
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electron-delocalization catalyzers for high performance, low-temperature Li–S batteries†
Jing
Zhang
a,
Lin
Li
a,
Mannan
Yang
 a,
Chen
Cheng
a,
Na
Tian
*a,
Yongzheng
Zhang
e,
Dongmao
Jiao
a,
Hongzhen
Lin
*b and
Jian
Wang
*cd
a,
Chen
Cheng
a,
Na
Tian
*a,
Yongzheng
Zhang
e,
Dongmao
Jiao
a,
Hongzhen
Lin
*b and
Jian
Wang
*cd
aSchool of Materials Science and Engineering & Faculty of Printing, Packaging Engineering and Digital Media Technology, Xi’an University of Technology, Xi’an 710048, China. E-mail: tianna@xaut.edu.cn
bi-Lab & CAS Key Laboratory of Nanophotonic Materials and Devices, Suzhou Institute of Nano-tech and Nano-bionics, Chinese Academy of Sciences, Suzhou, 215123, China. E-mail: hzlin2010@sinano.ac.cn
cHelmholtz Institute Ulm (HIU), Ulm D89081, Germany. E-mail: jian.wang@kit.edu; wangjian2014@sinano.ac.c
dKarlsruhe institute of Technology, Karlsruhe D76021, Germany
eState Key Laboratory of Chemical Engineering, East China University of Science and Technology, Shanghai 200237, China
First published on 26th October 2024
Abstract
The extremely depressive conversion kinetics of polysulfides due to sluggish Li+ diffusion kinetics remain to be resolved for low-temperature Li–S batteries (LT-LSB). Herein, the strategy for electron-delocalization of nanocatalysts has been designed through introducing oxygen defects on vanadium trioxide that was anchored on a porous carbon network (ODVO@PCN). The reconstructed active sites of the V2+ state tend to interact with sulfur species more easily due to the stronger hybridization between V2+ sites and S sites in sulfur species, allowing enhanced Li+ transformation kinetics across the electrolyte/electrode interface for a fast redox reaction in the low-temperature surrounding. Consequently, at a low temperature of 0 °C, the cell with the ODVO@PCN kinetic promotor exhibits 501 mA h g−1 at 1C and a long life time of up to 400 cycles at 0.5C. Reduced to ultralow −10 °C, the cell still provides a capacity of 706 mA h g−1 and stabilizes a remarkable capacity retention of 85% after 100 cycles.
The development of low-temperature lithium–sulfur (Li–S) batteries for next-generation energy storage systems has attracted increasing attention due to their high theoretical capacity (1675 mA h g−1) along with low cost and environmental friendliness, ensuring a relatively high energy density output under low temperature.1–4 Unfortunately, the practical application of low-temperature Li–S batteries (LT-LSBs) is significantly hindered by extremely sluggish conversion kinetics of polysulfides (LiPSs) due to depressive Li+ diffusion kinetics at low temperature, leading to low capacity with unsatisfactory sulfur utilization and inferior rate performance.5–9
To address the above challenges, current strategies are mainly concentrated on electrolyte engineering by tuning the freezing point of electrolytes or modulating the solvation shell structure of Li+ to increase the diffusion kinetics.10–13 However, for the specific commercial electrolytes, the sluggish Li+ diffusion kinetics is the dominance due to increased viscosity and enlarged solvation shell structure at a low operation temperature, resulting in an extremely high Li+ transport barrier.14–18 To this end, employing electrocatalysts to decrease energy barrier to promote interfacial and bulk Li+ kinetics become more viable.19,20 In comparison with perfect crystal catalysts, the electron-delocalization engineering has been put forward to adjust electronic density of catalysts, aiming at enhancing the numbers of catalytic sites and generating a higher density of free delocalized electrons for reactions.21–25 Meanwhile, it has been revealed that introducing defects into metal-based catalysts offers an effective way to regulate electron orbitals of central atoms, triggering abundant structure aberration for higher catalytic efficiency as well.21,26–29 However, such an electron-delocalization catalyzer with high activity toward Li+ kinetics to promote the redox reactions of LiPSs remains unknown at low temperature. Therefore, constructing catalysts with electron-delocalization tunability is being urgently pursued to enhance the sulfur utilization and electrochemical performance of LT-LSBs.
