
Utilizing 3-methyl-1-butene co-units to tailor phase transition behavior in butene-1 copolymers†
Jiazheng
Shen
,
Wei
Li
*,
Ruijun
Zhao
,
Yingzhuo
Liu
and
Zhe
Ma
 *
*
Tianjin Key Laboratory of Composite and Functional Materials, and School of Materials Science and Engineering, Tianjin University, Tianjin 300072, P. R. China. E-mail: weili_wq@tju.edu.cn; zhe.ma@tju.edu.cn
First published on 30th November 2023
Abstract
Copolymerization with extra co-units is often utilized to tailor the structure of crystalline polymers. In this work, a unique type of 3-methyl-1-butene (3M1B) co-monomer was chosen to introduce branched co-units into the polybutene-1 main chain. For this series of butene-1/3-methyl-1-butene random copolymers synthesized, the stretching-induced phase transition from the kinetically favored tetragonal form II into the thermodynamically stable trigonal form I was investigated using a combination of tensile tests and in situ wide-angle X-ray diffraction. The results revealed that for the II–I phase transition, not only the kinetics but also the orientation pathway shows strong dependence on the concentration of 3M1B co-units. For copolymer 3M1B1.27 with a low co-unit concentration of 1.27 mol%, form II transformed into form I as crystallites reached the off-axis orientation. With a high co-unit concentration of 3.12 mol%, form II of copolymer 3M1B3.12 could develop into a highly oriented state with c-axes parallel to the stretching direction, which subsequently transformed into highly oriented form I. More importantly, the stretching-induced II–I phase transition was suppressed by increasing 3M1B co-units of copolymers. Therefore, as the 3M1B co-unit concentration was increased in copolymers, the resistance to external deformation was decreased, resulting in decreased mechanical strength.
Introduction
At present, crystalline polymers account for more than two-thirds of synthetic polymer materials and have been widely applied in our daily life. However, polymorphism is a common phenomenon in semi-crystalline polymers and the different modifications endow materials with distinct properties.1–6 In addition, the crystallites generated may further experience phase transition during storage or/and use, especially with mechanical deformation, which changes the performance significantly.1,6–8 Thus, the formation and evolution of crystalline structures are crucial to polymer science and application.Polybutene-1 (PB) is a typical polymorphic polymer, which can adapt to diverse crystal modifications including trigonal form I/I′, tetragonal form II, and orthorhombic form III.9 Form II possesses a kinetics advantage, of which the growth rate is two orders of magnitude higher than that of the trigonal phase.10 However, form II is thermodynamically unstable and can spontaneously transform into the stable modification of trigonal form I.11–14 This phase transition from form II into form I could greatly change the mechanical properties.7,8,13 Thus, to effectively regulate the mechanical properties, the study of the phase transition process is attracting more and more attention.15–25 Over the past decades, various methods have been reported to tailor the phase transition process.15,25–46 Qiao et al. systematically investigated the correlation between temperature and phase transition kinetics. Their results found that for the II–I phase transition, two necessary steps of nucleation and growth have optimal temperatures of −10 and 40 °C, respectively, which leads to the maximum transition rate at room temperature.25 Recently, it was reported that when flow was applied in melt crystallization, the obtained form II crystallites exhibited obviously enhanced transition into form I.26–29 More interestingly, the external stretching deformation to solid form II is also effective in promoting phase transition.30–38 Liu et al. employed synchrotron wide-angle X-ray diffraction to study the stretching-induced II–I phase transition of polybutene-1 and found that stretching greatly accelerated the phase transition process. The mechanism of phase transition was explained as a direct crystal–crystal transition or the melting re-crystallization process, depending on the stretching temperature.30 Cavallo et al. revealed the quantitative correspondence between mechanical extension and phase transition. The results showed that the II–I phase transition of polybutene-1 was controlled by true stress for various stress rates and strain rates, which however was not available anymore for the fraction of form I over 0.5.31 Wang et al. explored the pathway for the II–I transition during stretching in detail. It was found that both the stretching rate and temperature varied the orientation pathway of phase transition, and the transformation kinetics was dependent on the orientation pathway.32 Therefore, understanding how the crystallite structure evolves during stretching is one important aspect to structure control and property design.
In addition to external stimuli, the modification of the molecular structure is able to regulate phase transition.15,32,39–46 As early as the 1960s, Turner–Jones investigated the crystallization and phase transition behaviors of butene-1 copolymers by incorporating linear or branched α-olefins with various numbers of carbon atoms. It was demonstrated that for butene-1 copolymers, copolymerizing the linear α-olefin with more than five carbon atoms and branched co-monomers retarded the II–I phase transition.39 Liu et al. recently found that as the six-membered cyclic co-units was introduced into butene-1 copolymers, the vinylcyclohexane co-units of the side chain showed a much larger influence on suppressing phase transition than the cyclohexene co-units located within the main chain.40 Furthermore, Zheng et al. investigated the combined influences of the internal macromolecular structure and the external stretching field on phase transition in butene-1/4-methyl-1-pentene (4M1P) copolymers.43 It was found that both factors together determined whether the stretching-induced phase transition can take place. For the applied experimental duration of 3000 h, a 4M1P concentration of 3.4 mol% was able to stop the phase transition under quiescent conditions. Differently, as form II crystallites were stretched, a larger threshold 4M1P concentration of 7.8 mol% should be met to impede the II–I phase transition.43 Moreover, with cyclic methylene-1,3-cyclopentane (MCP) co-units incorporated to significantly accelerate the II–I phase transition, Li et al. investigated the interplay between stretching and phase transition of butene-1 copolymers. It was interesting to find that after phase transition started with the mechanical yield, form II crystallites of the PB homopolymer and the MCP copolymers rotated and remained in the off-axis orientation during the following transition process. Moreover, the transformed I could increase the strain-hardening modulus by two orders of magnitude with respect to the initial form II.44 Clearly, the mutual effect of the stretching and the macromolecular structure on the phase transition cannot be simply inferred from the quiescent behaviors, which remains a complex and essential aspect for butene-1 copolymers.
In this work, to obtain comparable crystallinity, 3-methyl-1-butene (3M1B) of maximum 3.12 mol% was incorporated into the polybutene-1 main chain.39 This successfully excluded the potential effect of various crystallinities that often occur in copolymers.28,39,43,45 It was found that under quiescent conditions, the II–I phase transition was strongly suppressed by incorporation of the branched 3M1B co-units, in contrast to the accelerating effect of cyclic MCP co-units.44 Also interestingly, stretching can trigger the II–I phase transition after yielding, while the quantitative enhancement exhibited strong dependence on the concentration of incorporated 3M1B co-units. It was clearly demonstrated that increasing the content of branched 3M1B co-units could significantly change the orientation pathway of the stretching-induced phase transition, different from the persistent off-axis orientation in MCP copolymers.44 Based on these, the mutual effect of stretching and co-units on the phase transition kinetics was revealed, which helped understand the origin of different phase transition kinetics further.
