
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Controlled synthesis of CD2H-ketones†
Pankaj
Kumar
 and
Graham
Pattison
*
and
Graham
Pattison
*
Department of Chemistry, School of Natural Sciences, Joseph Banks Laboratories, University of Lincoln Green Lane, Lincoln, LN6 7DL, UK. E-mail: gpattison@lincoln.ac.uk
First published on 30th October 2024
Abstract
The synthesis of compounds containing partially deuterated groups such as CD2H lacks general methods. These compounds could be important for fine control of metabolic processes in drug discovery, or in the development of multifunctional probes for analysis by complementary spectroscopic techniques. Here, a convenient route to CD2H-methyl ketones is reported through coupling of esters with bis[(pinacolato)boryl]methane and trapping with D2O.
Recent approvals of the first two deuterated drugs Austedo (deutetrabenazine)1 and Sotyktu (deucravacitinib)2 by the US-FDA have cemented the merits of deuterium incorporation in medicinal chemistry.3 The kinetic isotope effect of the carbon-deuterium bond as compared to the carbon-hydrogen bond offers lower rates of metabolism and so longer half-life for deuterated drug candidates.4 Aside from their application in drug discovery, deuterated molecules have long been used as isotopic labels to elucidate reaction mechanisms5 and tracers to disclose metabolic pathways.6 Deuterium is an important probe atom in Raman spectroscopy,7 as well as in mass spectrometry.8
There is an important and growing need for molecules containing site-selective deuteration.9 The majority of methods for deuteration fall into 3 categories: (i) H/D exchange,10 (ii) addition of deuterium to unsaturated functionality,11 and (iii) coupling of reagents with pre-incorporated deuterium.12
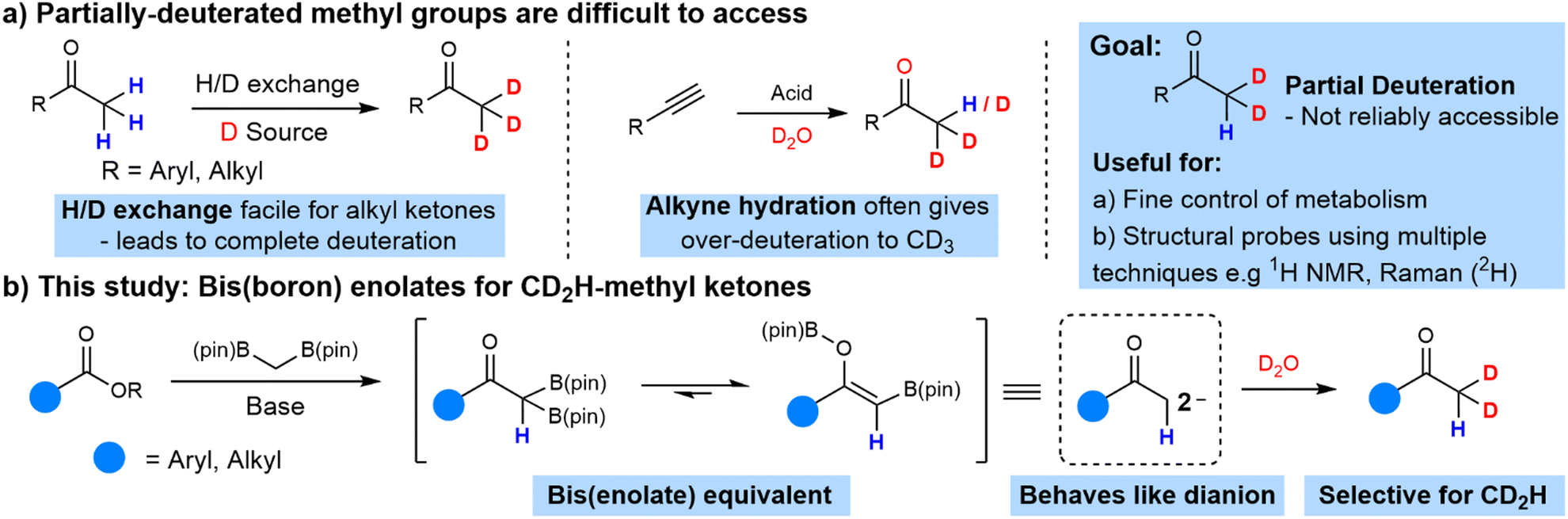
Ketones are some of the most common substrates for deuteration. H/D exchange using acid-, base- or transition-metal-catalysis via enolate or enol intermediates is the most common approach to access α-deuterated ketones (Scheme 1(a)).13
 | ||
| Scheme 1 Challenges in partial deuteration of ketones and our approach. | ||
It is extremely difficult to control the level of deuteration at the α-position of a ketone. Protocols that afford deuterated ketones give an all-or-nothing outcome, transforming a CH3 to a CD3 substituent and it is not possible to obtain products containing intermediate degrees of deuteration such as CDH2 or CD2H with control. There is therefore a demand to discover methods that would allow partial deuteration of methyl groups labelled with both 1H and 2H.