Herein, a strategy of electron-delocalization with high electron state tunability is proposed toward high-performance low-temperature-tolerant Li–S batteries through introducing oxygen defects on vanadium trioxide that anchored on a porous carbon network (ODVO@PCN). As certified by X-ray analysis with electrochemical evaluations, the dominant active sites of V2+ derived from electron-delocalization ODVO@PCN provide a more favorable interaction with sulfur species in virtue of the stronger orbital hybridization between V2+ sites and sulfur species, enabling strong adsorption on LiPSs and remarkable catalytic activity in Li+ transformation across the interface for fast redox kinetics. The cell with ODVO@PCN demonstrates a high rate capacity of 501 mA h g−1 at 1C and low decaying rate of 0.069% after 400 cycles at 0.5C under 0 °C. Decreasing to an ultralow temperature of −10 °C, the cell still stabilizes with a capacity retention of 85% after 100 cycles, indicating bright prospects for application of LT-LSBs.
The ODVO@PCN was obtained via hydrothermal synthesis with thermal reduction, forming the nanosized morphology and porous structure. As shown in Fig. 1A, characteristic peaks located at 28.0°, 37.2° and 55.6° can be easily identified as the planes of (401), (303) and (205) of the V2O3 (JCPDS 39-0774) phase. At the same time, the wide and broad diffraction peaks centered at 26.6°, 43.5° and 54.8° are attributed to the amorphous nanocarbon (JCPDS 75-2078) in the ODVO@PCN. Similar to VO@PCN, the ODVO@PCN nanocomposites shows an interconnected morphology without apparent agglomeration (Fig. 1B and Fig. S1, ESI†). The uniform and homogeneous distributions of elemental V, O and C in the ODVO@PCN are obtained from the energy-dispersive X-ray spectroscopy (EDX) mappings (Fig. 1C), verifying the homogeneous dispersion of V2O3−x nanoparticles within the porous carbon networks. In the TEM images (Fig. 1D and Fig. S2, ESI†), morphological similarity from V2O3 to V2O3−x is further disclosed without obvious morphological evolution in spite of experiencing reduction. These nanoparticles are tightly anchored on the carbon tube walls with a domain size of around 15 nm (Fig. 1E and Fig. S3, ESI†). Combined with high-resolution TEM (HRTEM) images in Fig. 1E and F, the lattice spacing of 0.202 nm is assigned to the (420) crystal plane of the ODVO@PCN, which is in good agreement with XRD results.
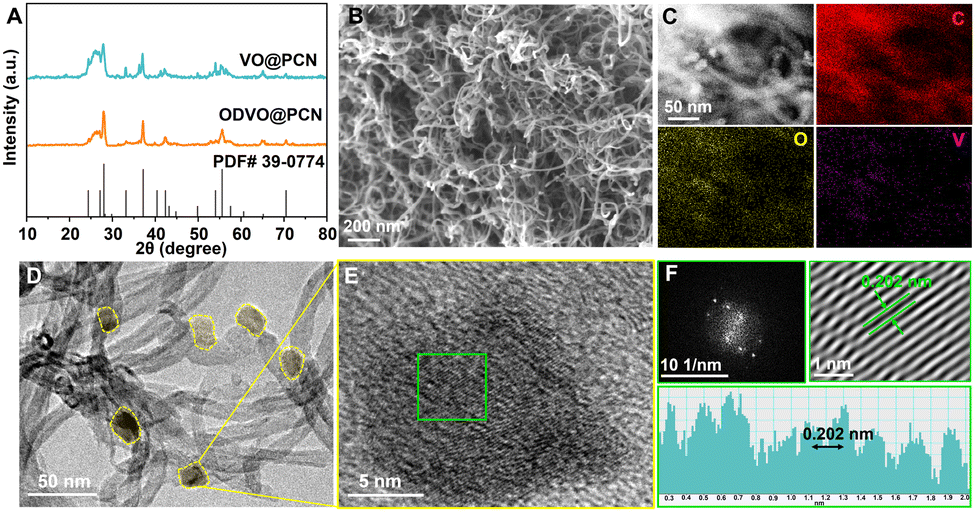 | ||
| Fig. 1 (A) XRD curves of VO@PCN and ODVO@PCN nanocomposites; (B) SEM image and (C) STEM image, and corresponding EDX elemental mappings; (D) TEM image, (E) HRTEM image, and (F) FFT and IFFT images of the highlighted area of the ODVO@PCN nanocomposite. | ||