Experimental section
Materials
A series of butene-1/3-methyl-1-butene random copolymers with different co-unit concentrations were prepared with a dimethylpyridylamidohafnium catalyst. The synthesis method has been reported previously.47 High-temperature gel permeation chromatography (PL-GPC 220) was used to characterize the molecular weight and distribution. NMR was used to determine the concentration of co-units in copolymers. The NMR results and analysis method are given in the ESI.†Table 1 summarizes the molecular characteristics.| Sample name | Co-unit concentration (mol%) | M w (kDa) | M w/Mn |
|---|---|---|---|
| PB | 0 | 620 | 1.8 |
| 3M1B1.27 | 1.27 | 770 | 2.0 |
| 3M1B2.34 | 2.34 | 720 | 2.2 |
| 3M1B3.12 | 3.12 | 909 | 1.9 |
Differential scanning calorimetry (DSC) measurement
DSC experiments were performed to characterize the thermal behaviors of the polymers using a TA Q2000 instrument, within a flowing N2 atmosphere at 50 mL min−1. The cooling and heating rates applied were 10 °C min−1. The crystallinity was estimated based on the cooling crystallization enthalpy, which is the ratio of absolute values between the crystallization enthalpy of the practical polymer and the melting enthalpy of 100% form II, i.e., ΔHc/ΔHom. For the crystallinity estimation, the employed melting enthalpy of 100% form II ΔHom was 62 J g−1.48Fourier transform infrared spectroscopy (FTIR) measurement
FTIR experiments were carried out with an IR-Tracer 100 infrared spectrometer (Shimadzu, Japan). For the FTIR experiments, the polymers were annealed at 180 °C for 10 min to clean thermal history and subsequently cooled at 10 °C min−1 using a Linkam hot-stage to obtain form II crystallites. The FTIR spectra were recorded from 400 to 4000 cm−1 at a resolution of 4 cm−1. Each FTIR spectrum was averaged over 10 scans. The fraction of transformed form I in the total crystallites was estimated (see Fig. S2†), according to the analysis method previously proposed by Zhong et al.49Tensile test
The tensile tests were performed using a Linkam TST350 tensile tester. The stretching temperature was 30 °C and the stretching rate was 10 μm s−1. For the tensile test, the polymers were annealed at 180 °C for 10 min to clean thermal history and subsequently cooled at 10 °C min−1 using a Linkam hot-stage to obtain form II crystallites.Wide angle X-ray diffraction (WAXD) measurement
The in situ WAXD experiments were performed to reveal the phase transition and the crystallite orientation. The X-ray wavelength was 0.154 nm. The two-dimensional (2D) WAXD patterns were obtained using a Pilatus 300 K detector with a resolution of 172 × 172 μm2. The distance between the detector and the sample was 163 mm and the image acquisition time was 30 s. The fraction of transformed form I (fI) was calculated with the below formula:where A(110)I and A(200)II are the integral areas of the diffraction peaks corresponding to form I (110)I and form II (200)II planes, respectively. To correct the diffraction intensity difference between different crystal planes, a parameter R of 0.36 was employed in this work.50
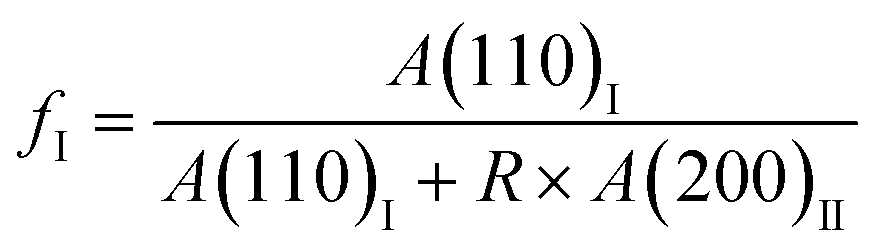
Results and discussion
The significantly slowed down quiescent phase transition
The influence of 3M1B co-units on the properties of copolymer crystallites was examined with differential scanning calorimetry. As shown in Fig. 1a, the introduction of 3M1B co-units does not significantly change the amount of generated crystallites and their thermal stabilities. Fig. 1b quantitatively presents that with increasing 3M1B co-unit concentration between 0 and 3.12 mol%, the crystallinity remains at around 51% and these crystallites also have comparable thermal stabilities. It was intensively reported that the presence of secondary co-units often disturbs the regularity of the main chain and significantly decreases the crystallization ability, resulting in slowed down kinetics and reduced crystallinity.28,43,45 Interestingly, as the secondary 3M1B co-units have a comparable size to the major repeating units of butene-1, these co-units could be included within the crystal cell.39 In this case, 3M1B co-units of maximum 3.12 mol% were utilized to incorporate the branched structure with comparable crystallinity.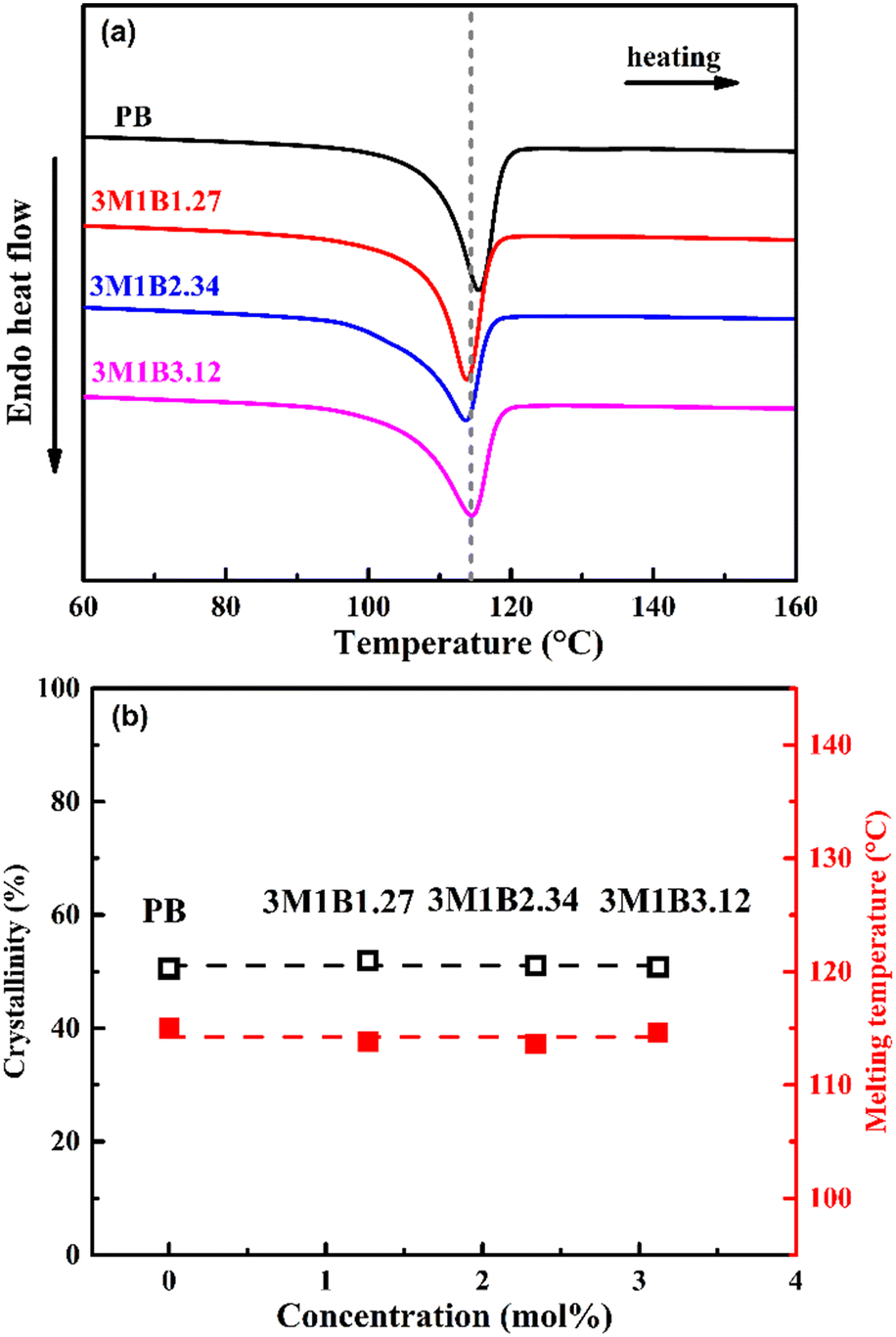 | ||
| Fig. 1 (a) DSC heating curves of PB and 3M1B copolymers and (b) the determined crystallinity and melting temperatures as a function of the concentration of 3M1B co-units. | ||
Next, the influence of 3M1B co-units on the phase transition from the kinetically favored form II into the thermodynamically stable form I was explored by FTIR. Taking 3M1B1.27 and 3M1B3.12 as examples, Fig. 2a and b show changes of the characteristic absorption peaks of form I and form II during the quiescent aging. It was found that as aging proceeds, the phase transition from form II to form I gradually proceeds in 3M1B1.27 but hardly happens in 3M1B3.12. Fig. 2c presents the quantitative evolution. It is clear that the 3M1B co-units have an obvious inhibiting effect on the II–I phase transition. For homopolymer PB, the II–I phase transition took around 80 h to reach an incomplete transition plateau with XI < 1.51 It was proposed that as the II–I phase transition proceeds, the transformed form I crystallites have normal elongation and lateral contraction compared with the initial form II. These adjustments change the environment surrounding the residual form II, including the extra normal pressure and lateral expansion, which slows down the phase transition.51 For the copolymer 3M1B1.27 with a relatively low co-unit concentration, the fraction of form I (XI) increased rapidly at the initial stage, but the plateau value dropped to a much lower value of only 0.7. As the concentration of 3M1B co-units was increased to 2.34 mol%, the thermodynamically stable form I exhibited a prolonged induction period of 168 h and constituted only around 25% of the total crystallites even after transition for 1440 h. As the concentration of 3M1B co-units reached 3.12 mol%, the II–I phase transition even hardly happened within the applied experimental period of 1440 h. This means that the majority of initial crystallites retain their tetragonal modification, although they tend to transform from the thermodynamic point of view. All results demonstrated that the 3M1B co-units could effectively vary the kinetics of quiescent phase transition.