This would be important for a couple of reasons. Firstly, partial deuteration would allow fine-tuned control of metabolic processes. In cases where metabolism of a CH3 group was too fast and a CD3 group metabolized too slow, intermediate levels of deuteration could give ideal rates of metabolism. A second application of molecules containing CD2H and CDH2 groups could be as mechanistic probes containing both 1H and 2H, allowing the deuterated site to be simultaneously studied by multiple complementary techniques including 1H NMR and Raman spectroscopy (2H), as well as mass spectrometry.
However, approaches to achieve the synthesis of partially-deuterated methyl groups are extremely rare. Silicates derived from CDH2I and CD2HI allow the addition of deuterated methyl groups to (hetero)arenes.14 However, CDH2I and CD2HI are both extremely costly and difficult to access. Another approach to access ketones containing partially-deuterated methyl groups would be from the hydration of terminal alkynes using a source of deuterium (Scheme 1(a)). Isolated examples of the synthesis of CD2H ketones have been reported using this approach, but the majority of cases suffer over-deuteration, with CD3-ketones formed commonly, particularly under acid catalysis.15
Our goal at the outset of this project was the development of an efficient and selective methodology for the synthesis of ketones containing a partially-deuterated CD2H methyl substituent. Our group and others have demonstrated the reaction of lithiated geminal bis(boron) compounds16 with esters and related derivatives to generate α,α-bis(enolate) equivalents that can be trapped with a variety of electrophiles to afford geminally difunctionalized ketones.17 Each boron atom could be trapped successively with an electrophilic deuterium source to construct a di-deuterated methyl ketone (Scheme 1(b)). D2O would be our ideal source of deuterium due to its low cost and ready availability.
The biggest challenge we anticipated would be control over the degree of deuteration. In our previous chemistry17a we trapped bis(boron)-enolate intermediates with either NFSI or iodomethane to provide gem-difluoro and gem-dimethyl ketones respectively.17a In those cases, further reaction to give a trifluoromethyl or tert-butyl ketone might be difficult. Fluorination lowers the reactivity of enolates due to its electron-withdrawing effect,18 and electrophilic fluorination of a CF2H-ketone to give a CF3-ketone is not a known reliable transformation. Further alkylation of a CH(CH3)2-ketone to give a C(CH3)3-ketone is likely to be slow due to steric hindrance. However, in a di-deuteration process, exchange of hydrogen for deuterium could be facile and controlling the degree of deuteration may prove to be a challenge under the basic conditions used to generate bis(boron) enolates.
Here we report our protocol for the synthesis of CD2H-substituted ketones. We used similar conditions to our recently reported difluorination and dimethylation methods, but found that reducing the amount of solvent used and increasing the amount of D2O for trapping was required for high selectivity towards the di-deuterated compound over other isotopomers. After optimization,19 we standardized the reaction conditions to those with the highest combination of both conversion and selectivity towards the di-deuterated product, i.e., 1.5 eq. NaHMDS, 2.0 eq. bis[(pinacolato)boryl]methane in 0.3 mL anhydrous THF and 10 eq. of D2O. We then examined the generality of this protocol with non-enolizable esters.