Accordingly, ODVO@PCN affords the specific surface area of 121 m2 g−1 with hierarchical pore structures within the pore size of 2–50 nm to facilitate the physical and chemical adsorption to LiPSs (Fig. 2A and Fig. S4, ESI†). Such micropores (2–5 nm) with mesopores (10–50 nm) in the ODVO@PCN is advantageous for the simultaneous confinement and conversion of short-chain and long-chain LiPSs. Therefore, the ODVO@PCN-S after sulfur infiltration retains the specific surface area of 14 m2 g−1 with partial pore structures centered around the pore size of 50 nm (Fig. S5, ESI†). Beneficially, thermogravimetric analysis confers a V2O3−x content of 20.6 wt% in ODVO@PCN and high sulfur content of 75 wt% in ODVO@PCN-S (Fig. 2B and Fig. S6, ESI†), respectively. The typical peaks in the Raman spectra of both defective ODVO@PCN and perfect VO@PCN are recognized at around 282.6, 404.4, 509.8, 690.6, 860.5, and 991.5 cm−1 (Fig. 2C),30,31 which belong to various modes of V–O bond vibrations. Upon the electron-delocalization of defects in V2O3−x, the redshift of the V–O bond vibration peaks at 282.6 and 860.5 are created and observed, respectively. Also, ID/IG varies from 1.12 of VO@PCN to 1.23 of ODVO@PCN (Fig. S7, ESI†), suggesting the reformation of the electronic state in the carbon.32 The ODVO@PCN in the EPR spectra of Fig. 2D shows a more pronounced response than the VO@PCN nanocomposite around a g-factor of ≈1.97 between the values of the V-defect and the O-defect, which verifies the leading electron-delocalizing state of the V2+ species formed by removing some of the oxygen atoms within the ODVO@PCN sample.33 Furthermore, the characteristic peaks of elemental V, O, C and N were exhibited in the wide XPS spectra (Fig. S8, ESI†). The high-resolution O 1s spectra of these three composites in Fig. 2E mainly exhibit two main characteristic peaks at 530.2 and 532.5 eV, assigned to lattice oxygen (OL) and surface defect oxygen (OD), respectively.21 The intensity ratio of IOD/IOL in ODVO@PCN is calculated to be 0.72 while that in VO@PCN is 0.59, suggesting that ODVO@PCN composites contain more oxygen defects. After loading of sulfur, there is a slight decrease of the IOD/IOL value to 0.68 with a red shift of both the OL and OD peaks due to the effective interaction with the sulfur. Meanwhile, the two main peaks centered around 516.1 and 517.4 eV in the high-resolution V 2p spectrum are attributed to the two species of V 2p3/2 (V2+) and V 2p3/2 (V3+) (Fig. 2F), respectively.34 Accordingly, the evolution in the intensity ratio of III/II from 0.79 for VO@PCN to 0.88 for ODVO@PCN well demonstrates the electron-delocalization state, caused the valence changes and the emergence of the V 2p1/2 (V2+) peak at 523.3 eV with an apparent red shift for peaks of the V species. A surge in III/II to 1.1 for the ODVO@PCN-S nanocomposite further confirms the dominant active sites built by V2+ position due to stronger hybridization of electron orbitals between V2+ sites and sulfur species (Fig. S9, ESI†). These results well confirm the successfully constructed electron-delocalization state within the ODVO@PCN nanocomposite via the formation of oxygen defect.
