 4, 316–322 CrossRef CAS.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006–1011 CrossRef CAS PubMed.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
- J. J. Mckinnon, D. Jayatilaka and M. A. Spackman, Chem. Commun., 2007, 3814–3816 RSC.
- M. S. Grigoriev, K. E. German and A. Y. Maruk, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, m2061 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2299030 and 2299031. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ce01164j |
| This journal is © The Royal Society of Chemistry 2024 |