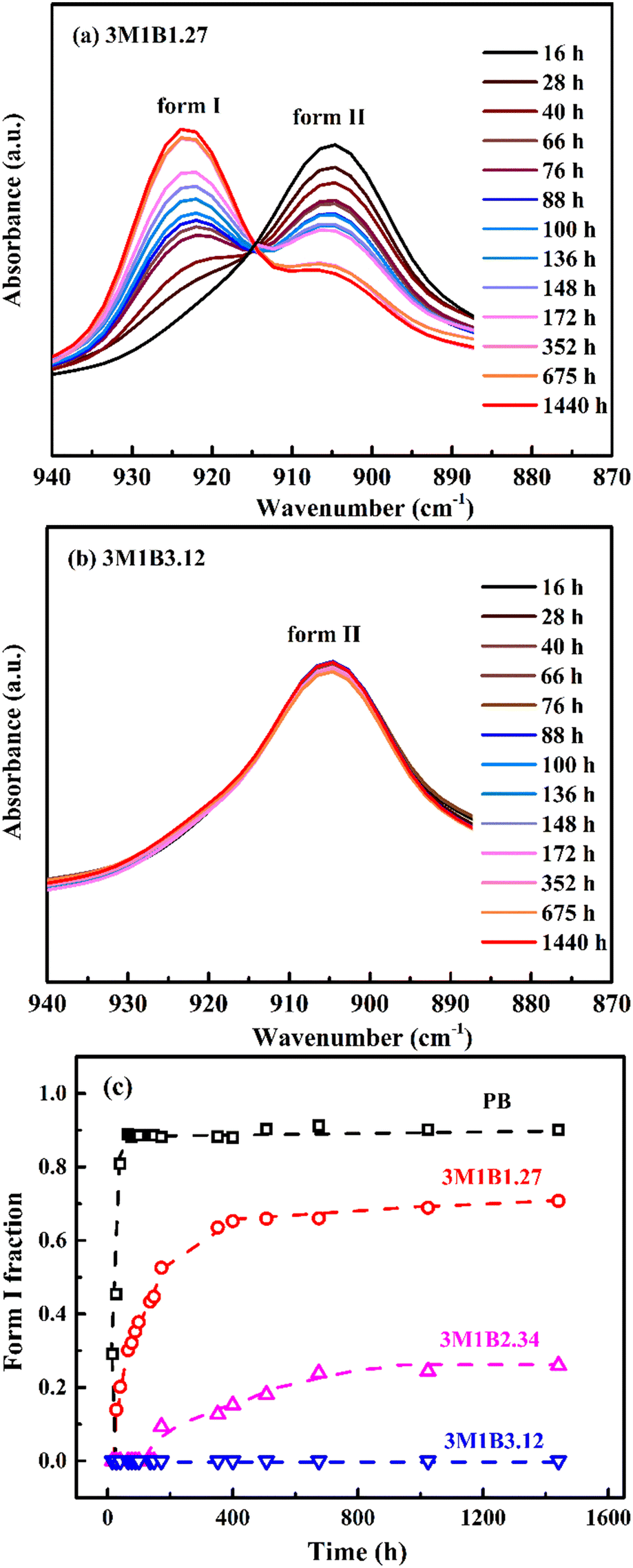 | ||
| Fig. 2 The FTIR spectra of aging in (a) 3M1B1.27 and (b) 3M1B3.12. (c) The evolution of the form I fraction (XI) in PB and 3M1B copolymers during transition at room temperature. | ||
The influence of 3M1B co-units on stretching-induced II–I phase transition
It has been recognized that for butene-1 copolymers, the mechanical stretching can effectively accelerate the phase transition from form II to form I.30–38 Recently, it was reported that yielding was mainly determined by the initial crystallites, while the stretching strength was associated with the evolution degree of II–I phase transition.30 To reveal the structure origin of macroscopic mechanical properties, the in situ WAXD method was employed to track the evolution of microscopic crystallites. Fig. 3 shows the typical 2D WAXD patterns recorded during stretching, of which the specific values of the corresponding strain and stress were marked in the mechanical curve. The copolymers 3M1B1.27 and 3M1B3.12 were selected as representatives of low and high co-unit concentrations, respectively. Clearly, for both 3M1B1.27 and 3M1B3.12, the initial form II gradually transformed into form I during stretching. Moreover, as shown in Fig. 3c and d, the II–I phase transition began after yielding. It was proposed that yielding was required to trigger the stretching-induced II–I phase transition, which may be related to the generation of new tie chains by lamellar collapse.52 Additionally, it was reported that the transformed form I exhibits much larger hardness and strength than form II.7,8,13 This means that the transformed form I crystallites exhibit a larger resistance to deformation. In this case, the continuous stretching requires rapidly increased stress to implement the deformation.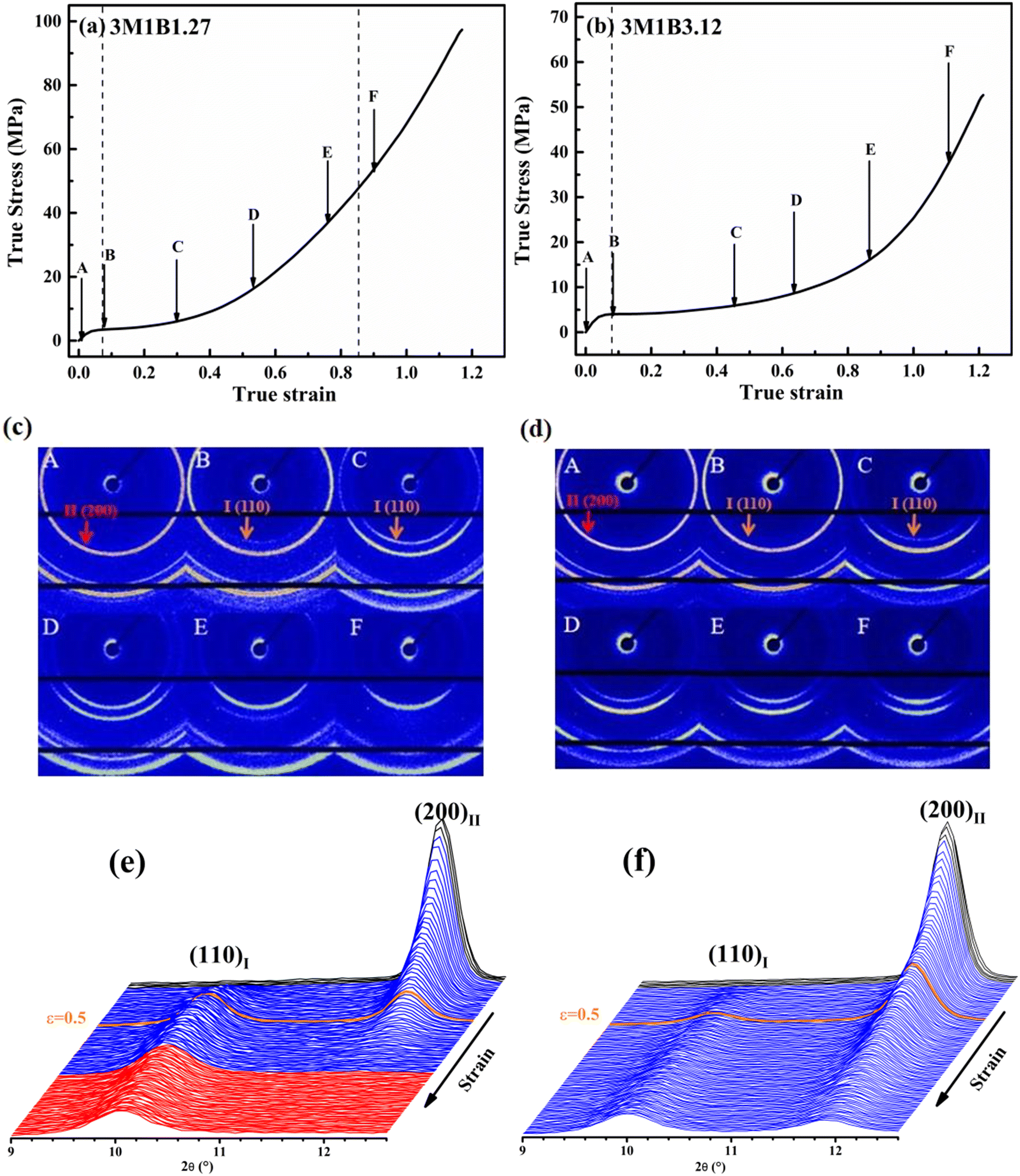 | ||
| Fig. 3 The true stress–true strain curves (a and b) and the selected in situ 2D WAXD patterns (c and d) obtained during stretching and the integrated 1D diffraction intensity profiles (e and f) of 3M1B1.27 and 3M1B3.12. The blue lines represent the process of II–I phase transition and the red ones represent the stretching period after phase transition in 3M1B1.27. The stretching direction is horizontal. | ||
To provide more details about stretching-induced phase transition, the integrated 1D diffraction intensity profiles of 3M1B1.27 and 3M1B3.12 are given in Fig. 3e and f (the results of PB and 3M1B2.34 are presented in Fig. S3†). Different from 3M1B1.27, 3M1B3.12 with a relatively higher co-unit concentration not only showed lower levels of transformed form I (see strain = 0.5), but also reached incomplete transition before fracture (the last curves). It was first suggested that stretching can effectively accelerate phase transition for all polymers studied in this work, since 3M1B1.27 and 3M1B3.12 took much longer induction periods of 28 and 168 h, respectively, to start the phase transition (Fig. 2c). Furthermore, the quantitative enhancement by stretching was dependent on the concentration of 3M1B co-units. After the 3M1B co-unit concentration was increased from 1.27 to 3.12 mol%, the enhanced II–I phase transition changed from 100% transition to only partial transition.