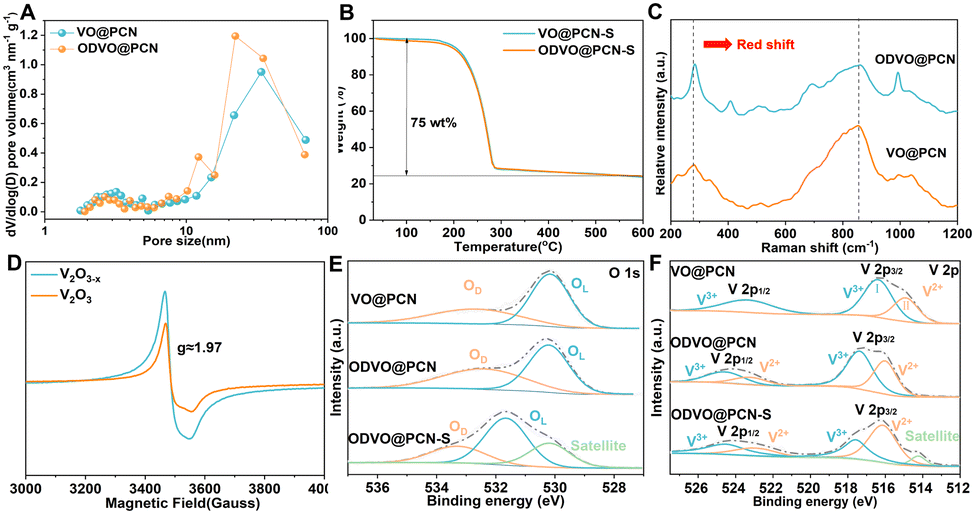 | ||
| Fig. 2 (A) Pore size distribution of ODVO@PCN and VO@PCN; (B) thermogravimetric analysis (TGA) curves of ODVO@PCN-S and VO@PCN-S nanocomposites; (C) characteristic Raman spectra of the ODVO@PCN and VO@PCN nanocomposites; (D) EPR spectra of the as-synthesized ODVO@PCN and VO@PCN nanocomposites; high-resolution spectra of (E) O 1s and (F) V 2p of the ODVO@PCN, ODVO@PCN-S and VO@PCN nanocomposites. | ||
Benefiting from the abundant catalytic sites created by electron-delocalization state, the ODVO@PCN displays better ion conductivity than that of VO@PCN (1.78 vs. 0.82) to allow faster Li+ diffusion kinetics (Fig. 3A and Fig. S10, S11, ESI†). In comparison to the electrochemical impedance spectroscopy (EIS) of the VO@PCN-S cathode (Fig. 3B), the ODVO@PCN-S cathode possesses a remarkably smaller charge transfer resistance (Rct) (53 vs. 272 Ω) and a higher ion diffusion coefficient with a lower fitting slope (11.3 vs. 13.1) (Fig. 3B and C), so is the ODVO@PCN-S cathode after cycling (Fig. S12, ESI†), suggesting a much higher kinetics and faster lithium-ion mobility. In the cyclic voltammetry (CV) curves, the ODVO@PCN-S cathode exhibits a lower potential polarization (300 vs. 350 mV) and a higher response current than the VO@PCN-S cathode, demonstrating that ODVO@PCN can effectively strengthen the redox reaction of LiPSs (Fig. 3D). In the following three cycles, the ODVO@PCN-S cathode displays excellent repeatability, certifying the good electrochemical reversibility (Fig. S13, ESI†). Meanwhile, the Tafel slopes in Fig. S14 (ESI†), simulated from the second reduction peak in the CV curve of the ODVO@PCN-S cathode, are obviously smaller than those of the VO@PCN-S cathode (16.7 vs. 21.1 mV dec−1). In addition, Li-ion diffusion capability can be evaluated on the scan rate dependent CV curves from the Randles–Sevcik equation:  . The simulated slope of ODVO@PCN-S is higher than that of VO@PCN-S (4.7 vs. 3.4), further proving that ODVO@PCN-S does possess faster redox conversion kinetics (Fig. 3E, F and Fig. S15, ESI†).
. The simulated slope of ODVO@PCN-S is higher than that of VO@PCN-S (4.7 vs. 3.4), further proving that ODVO@PCN-S does possess faster redox conversion kinetics (Fig. 3E, F and Fig. S15, ESI†).