Ratios of deuterated products were measured by 1H NMR and validated by mass spectrometry. No CD3-ketone product was ever observed in 13C NMR, and could only be observed in trace amounts (<5%) in a high resolution mass spectrum.
para-Substituted benzoates (Scheme 2, 1a–f) reacted smoothly under the optimized conditions affording the desired ketone (3a–3f) in moderate to good yields (44–75%) and high selectivity for di-deuteration (85–94%). The reaction was equally successful when unsubstituted (3g), as well as meta- and ortho-substituted benzoate esters were used (3h–3l). The strategy also excelled with heteroaromatic derivatives, with systems containing thiophene, pyridine and pyrazole rings (3m–3p). An 8 mmol scale reaction giving 3i proceeded with comparable yield and isotopic selectivity to its smaller scale performance.
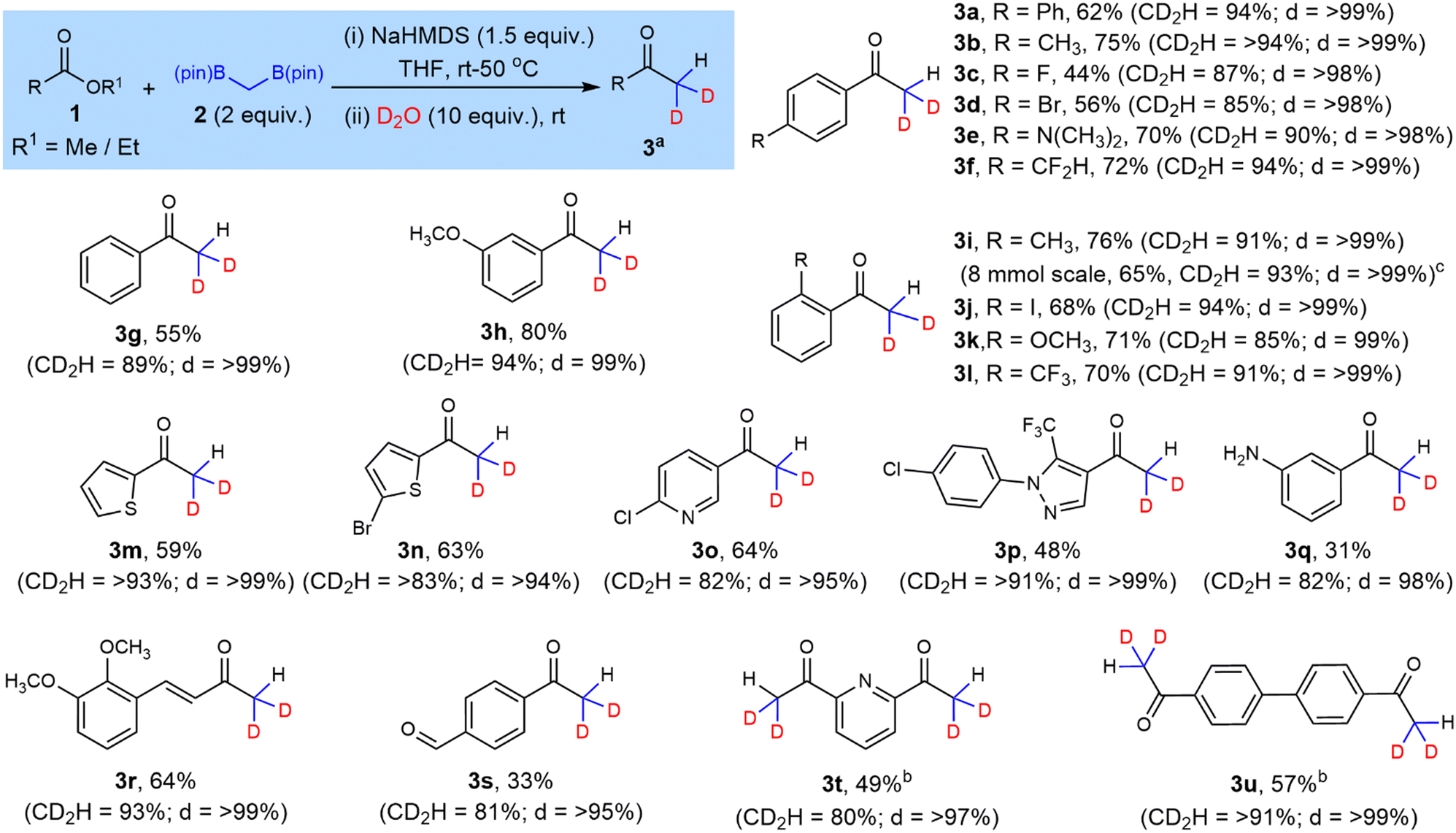 | ||
| Scheme 2 Substrate scope for di-deuteration of non-enolizable esters (a) CD2H = % dideuteration; d = overall% deuteration (mono + di); (b) reaction performed with 4 equiv. 2, 3 equiv. NaHMDS and 20 equiv. D2O; (c) reaction performed with 8 mmol 1i. | ||
Next, we tested the tolerance of the protocol towards some reactive functional groups. Methyl 3-aminobenzoate resulted in a mixture of products with the di-deuterated ketone 3q as the major product in 31% isolated yield and 82% di-deuteration. Methyl 4-hydroxybenzoate proved unreactive likely due to the interaction of the acidic phenol with NaHMDS. A cinnamate ester derivative (1r) showed highly selective reaction at the ester rather than the β-position of the alkene to furnish 3r. Interestingly, the strategy could also tolerate to some extent an aldehyde, giving 3s in 33% isolated yield. The boron-Wittig olefination product16a,d was also obtained via reaction at the aldehyde but this by-product was fully separable from the desired acetophenone.
Symmetric aromatic diesters when subjected to double the standard stoichiometry produced tetra-deuterated diketones 3t and 3u with moderate yields (Scheme 2). The slightly lower yields of these tetra-deuterated diketones was attributed to small amounts of reaction at only one of the ester sites.
We were interested in the stability of the products we had prepared, particularly with regard to scrambling of deuterium. Pleasingly, the di-deuterated compounds showed no signs of change in deuteration ratios even after a year of storage at room temperature. However, compound 3q, containing an amine did show deterioration over time in the ratio of deuterated products when stored in solution. We also carried out exchange experiments in buffers of different pH which showed that the ketones showed little deuterium exchange at acidic or neutral pH after 24 hours, but showed a small amount of exchange at pH 10 and significant exchange at pH 12.19