The influence of 3M1B co-units on the crystal orientation
Recently, Cavallo et al. found that in polybutene-1, the II–I phase transition follows a stress-controlled mechanism, which is available for a broad transition range with an upper limit of form I fraction = 0.4–0.5.31 Similarly, Wang et al. reported that for the II–I phase transition, the stress-controlled kinetics showed a close correlation with the crystallite orientation.32 Next, to understand the above phase transition varying among copolymers (Fig. 3), the exploration of the crystallite orientation is necessary. Firstly, Fig. 4a shows the azimuthal scans of form II (200)II planes for 3M1B1.27. For the initial form II crystallites, stretching induced the isotropic texture to develop into oriented structures, which have the preferred alignment direction but still have a certain deviation angle from the stretching direction. The azimuthal scans of form II (200)II planes for PB and 3M1B2.34 are shown in Fig. S4.† Similar to 3M1B1.27, the form II crystallites of PB and 3M1B2.34 remained in the off-axis orientation but their c-axes gradually became more parallel to the stretching direction. However, 3M1B3.12 showed obviously different orientation evolution with respect to the aforementioned 3M1B1.27, even though their form II crystallites did have a similar initial orientation state in the early stretching stage. Fig. 4b presents that as the phase transition proceeded in 3M1B3.12, the form II crystallites gradually developed into a highly oriented state, as indicated by the azimuthal peak at 180°. Additionally, the azimuthal scans of form I (110)I planes for PB and 3M1B copolymers are summarized in Fig. S5.† These scans presented that for copolymers with low co-unit concentration, like 3M1B1.27, form I crystallites consistently presented an off-axis orientation throughout the whole process of II–I phase transition. However, for 3M1B3.12, form I crystallites developed into a highly oriented state just after the start of phase transition. It was demonstrated that increasing the 3M1B co-unit concentration is able to greatly vary the orientation pathway.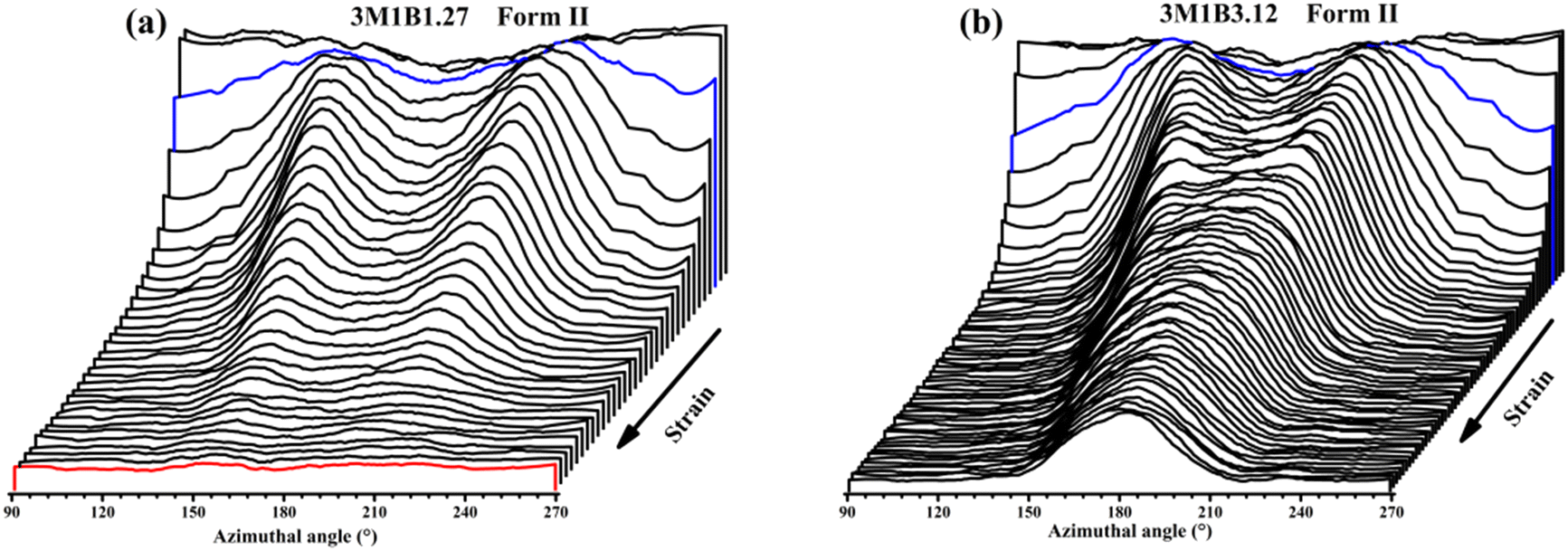 | ||
| Fig. 4 The intensity azimuthal scans of the form II (200)II plane of (a) 3M1B1.27 and (b) 3M1B3.12. The blue line represents the start of phase transition. The red line observed in 3M1B1.27 represents the end of phase transition. | ||
The changes of the form II peak azimuth were determined to quantitatively present the evolution of the crystallite orientation. Fig. 5 shows the evolution of form II crystallites together with the II–I phase transition kinetics. The initial peak azimuths that can be distinguished in the early stretching stage after yielding were quite close for all polymers, showing the similar initial form II orientation. Differently, the stretching-induced orientation evolution, which occurs after yielding, varies largely with increasing co-unit concentration. In PB and 3M1B1.27, the peak azimuth just