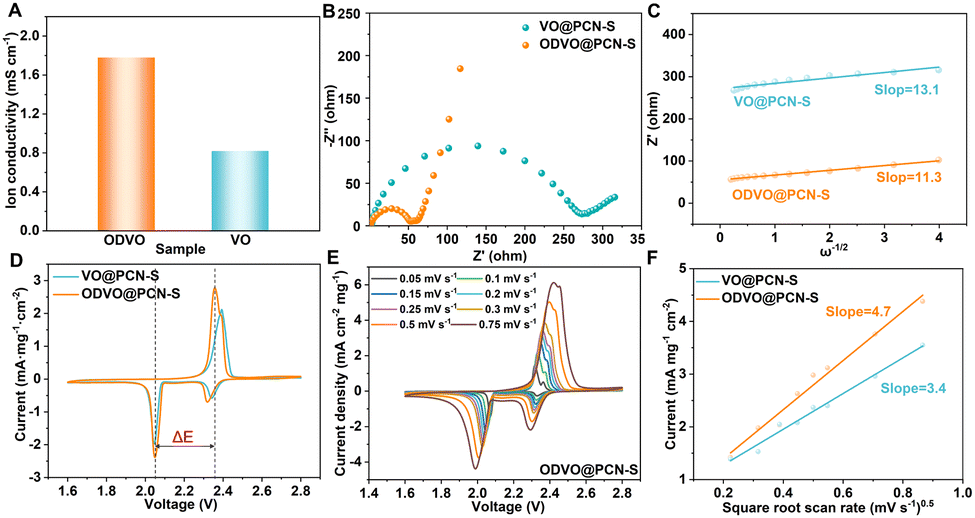 | ||
| Fig. 3 (A) Ionic conductivity test at the electrode/electrolyte interface; (B) comparison of EIS between ODVO@PCN-S and VO@PCN-S electrodes and (C) the corresponding Li ion transport; (D) the CV curves of the two cathodes tested at 0.1 mV s−1; (E) scan rate-dependent CV curves on the ODVO@PCN-S cathode; (F) current density at the lower cathodic peaks of the cathodes versus the square root of the scan rate. | ||
Thanks to the enhanced redox conversion kinetics of the ODVO@PCN catalyzer, the ODVO@PCN@S cathode acquires higher sulfur utilization with improved initial capacity of 1240 mA h g−1 at 0.05C under 0 °C (Fig. 4A). As is well known, the cycling and rate reversibility of sulfur cathode is markedly determined by the ion diffusion kinetics due to the ionic/electronic insulating properties of Li2S at low temperature.35–37 The ODVO@PCN@S cathode displays the smaller delithiation potential barrier of 2.24 V against 2.25 V of VO@PCN@S at 0 °C, directly reflecting the catalytic effect for the increased Li ion transport in Li2S (Fig. 4B). Upon further cycling at 0.2C, the ODVO@PCN@S cathode delivers an initial capacity of 890 mA h g−1 and stabilizes at 661 mA h g−1 with high coulombic efficiency of 99% after 200 cycles (Fig. 4C), while the cell with the VO@PCN@S cathode only provides 695 mA h g−1 and even failed within 100 cycles at 0 °C. Increasing to 0.5C, the ODVO@PCN@S cathode can maintain a high reversible capacity of 512 mA h g−1 with a low decay rate of 0.069% at CE approaching 100% after 400 cycles (Fig. 4D and Fig. S16, ESI†). Lowering the operation temperature to −10 °C, the ODVO@PCN@S cathode still exhibits an initial capacity of 706 mA h g−1 and displays a remarkable capacity retention rate of 85% with stable discharge plateaus after 100 cycles (Fig. 4D and Fig. S17, ESI†), verifying the excellent cycling stability of the catalyzed ODVO@PCN@S cathode at ultra-low temperature. Switching the current rate from 0.05 to 1C under 0 °C, the ODVO@PCN@S cathode exhibits stable rate performance with obvious discharge plateaus (Fig. 4E and Fig. S18, S19, ESI†), much lower than overpotentials at each current rate of VO@PCN@S (Fig. 4F). At 1C, the ODVO@PCN@S maintains a capacity of 501 mA h g−1, while the capacity of VO@PCN@S declines rapidly to as low as 234 mA h g−1, suggesting that highly active ODVO@PCN can catalyze the Li+ diffusion kinetics to realize fast redox kinetics of LiPSs even at low temperature. Increasing to room temperature, the ODVO@PCN catalyzed sulfur cathode displays high-rate performance to 6C (727 mA h g−1) with a continuously low overpotential (Fig. S20, ESI†). In addition, the cell with the ODVO@PCN catalyzer stabilizes 721 mA h g−1 after 200 cycles at 1C (Fig. S21, ESI†). Even at 5C, a high reversible capacity of 421 mA h g−1 after 1000 cycles with a decay rate of as low as 0.048% per cycle is realized (Fig. S22, ESI†), thereby verifying a remarkable electrochemical stability of the ODVO@PCN catalyzed sulfur cathode.