Once the substrate scope with non-enolizable esters was established, we then explored the behaviour of enolizable esters. Unfortunately, using the same conditions as in Scheme 2 gave poor yields due to self-Claisen and competing processes. In our previous work we found LiTMP to be a useful base to avoid self-Claisen byproducts.17a As a stronger base than NaHMDS20 it allowed complete lithiation of the geminal bis(boron) compound before addition to the ester, meaning formation of the bis(boron) enolate occurred before any self-Claisen processes as the reagents were added sequentially. However, in this case whilst the use of LiTMP did improve the yield, it gave very poor selectivity for di-deuteration.19
This failure to improve selectivity with LiTMP prompted us to carry out some mechanistic work to examine the effects of LiTMP on di-deuteration. We began by looking at the use of LiTMP with non-enolizable esters, which had proved very successful with NaHMDS. Methyl 2-methylbenzoate (1i) was subjected to dideuterative coupling in the presence of 2 equivalents of LiTMP, which resulted in the synthesis of 3i (65%) with 38% di-deuteration and 75% overall deuteration. This was much less selective than when NaHMDS was used (Scheme 3(a)). For further evidence, we treated pre-prepared selectively di-deuterated 3i (93.4% CD2H) with 2 equivalent LiTMP for 45 minutes which was then quenched with D2O. This led to a decrease in di-deuteration to 75% (Scheme 3(b)). Evidently, the use of LiTMP allowed us to avoid any self-Claisen reaction, but gave significant scrambling and low selectivity in deuteration.
 | ||
| Scheme 3 Mechanistic studies on exchange processes using LiTMP. | ||
We therefore returned to NaHMDS as our base of choice. A change of solvent to anhydrous toluene combined with delayed addition of ester gave some improvement in yield and deuteration selectivity.19 As our priority was selectivity for di-deuteration rather than isolated yield these conditions were selected to further explore the substrate scope of the reaction with enolizable esters (Scheme 4). A series of 3-phenylpropionate esters were transformed to the CD2H-ketones in low to moderate yield (28–45%), but with very good purity and selectivity for the di-deuterated product (83–87%, Scheme 4, 3v–3x). Dimethyl cyclohexane-1,4-dicarboxylate 1y with double the quantities of reagents, gave the symmetric tetra-deuterated diketone 3y. No deuterium was introduced at the non-methyl α-position, confirming the high selectivity of this process, and any self-Claisen by-product was separable.