increased slightly from around 140° to 160° during the whole process of II–I phase transition. For these two polymers with relatively fast phase transition, all form II crystallites finish the complete transition into form I with the off-axis orientation. Afterwards, the obtained form I crystallites further rotated to make c-axes more parallel to the stretching direction (see Fig. S5†), while the residual form II remained in the off-axis orientation. Differently, in 3M1B2.34, the peak azimuth gradually developed into around 175° until the fracture, where there were still some form II crystallites left. What is more interesting is that in 3M1B3.12, simultaneously with the II–I phase transition, form II quickly rotated into the highly oriented state, as demonstrated by the peak azimuth of 180°. At a form I fraction fI = 0.60, all residual form II became highly oriented, which afterwards transformed into the highly oriented form I. These results suggested that actually, the II–I phase transition follows various orientation pathways, where increasing the co-unit concentration causes the copolymer more likely to transform within the highly oriented crystallites. To show the orientation evolution of the stretching-induced phase transition, a schematic representation of distinct pathways is presented in Fig. 6.
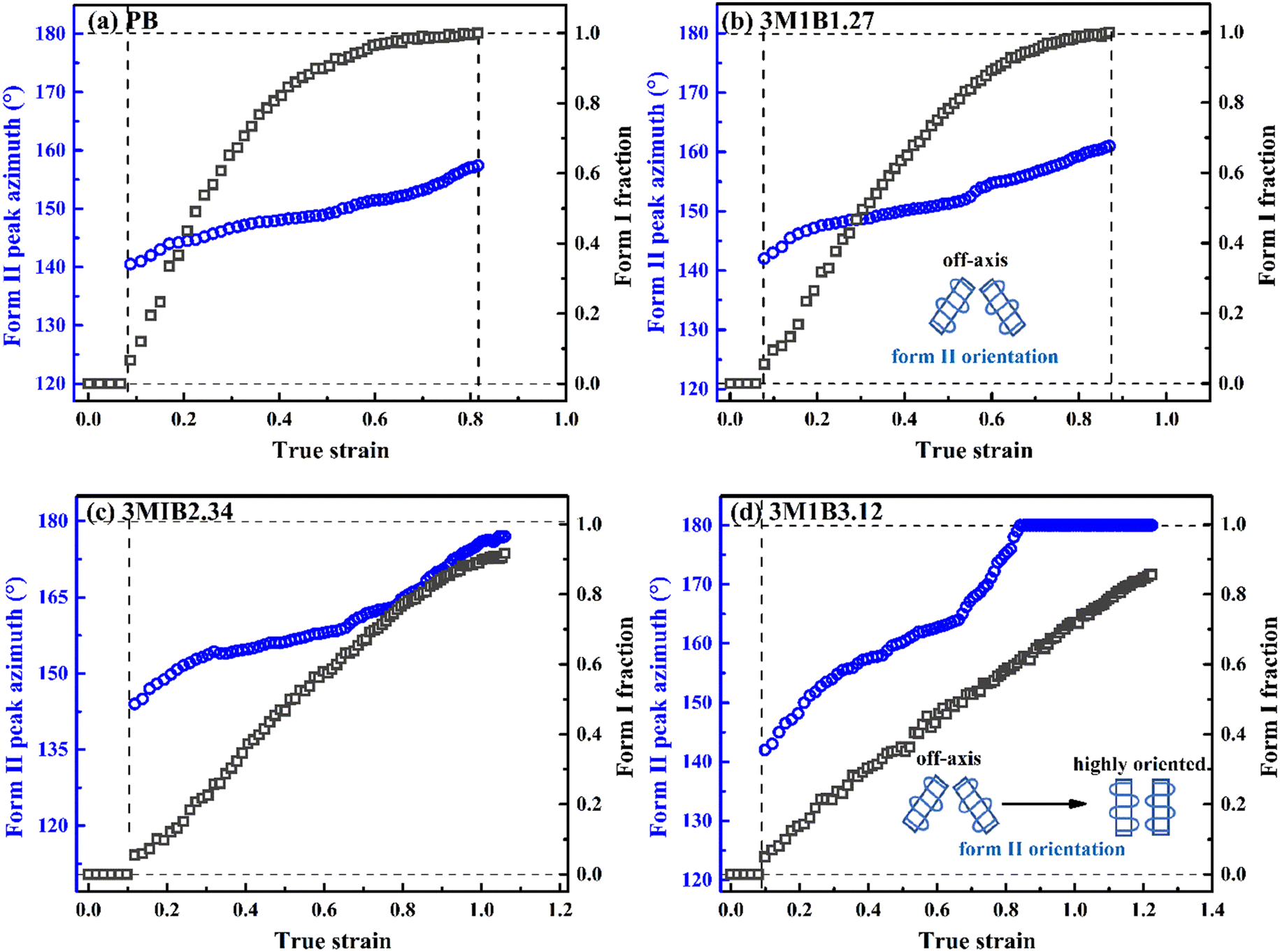 | ||
| Fig. 5 Stretching-induced evolution of the form II peak azimuth in copolymers (a) 3M1B1.27 and (b) 3M1B3.12 with the phase transition (i.e., fI determined from WAXD data). | ||
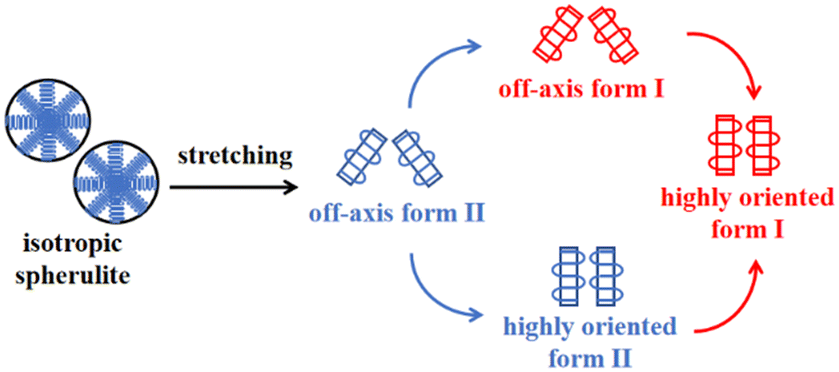 | ||
| Fig. 6 The schematic representation of structural changes of the stretching-induced phase transition affected by 3M1B co-units. | ||
The phase transition kinetics reflected how fast the crystalline stems adjust their packing inside the lattice cell, which may involve both of the conformational and positional ordering depending on the type of polymer. It has been recognized in a general sense that crystallite orientation can happen with mechanical stretching, but its correlation with phase transition was often easy to be ignored. Recently, Wang et al. found that the orientation pathways during phase transition strongly depend on the stretching conditions including both temperature and stretching speed.32 Concerning the relationship of the orientation pathway with the co-unit concentration, Li et al. employed cyclic methylene-1,3-cyclopentane (MCP) co-units to study the stretching-induced phase transition. It was found that after incorporating these cyclic MCP co-units capable of accelerating quiescent phase transition, the crystallinity was obviously decreased, while the stretching-induced phase transition remained in the off-axis orientation.44 Interestingly, the results presented in this work demonstrated that the orientation pathway can also be varied by regulating the concentration of 3M1B co-units, though this co-unit type even does not significantly decrease the crystallinity. For the 3M1B concentration of 1.27 mol%, the form II crystallites seem to implement the II–I phase transition with the stable off-axis orientation (Fig. 5b and 7), which just slightly evolved during stretching. The orientation pathway observed in the relatively low 3M1B concentration is similar to that with relatively low temperature and stretching speed.32 As the 3M1B concentration was increased to 2.34 mol%, the phase transition proceeded simultaneously with distinct orientation evolution approaching the highly oriented state. When the 3M1B concentration was 3.12 mol%, a large part of the II–I phase transition was implemented within the highly oriented crystallites, which was observed with a relatively high temperature, like 70 °C for the butene-1/ethylene copolymer with 1.5 mol% ethylene co-units.32
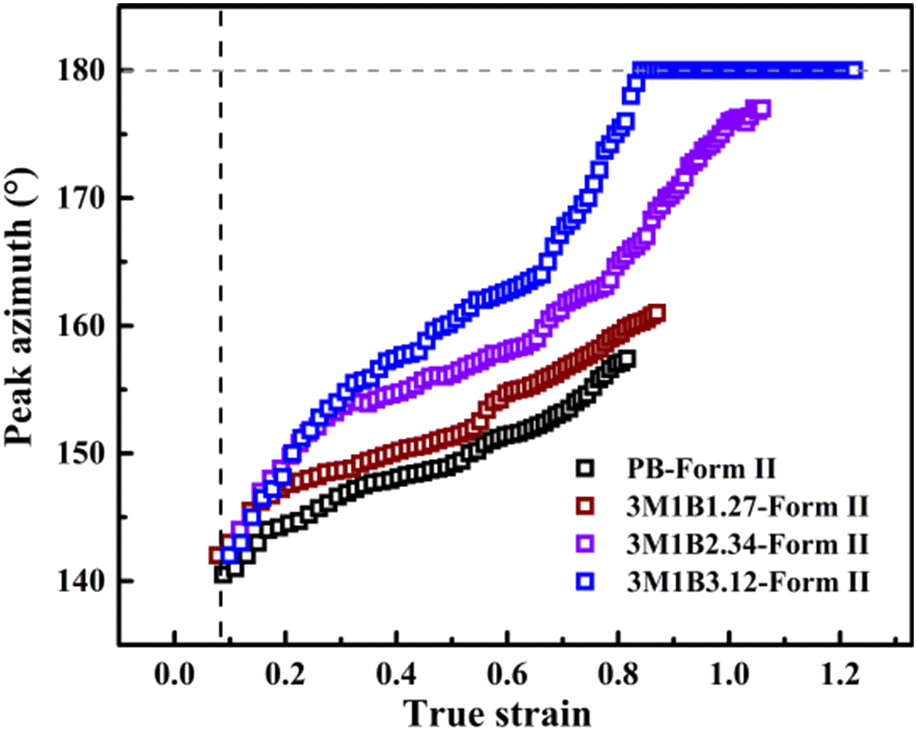 | ||
| Fig. 7 The evolution of the peak azimuth of the form II (200)II plane of PB and 3M1B copolymers. | ||
The influence of 3M1B co-units on the phase transition kinetics and mechanical properties
The phase transition and orientation reflect the simultaneous structural evolution at different length scales, which might couple with each other. For form II crystallites, the orientation could influence the microscopic stress applied on the crystal lattice and as a consequence affect the transition kinetics.43 Thus, the determined kinetics of the stretching-induced phase transition was compared for different polymers as plotted in Fig. 8. These results showed that although incorporation of 3M1B co-units does not significantly decrease the crystallinity of form II, it clearly suppresses the phase transition into more stable form I even with the aid of mechanical stretching. As the concentration of 3M1B co-units was increased to 2.34 mol% and higher, only some of the form II can transform into form I before sample fracture.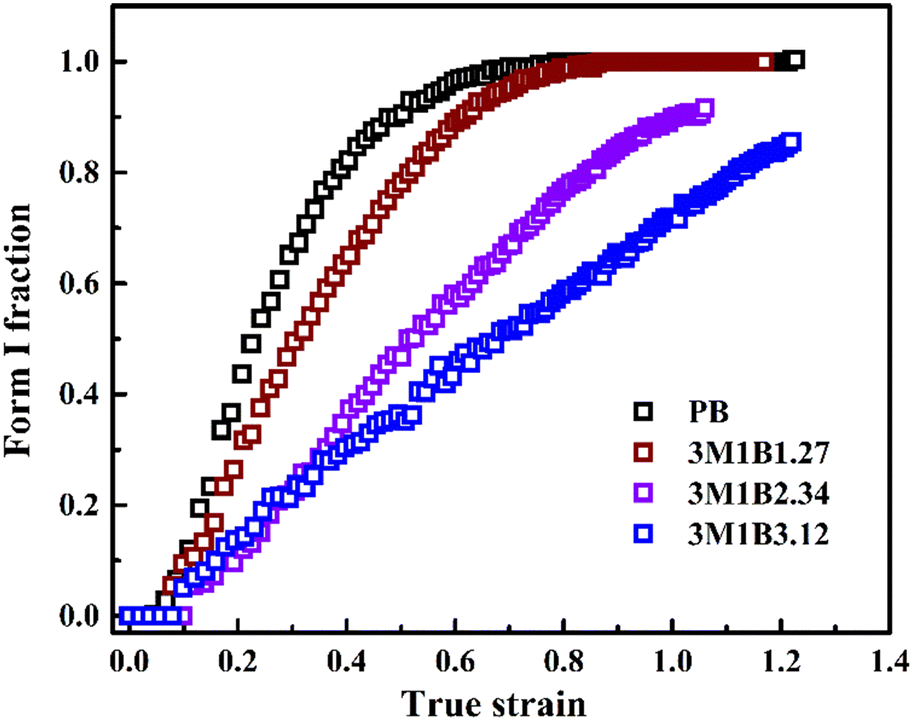 | ||
| Fig. 8 Evolution of the form I fraction with true strain of PB and 3M1B copolymers. | ||
Zheng et al. proposed a reasonable mechanism that the microscopic stress component perpendicular to the c-axis direction (σ⊥) might facilitate the lateral adjustment of crystalline stems within the unit cell, leading to the accelerated phase transition.43 After yielding, the broken crystallites of initial form II crystallites rotate and consequently the c-axes of the crystal lattice tend to become parallel to the stretching direction (i.e., highly oriented state with a peak azimuth of 180°). Thus, the component stress σ⊥ is relatively smaller in the highly oriented state than that of the off-axis orientation. In this case, the stretching-induced phase transition becomes slower as the 3M1B co-unit concentration increases. Thus, it is reasonable to understand the mechanical strength of various polymers studied in this work. Fig. 9 shows the true stress–strain curves of PB and 3M1B copolymers. Both PB and 3M1B copolymers undergo a multi-step deformation process, including elastic deformation, yielding, and strain hardening.53 The different deformation stages exhibit distinct dependencies on the presence of 3M1B co-units. It was found that the introduction of 3M1B co-units does not significantly change the elastic deformation and yielding.