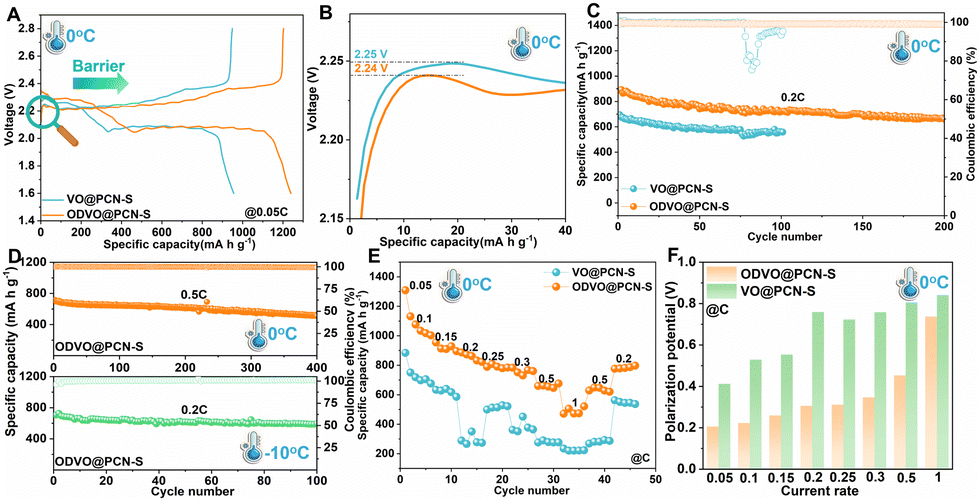 | ||
| Fig. 4 (A) Initial charge–discharge profiles of the ODVO@PCN-S and VO@PCN-S cathodes at 0.05C; (B) comparison of the corresponding decomposition potential of Li2S on the two cathodes; (C) cycle performance of the cathodes at 0.2C; (D) cycle performance of the ODVO@PCN-S cathodes at 0.5C (0 °C) and at 0.2C (–10 °C), respectively; (E) rate performance of the cathodes; (F) comparison of the overpotential at rates varying from 0.05 to 1C under low temperature. | ||
In summary, we have successfully synthesized the electron-delocalized ODVO@PCN nanocatalyst as sulfur hosts for low-temperature Li–S battery, featuring strong electron regulation capability for high catalytic efficiency toward the polysulfide reaction and Li+ transformation kinetics for fast low temperature redox reaction. At a low temperature of 0 °C, the cell with the ODVO@PCN-S cathode exhibits a high-rate capacity of 501 mA h g−1 at 1C and high CE approaching 100% after 400 cycles at 0.5C. Impressively, the cell still provides a high initial capacity of 706 mA h g−1 and a stabilized capacity retention rate of 85% after 100 cycles at 0.2C even under ultralow temperature of −10 °C, much superior to that in most of the literature reports (Table S1, ESI†). Our ODVO@PCN nanocatalyst strongly shows the bright prospects of using electron-delocalized catalysts for high-performance LT-LSBs.
The authors are grateful for financial support from the National Key R&D Program of China (2021YFA1201503), the National Natural Science Foundation of China (No. 22309144, 52371198, 21972164, and 22279161), and the Natural Science Foundation of Jiangsu Province (BK. 20210130), and the Opening funding from Key Laboratory of Engineering Dielectrics and Its Application (Harbin University of Science and Technology) (No. KFM202507, Ministry of Education). We thank the support from Nano-X, Suzhou Institute of Nano-tech and Nano-bionics.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- L. Ma, Y. Wang, Z. Wang, J. Wang, Y. Cheng, J. Wu, B. Peng, J. Xu, W. Zhang and Z. Jin, ACS Nano, 2023, 17, 11527–11536 CrossRef CAS PubMed.