 | ||
| Scheme 4 Synthesis of CD2H-ketones from enolizable esters; (a) CD2H = % dideuteration; d = overall % deuteration (mono + di); (b) reaction performed with 4 equiv. 2, 3 equiv. NaHMDS and 20 equiv. D2O. | ||
We next wanted to test our products in some standard carbonyl transformations to see if the deuteration was retained. Reactions that involved nucleophilic addition to the carbonyl group, including reduction with NaBH4 (4i), Grignard addition (5i, 6i) and tosyl-hydrazone formation (7i) all proceeded in good yield and with very high levels of retention of deuteration (Scheme 5). However, reactions involving enolate formation perhaps predictably led to significant erosion of deuteration.
 | ||
| Scheme 5 Transformations of CD2H ketones with retention of deuterium. | ||
To summarize, a protocol has been developed to access partially deuterated methyl ketones through coupling of an ester with bis[(pinacolato)boryl]methane in the presence of base. The strategy gives moderate to good yields of the di-deuterated methyl ketones with deuteration only at the positions where boron was present in the bis(boron) enolate intermediate. Gram-scale synthesis and further transformations of the di-deuterated methyl ketones with retention of deuterium validates the method for use in future pharmaceutical and industrial explorations.
We thank EPSRC (grant ref: EP/W012626/1) for funding this work. This work was carried out using JBL (Joseph Banks Laboratories) Science facilities at the University of Lincoln.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- C. Schmidt, Nat. Biotechnol., 2017, 35, 493–494 CrossRef CAS PubMed.
- T. M. Truong, G. N. Pathak, A. Singal, V. Taranto and B. K. Rao, Ann. Pharmacother., 2024, 58, 416–427 CrossRef CAS.
- (a) J. Atzrodt, V. Derdau, W. J. Kerr and M. Reid, Angew. Chem., Int. Ed., 2018, 57, 1758–1784 CrossRef CAS PubMed; (b) R. M. C. Di Martino, B. D. Maxwell and T. Pirali, Nat. Rev. Drug Discovery, 2023, 22, 562–584 CrossRef CAS PubMed.
- (a) K. B. Wiberg, Chem. Rev., 1955, 55, 713–743 CrossRef CAS; (b) S. Scheiner and M. Čuma, J. Am. Chem. Soc., 1996, 118, 1511–1521 CrossRef CAS.
- W. W. Cleland, Crit. Rev. Biochem., 1982, 13, 385–428 CrossRef CAS PubMed.
- J. Kim, S. Seo and T.-Y. Kim, Anal. Chim. Acta, 2023, 1242, 340722 CrossRef CAS PubMed.
- S. Egoshi, K. Dodo and M. Sodeoka, Curr. Opin. Chem. Biol., 2022, 70, 102181 CrossRef CAS PubMed.
- E. I. James, T. A. Murphree, C. Vorauer, J. R. Engen and M. Guttman, Chem. Rev., 2022, 122, 7562–7623 CrossRef CAS PubMed.
- (a) J. Atzrodt, V. Derdau, W. J. Kerr and M. Reid, Angew. Chem., Int. Ed., 2018, 57, 3022–3047 CrossRef CAS PubMed; (b) S. Kopf, F. Bourriquen, W. Li, H. Neumann, K. Junge and M. Beller, Chem. Rev., 2022, 122, 6634–6718 CrossRef CAS PubMed.
- A. Sattler, ACS Catal., 2018, 8, 2296–2312 CrossRef CAS.
- Z. P. Vang, S. J. Hintzsche and J. R. Clark, Chem. – Eur. J., 2021, 27, 9988–10000 CrossRef CAS PubMed.