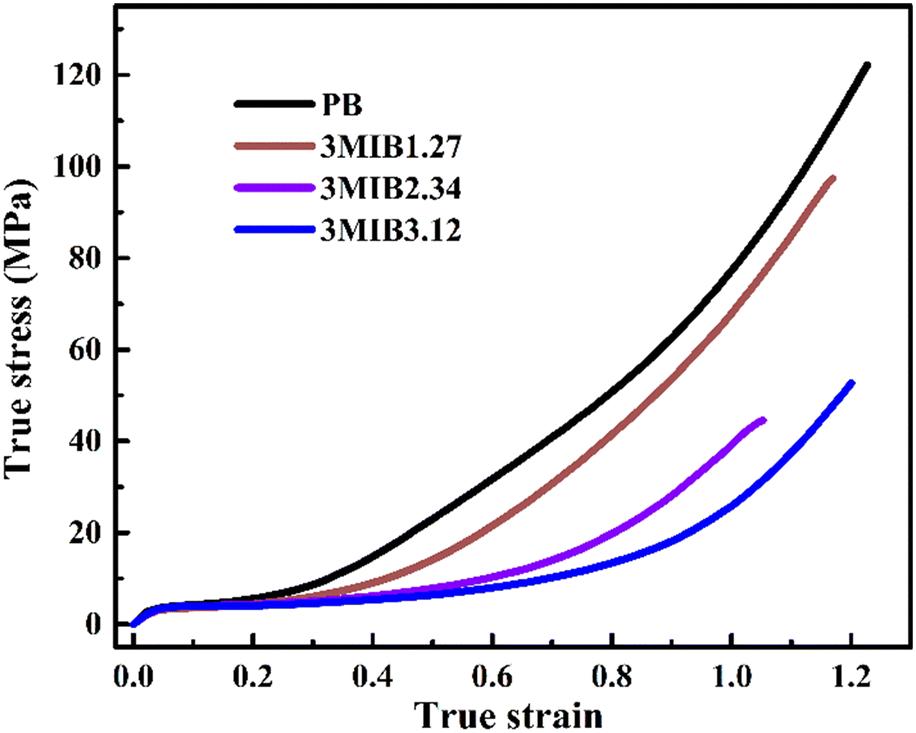 | ||
| Fig. 9 The stress–strain curves of PB and 3M1B copolymers. | ||
However, in the late stretching stage beyond yielding, the copolymers started to exhibit lower stress than PB. As the concentration of 3M1B co-units was further increased, the stress continued to decrease, which is related to the degree of phase transition happening during stretching. The suppressed phase transition obviously decreases the resistance to the external deformation, and consequently varies the mechanical strength.
Conclusions
With a dimethylpyridylamidohafnium catalyst, butene-1/3-methyl-1-butene (3M1B) random copolymers with co-unit concentrations ranging from 1.27 to 3.12 mol% were designed and synthesized. 3M1B co-units of maximum 3.12 mol% were utilized to introduce branched co-units into copolymers with comparable crystallinity. The presence of 3M1B co-units changes the phase transition behaviours under quiescent and stretching-induced conditions. Firstly, the introduction of 3M1B co-units not only slowed down the kinetics of II–I phase transition but also reduced the form I fraction of the incomplete transition plateau.Secondly, the stretching-induced II–I phase transition was studied with in situ wide-angle X-ray diffraction. For the II–I phase transition, not only the kinetics but also the orientation pathway was explored. It was found that stretching greatly accelerates the II–I phase transition. Moreover, it was interesting to find that increasing the 3M1B co-unit concentration significantly affected the pathway of the stretching-induced orientation, different from the persistent off-axis orientation in copolymers with cyclic methylene-1,3-cyclopentane co-units incorporated.44 For 3M1B1.27 with a relatively lower 3M1B co-unit concentration, form II mainly transformed into form I with an off-axis orientation, where the lamellar normal direction did not become parallel to the stretching direction yet. Differently, for 3M1B2.34, the orientation of form II developed largely from the off-axis state to a highly oriented state. As the co-unit concentration reached 3.12 mol%, the form II of 3M1B3.12 quickly evolved into a highly oriented state along the stretching direction, and subsequently transformed into form I. Thus, the increase of 3M1B co-units also changes the orientation pathway for phase transition.
Author contributions
Jiazheng Shen: conceptualization, methodology, formal analysis, investigation, data curation, writing – original draft, writing – review and editing. Wei Li: methodology, software, investigation, resources, data curation. Ruijun Zhao: methodology, investigation, formal analysis. Yingzhuo Liu: investigation, formal analysis. Zhe Ma: conceptualization, methodology, supervision, writing – original draft, writing – review and editing, project administration, funding acquisition.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (52022065).References
- C. De Rosa and F. Auriemma, J. Am. Chem. Soc., 2006, 128, 11024–11025 CrossRef CAS.
- J. Karger-Kocsis, Polym. Eng. Sci., 1996, 36, 203–210 CrossRef CAS.
- R. Imamura, A. B. Silva and R. Gregorio, J. Appl. Polym. Sci., 2008, 110, 3242–3246 CrossRef CAS.
- M. F. Butler and A. M. Donald, Macromolecules, 1995, 28, 6383–6393 CrossRef CAS.
- J. Zhou, Y. Zheng, G. Shan, Y. Bao, W. Wang and P. Pan, Macromolecules, 2019, 52, 1334–1347 CrossRef CAS.
- J. Zhao, S. Feng, W. Zhang, W. Chen, J. Sheng, W. Yu and L. Li, Macromolecules, 2021, 54, 9204–9216 CrossRef CAS.
- F. Azzurri, A. Flores, G. C. Alfonso and F. J. B. Calleja, Macromolecules, 2002, 35, 9069–9073 CrossRef CAS.
- F. Danusso and G. Gianotti, Makromol. Chem., 1965, 88, 149–158 CrossRef CAS.
- R. L. Miller and V. F. Holland, J. Polym. Sci., Part B: Polym. Lett., 1964, 2, 519–521 CrossRef CAS.