- L. Zhou, D. L. Danilov, F. Qiao, J. Wang, H. Li, R. A. Eichel and P. H. L. Notten, Adv. Energy Mater., 2022, 12, 2202094 CrossRef CAS.
- X. Li, Q. Guan, Z. Zhuang, Y. Zhang, Y. Lin, J. Wang, C. Shen, H. Lin, Y. Wang, L. Zhan and L. Ling, ACS Nano, 2023, 17, 1653–1662 CrossRef CAS PubMed.
- J. Zhang, C. You, H. Lin and J. Wang, Energy Environ. Mater., 2022, 5, 731–750 CrossRef CAS.
- J. Wang, J. Zhang, Y. Zhang, H. Li, P. Chen, C. You, M. Liu, H. Lin and S. Passerini, Adv. Mater., 2024, 36, 2402792 CrossRef CAS PubMed.
- J. Li, L. Liu, J. Wang, Y. Zhuang, B. Wang and S. Zheng, ACS Sustainable Chem. Eng., 2023, 11, 3657–3663 CrossRef CAS.
- Z. Yu, J. Zhang, C. Wang, R. Hu, X. Du, B. Tang, H. Qu, H. Wu, X. Liu, X. Zhou, X. Yang and G. Cui, J. Energy Chem., 2020, 51, 154–160 CrossRef.
- H. Zhang, J. Chen, Z. Li, Y. Peng, J. Xu and Y. Wang, Adv. Funct. Mater., 2023, 33, 2304433 CrossRef CAS.
- J. Xu, Y. Qiu, J. Yang, H. Li, P. Han, Y. Jin, H. Liu, B. Sun and G. Wang, Adv. Funct. Mater., 2023, 34, 2306206 CrossRef.
- Z. Chang, H. Yang, Y. Qiao, X. Zhu, P. He and H. Zhou, Adv. Mater., 2022, 34, 2201339 CrossRef CAS PubMed.
- Z. Li, L. P. Hou, N. Yao, X. Y. Li, Z. X. Chen, X. Chen, X. Q. Zhang, B. Q. Li and Q. Zhang, Angew. Chem., Int. Ed., 2023, 62, 202309968 CrossRef PubMed.
- L. Luo, K. Chen, H. Chen, H. Li, R. Cao, X. Feng, W. Chen, Y. Fang and Y. Cao, Adv. Mater., 2023, 36, 2308881 CrossRef PubMed.
- N. Sun, R. Li, Y. Zhao, H. Zhang, J. Chen, J. Xu, Z. Li, X. Fan, X. Yao and Z. Peng, Adv. Energy Mater., 2022, 12, 2200621 CrossRef CAS.
- X. Zhang, X. Li, Y. Zhang, X. Li, Q. Guan, J. Wang, Z. Zhuang, Q. Zhuang, X. Cheng, H. Liu, J. Zhang, C. Shen, H. Lin, Y. Wang, L. Zhan and L. Ling, Adv. Funct. Mater., 2023, 33, 2302624 CrossRef CAS.
- X. Pang, B. An, S. Zheng and B. Wang, Chem. Eng. J., 2023, 458, 141445 CrossRef CAS.
- Z. Yu, B. Wang, X. Liao, K. Zhao, Z. Yang, F. Xia, C. Sun, Z. Wang, C. Fan, J. Zhang and Y. Wang, Adv. Energy Mater., 2020, 10, 2000907 CrossRef CAS.
- J. Wang, J. Zhang, S. Duan, L. Jia, Q. Xiao, H. Liu, H. Hu, S. Cheng, Z. Zhang, L. Li, W. Duan, Y. Zhang and H. Lin, Nano Lett., 2022, 22, 8008–8017 CrossRef CAS PubMed.
- Y. Feng, B. Zhong, R. Zhang, M. Peng, Z. Hu, Z. Wu, N. Deng, W. Zhang and K. Zhang, Adv. Energy Mater., 2023, 13, 2203912 CrossRef CAS.
- Y. Song, L. Zou, C. Wei, Y. Zhou and Y. Hu, Carbon Energy, 2022, 5, 286 CrossRef.
- J. Wang, L. Li, H. Hu, H. Hu, Q. Guan, M. Huang, L. Jia, H. Adenusi, K. V. Tian, J. Zhang, S. Passerini and H. Lin, ACS Nano, 2022, 16, 17729–17760 CrossRef CAS PubMed.