- (a) J. Steverlynck, R. Sitdikov and M. Rueping, Chem. – Eur. J., 2021, 27, 11751–11772 CrossRef CAS PubMed; (b) K. Ban, K. Imai, S. Oyama, J. Tokunaga, Y. Ikeda, H. Uchiyama, K. Kadota, Y. Tozuka, S. Akai and Y. Sawama, Angew. Chem., Int. Ed., 2023, 62, e202311058 CrossRef CAS PubMed; (c) M. Jiao, J. Zhang, M. Wang, H. Lu and Z. Shi, Nat. Commun., 2024, 15, 5067 CrossRef CAS PubMed.
- (a) J. Eames, G. S. Coumbarides, M. J. Suggate and N. Weerasooriya, Eur. J. Org. Chem., 2003, 634–641 CrossRef CAS; (b) X. Ariza, G. Asins, J. Garcia, F. G. Hegardt, K. Makowski, D. Serra and J. Velasco, J. Labelled Compd. Radiopharm., 2010, 53, 556–558 CrossRef CAS; (c) H.-k Fan, S. Yang, J.-h Li, Q.-q Teng and M. Chen, Eur. J. Org. Chem., 2022, e202201218 CrossRef CAS.
- M. Abdellaoui, T. Deis, M. A. Wiethoff, C. Bahri, G. Lemière, C. Ollivier and L. Fensterbank, Adv. Synth. Catal., 2023, 365, 884–891 CrossRef CAS.
- (a) Z. Nairoukh, D. Avnir and J. Blum, ChemSusChem, 2013, 6, 430–432 CrossRef CAS PubMed; (b) Y. Liu, J. Guo, Y. Liu, X. Wang, Y. Wang, X. Jia, G. Wei, L. Chen, J. Xiao and M. Cheng, Chem. Commun., 2014, 50, 6243–6245 RSC.
- (a) D. S. Matteson, R. J. Moody and P. K. Jesthi, J. Am. Chem. Soc., 1975, 97, 5608–5609 CrossRef CAS; (b) K. Endo, M. Hirokami and T. Shibata, J. Org. Chem., 2010, 75, 3469–3472 CrossRef CAS PubMed; (c) K. Hong, X. Liu and J. P. Morken, J. Am. Chem. Soc., 2014, 136, 10581–10584 CrossRef CAS PubMed; (d) J. R. Coombs, L. Zhang and J. P. Morken, Org. Lett., 2015, 17, 1708–1711 CrossRef CAS PubMed; (e) S. A. Murray, M. Z. Liang and S. J. Meek, J. Am. Chem. Soc., 2017, 139, 14061–14064 CrossRef CAS PubMed; (f) T. C. Stephens and G. Pattison, Org. Lett., 2017, 19, 3498–3501 CrossRef CAS PubMed; (g) O. Salvado, R. Gava and E. Fernández, Org. Lett., 2019, 21, 9247–9250 CrossRef CAS PubMed.
- (a) C. E. Iacono, T. C. Stephens, T. S. Rajan and G. Pattison, J. Am. Chem. Soc., 2018, 140, 2036–2040 CrossRef CAS PubMed; (b) W. Sun, L. Wang, C. Xia and C. Liu, Angew. Chem., Int. Ed., 2018, 57, 5501–5505 CrossRef CAS PubMed; (c) Y. Hu, W. Sun and C. Liu, Synlett, 2019, 1105–1110 CAS; (d) G. R. A. Garratt and G. Pattison, Synlett, 2020, 1656–1662 CAS; (e) B. Lee and P. J. Chirik, J. Am. Chem. Soc., 2020, 142, 2429–2437 CrossRef CAS PubMed; (f) W. Sun, L. Wang, Y. Hu, X. Wu, C. Xia and C. Liu, Nat. Commun., 2020, 11, 3113 CrossRef CAS PubMed; (g) W. Sun, L. Xu, Y. Qin and C. Liu, Nat. Synth., 2023, 2, 413–422 CrossRef.
- (a) D. J. Leng, C. M. Black and G. Pattison, Org. Biomol. Chem., 2016, 14, 1531–1535 RSC; (b) G. Pattison, Eur. J. Org. Chem., 2018, 3520–3540 CrossRef CAS.
- For full details please see the ESI†.
- D. S. Matteson and R. J. Moody, Organometallics, 1982, 1, 20–28 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc04819a |
| This journal is © The Royal Society of Chemistry 2024 |