- M. Yamashita, A. Hoshino and M. Kato, J. Polym. Sci., Part B: Polym. Phys., 2007, 45, 684–697 CrossRef CAS.
- V. Petraccone, B. Pirozzi, A. Frasci and P. Corradini, Eur. Polym. J., 1976, 323–327 CrossRef CAS.
- P. Corradini, R. Napolitano, V. Petraccone and B. Pirozzi, Eur. Polym. J., 1984, 931–935 CrossRef CAS.
- T. Miyoshi and A. Mamun, Polym. J., 2012, 44, 65–71 CrossRef CAS.
- T. Miyoshi, A. Mamun and D. Reichert, Macromolecules, 2010, 43, 3986–3989 CrossRef CAS.
- Y. Wang, Y. Lu, J. Zhao, Z. Jiang and Y. Men, Macromolecules, 2014, 47, 8653–8662 CrossRef CAS.
- B. Wang, K. He, Y. Lu, Y. Zhou, J. Chen, C. Shen, J. Chen, Y. Men and B. Zhang, Macromolecules, 2020, 53, 6476–6485 CrossRef CAS.
- X. Li, P. Chen, M. Xu, J. Ding, L. Chen, X. Zhang and X. Tian, Macromolecules, 2021, 54, 8751–8769 CrossRef CAS.
- W. Wang, B. Wang, S. F. S. P. Looijmans, E. Carmeli, M. Rosenthal, G. Liu and D. Cavallo, Macromolecules, 2021, 54, 9663–9669 CrossRef CAS.
- Y. Lu, D. Lyu, C. Han, X. Yang, P. C. Lee and Y. Men, Macromolecules, 2022, 55, 10534–10542 CrossRef CAS.
- L. Ni, S. Xu, C. Sun, Y. Qin, Y. Zheng, J. Zhou, C. Yu and P. Pan, ACS Macro Lett., 2022, 11, 257–263 CrossRef CAS PubMed.
- R. Xin, Y. Li, H. Shen, J. Hu, S. Wang, H. Zhang and S. Yan, Macromolecules, 2022, 55, 8203–8209 CrossRef CAS.
- J. Chen, B. Wang, C. Shen, J. Chen, G. Reiter and B. Zhang, Macromolecules, 2023, 56, 5050–5057 CrossRef CAS.
- L. Ni, C. Sun, S. Xu, W. Xiang, Y. Pan, B. Wang, Y. Zheng, C. Yu and P. Pan, Macromolecules, 2023, 56, 1973–1982 CrossRef CAS.
- Y. Qiao and Y. Men, Macromolecules, 2017, 50, 5490–5497 CrossRef CAS.
- Y. Qiao, Q. Wang and Y. Men, Macromolecules, 2016, 49, 5126–5136 CrossRef CAS.
- J. Chen, B. Wang, T. Sun, J. Xu, J. Chen and B. Zhang, Polymer, 2019, 185, 121966 CrossRef.
- F. Su, X. Yang, B. Dong, J. Zhao, F. Lv, Y. Ji, C. Liu and L. Li, Polymer, 2019, 179, 121719 CrossRef CAS.
- Y. Lou, L. Liu, W. Li, R. Zhao and Z. Ma, Macromolecules, 2021, 54, 10150–10162 CrossRef CAS.
- L. Liu, Z. Chu, Y. Liao, Z. Ma and Y. Li, Macromolecules, 2020, 53, 8476–8486 CrossRef CAS.
- Y. Liu, K. Cui, N. Tian, W. Zhou, L. Meng, L. Li, Z. Ma and X. Wang, Macromolecules, 2012, 45, 2764–2772 CrossRef CAS.
- D. Cavallo, M. J. W. Kanters, H. J. M. Caelers, G. Portale and L. E. Govaert, Macromolecules, 2014, 47, 3033–3040 CrossRef CAS.
- W. Wang, C. Shao, L. Zheng, B. Wang, L. Pan, G. Ma, Y. Li, Y. Wang, C. Liu and Z. Ma, J. Polym. Sci., Part B: Polym. Phys., 2018, 57, 116–126 CrossRef.
- Y. Wang, Z. Jiang, Z. Wu and Y. Men, Macromolecules, 2012, 46, 518–522 CrossRef.
- W. Chen, X. Li, H. Li, F. Su, W. Zhou and L. Li, J. Mater. Sci., 2013, 48, 4925–4933 CrossRef CAS.
- Z. Zhang, X. Chen, C. Zhang, C.-T. Liu, Z. Wang and Y.-P. Liu, Chin. J. Polym. Sci., 2020, 38, 888–897 CrossRef CAS.
- Y. Liu, Y. Wang, Q. Yu, L. Ren, Y. Wu, Z. Wang, Q. Li and B. S. Hsiao, Polymer, 2021, 226, 123833 CrossRef CAS.
- S. Feng, J. Zhu, W. Yu, H. Guo, W. Chen, A. Lu and L. Li, Macromolecules, 2022, 55, 2333–2344 CrossRef CAS.
- W. Li, D. Zhang, C. Qv, R. Zhao and Z. Ma, Macromolecules, 2023, 56, 2761–2771 CrossRef CAS.
- A. Turner-Jones, Polymer, 1966, 7, 23–59 CrossRef.
- L. Liu, Y. Lou, C. Qv, Z. Ma and Y. Li, Chin. J. Chem., 2022, 40, 1429–1436 CrossRef CAS.
- C. De Rosa, O. R. de Ballesteros, F. Auriemma, R. Di Girolamo, C. Scarica, G. Giusto, S. Esposito, S. Guidotti and I. Camurati, Macromolecules, 2014, 47, 4317–4329 CrossRef CAS.
- L. He, B. Wang, F. Yang, Y. Li and Z. Ma, Macromolecules, 2016, 49, 6578–6589 CrossRef CAS.
- L. Zheng, L. Liu, C. Shao, W. Wang, B. Wang, L. Pan, Y. Li and Z. Ma, Macromolecules, 2019, 52, 1188–1199 CrossRef CAS.
- W. Li, L. Liu, L. Zheng, Y. Lou, Z. Ma and Y. Li, Macromolecules, 2020, 53, 2145–2156 CrossRef CAS.
- Y. Liao, L. Liu, Z. Ma and Y. Li, Macromolecules, 2020, 53, 2088–2100 CrossRef CAS.
- Y. Lou, W. Li, C. Qv and Z. Ma, Chin. J. Polym. Sci., 2022, 41, 414–421 CrossRef.
- X. Wang, Y. Wang, X. Shi, J. Liu, C. Chen and Y. Li, Macromolecules, 2014, 47, 552–559 CrossRef CAS.
- G. C. Alfonso, F. Azzurri and M. Castellano, J. Therm. Anal. Calorim., 2001, 66, 197–207 CrossRef CAS.
- Z. Zhong, H. Ge and Z. Su, Polymer, 2018, 156, 30–38 CrossRef CAS.
- C. De Rosa, F. Auriemma, O. R. de Ballesteros, F. Esposito, D. Laguzza, R. Di Girolamo and L. Resconi, Macromolecules, 2009, 42, 8286–8297 CrossRef CAS.
- Y. Qiao, H. Wang and Y. Men, Macromolecules, 2018, 51, 2232–2239 CrossRef CAS.
- W. Wang, L. Zheng, L. Liu, W. Li, Y. Li and Z. Ma, Polym. Cryst., 2019, 2, e10052 Search PubMed.
- K. Nitta and H. Nomura, Polymer, 2014, 55, 6614–6622 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S5. See DOI: https://doi.org/10.1039/d3ce01131c |
| This journal is © The Royal Society of Chemistry 2024 |