- J. Zhang, R. He, Q. Zhuang, X. Ma, C. You, Q. Hao, L. Li, S. Cheng, L. Lei, B. Deng, X. Li, H. Lin and J. Wang, Adv. Sci., 2022, 9, 2202244 CrossRef CAS PubMed.
- P. Chen, T. Wang, D. He, T. Shi, M. Chen, K. Fang, H. Lin, J. Wang, C. Wang and H. Pang, Angew. Chem., Int. Ed., 2023, 62, 202311693 CrossRef PubMed.
- J. Wang, J. Zhang, J. Wu, M. Huang, L. Jia, L. Li, Y. Zhang, H. Hu, F. Liu, Q. Guan, M. Liu, H. Adenusi, H. Lin and S. Passerini, Adv. Mater., 2023, 35, 2302828 CrossRef CAS PubMed.
- J. Wang, J. Zhang, S. Cheng, J. Yang, Y. Xi, X. Hou, Q. Xiao and H. Lin, Nano Lett., 2021, 21, 3245–3253 CrossRef CAS PubMed.
- J. Li, L. Gao, F. Pan, C. Gong, L. Sun, H. Gao, J. Zhang, Y. Zhao, G. Wang and H. Liu, Nano-Micro Lett., 2023, 16, 35 CrossRef PubMed.
- S. Cheng, J. Wang, S. Duan, J. Zhang, Q. Wang, Y. Zhang, L. Li, H. Liu, Q. Xiao and H. Lin, Chem. Eng. J., 2021, 417, 128172 CrossRef CAS.
- Z. Liang, J. Shen, X. Xu, F. Li, J. Liu, B. Yuan, Y. Yu and M. Zhu, Adv. Mater., 2022, 34, 2200102 CrossRef CAS PubMed.
- Y. Wang, R. Zhang, J. Chen, H. Wu, S. Lu, K. Wang, H. Li, C. J. Harris, K. Xi, R. V. Kumar and S. Ding, Adv. Energy Mater., 2019, 9, 1900953 CrossRef.
- J. Zhang, L. Jia, H. Lin and J. Wang, Adv. Energy Sustainability Res., 2022, 3, 2100187 CrossRef CAS.
- Y. Bai, Y. Tang, L. Liu, X. Li and Y. Gao, ACS Sustainable Chem. Eng., 2018, 6, 14614–14620 CrossRef CAS.
- G. P. Pandey, T. Liu, E. Brown, Y. Yang, Y. Li, X. S. Sun, Y. Fang and J. Li, ACS Appl. Mater. Interfaces, 2016, 8, 9200–9210 CrossRef CAS PubMed.
- J. Zhang, C. You, J. Wang, H. Xu, C. Zhu, S. Guo, W. Zhang, R. Yang and Y. Xu, Chem. Eng. J., 2019, 368, 340–349 CrossRef CAS.
- W.-D. Liu, X. Tang, J.-A. Feng, C.-Y. Zhang, H. Liu, C. Shi, X.-X. Zhao and J.-J. Song, Rare Met., 2023, 43, 455–477 CrossRef.
- S. Petnikota, J. J. Toh, J. Y. Li, R. Chua and M. Srinivasan, ChemElectroChem, 2018, 6, 493–503 CrossRef.
- J. Wang, L. Jia, S. Duan, H. Liu, Q. Xiao, T. Li, H. Fan, K. Feng, J. Yang, Q. Wang, M. Liu, J. Zhong, W. Duan, H. Lin and Y. Zhang, Energy Storage Mater., 2020, 28, 375–382 CrossRef.
- J. Wang, L. Jia, J. Zhong, Q. Xiao, C. Wang, K. Zang, H. Liu, H. Zheng, J. Luo, J. Yang, H. Fan, W. Duan, Y. Wu, H. Lin and Y. Zhang, Energy Storage Mater., 2019, 18, 246–252 CrossRef.
- J. Zhang, S. Duan, C. You, J. Wang, H. Liu, S. Guo, W. Zhang and R. Yang, J. Mater. Chem. A, 2020, 8, 22240–22250 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc04372c |
| This journal is © The Royal Society of Chemistry 2024 |