
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and crystal structure of acentric anhydrous beryllium carbonate Be(CO3)†
Dominik
Spahr
 *a,
Lkhamsuren
Bayarjargal
a,
Elena
Bykova
a,
Maxim
Bykov
b,
Tim H.
Reuter
a,
Lukas
Brüning
b,
Pascal L.
Jurzick
c,
Lena
Wedek
a,
Victor
Milman
d,
Björn
Wehinger
e and
Björn
Winkler
a
*a,
Lkhamsuren
Bayarjargal
a,
Elena
Bykova
a,
Maxim
Bykov
b,
Tim H.
Reuter
a,
Lukas
Brüning
b,
Pascal L.
Jurzick
c,
Lena
Wedek
a,
Victor
Milman
d,
Björn
Wehinger
e and
Björn
Winkler
a
aGoethe University Frankfurt, Institute of Geosciences, Altenhöferallee 1, 60438 Frankfurt, Germany. E-mail: d.spahr@kristall.uni-frankfurt.de
bGoethe University Frankfurt, Institute of Inorganic and Analytical Chemistry, Max-von-Laue-Straße 7, 60438 Frankfurt, Germany
cUniversity of Cologne, Institute of Inorganic Chemistry, Greinstraße 6, 50939 Cologne, Germany
dDassault Systèmes BIOVIA, 334 Cambridge Science Park, CB4 0WN Cambridge, UK
eEuropean Synchrotron Radiation Facility ESRF, 71 avenue des Martyrs, CS40220, 38043 Grenoble Cedex 9, France
First published on 29th August 2024
Abstract
The anhydrous alkaline earth metal carbonate Be(CO3) was synthesized in a laser-heated diamond anvil cell at moderate pressures and temperatures (20(2) GPa and 1500(200) K) by a reaction of BeO with CO2. It crystallizes in the acentric, trigonal space group P3121 with Z = 3. The crystal structure was obtained from synchrotron single crystal X-ray diffraction data and confirmed by density functional theory-based calculations in combination with Raman spectroscopy. Second harmonic generation measurements were employed to verify the acentric space group symmetry. The crystal structure of Be(CO3) is characterized by the presence of isolated [CO3]2−-groups and BeO4-tetrahedra. This is a new structure type and such a topology has not been observed in carbonates before. Be(CO3) can be recovered to ambient conditions and is not undergoing a phase transition during decompression.
Carbonates hosting alkaline earth metal cations are ubiquitous in nature, as calcite (Ca(CO3)), aragonite (Ca(CO3)), dolomite ((Ca,Mg)(CO3)2) and magnesite (Mg(CO3)) account for >90% of the carbonates present in the Earth's crust.1 These “conventional” carbonates are characterized by the presence of trigonal, nearly planar [CO3]2−-groups, in which sp2-hybrid orbitals are present between the central carbon atom and the surrounding oxygen atoms.1,2 Due to their abundance, the phase diagrams of Ca(CO3), (Ca,Mg)(CO3)2, Mg(CO3) or Fe(CO3) have been studied in great detail in the last decades.3–6
For anhydrous carbonates with M2+(CO3) it was found that the ionic radius of the M2+ cation is a major factor determining if the corresponding carbonate crystallizes in the calcite (CN = 6) or aragonite (CN = 9) structure-type, where the ionic radius of Ca2+ (1.00 Å in 6-fold coordination), is a border case.7,8 Ca(CO3) is found with both structure types.9,10 Carbonates hosting larger cations (Sr2+, Ba2+) typically crystallize in the aragonite structure type, while carbonates with small cations such as Mg2+ or with 3d transition metal cations (Mn2+, Fe2+, Co2+, Ni2+, Cu2+ and Zn2+) occur in calcite structure at ambient pressures.8–12
In the family of alkaline earth metals, beryllium exhibits an extraordinary position. In contrast to the next member magnesium (r(Mg2+) = 0.72 Å in 6-fold coordination) the ionic radius is significantly smaller (r(Be2+) = 0.45 Å).7 It is surprising that except for beryllium for all the other alkaline earth metals (Mg, Ca, Sr, Ba) crystal structures of carbonate phases had been reported.9,10 Even for Ra(CO3) an aragonitic and a disordered carbonate phase have been described.13,14 Due to the small cation radius, it is most likely that a Be-carbonate would either crystallize in the calcite type structure or in a new structure type, but not in the aragonite type structure. Counterintuitively, theoretical density functional theory (DFT) based calculations reveal a higher stability of Be(CO3) having aragonite structure type.15 In contrast to the alkaline earth metals, the crystal structure of Li2(CO3), the lightest member of the alkali metal carbonates is well established.16 In Li2(CO3) the monovalent Li+ cations (r(Li+) = 0.76 Å) are in 6-fold coordination and the ionic radius is larger than the one of the divalent Be2+.
It should be noted that in some textbooks the synthesis of unstable Be(CO3), which should decompose in air, is described from BeO and CO2 at high temperatures.17 Nevertheless, we cannot find any reported crystal structure on Be(CO3). The discovery of the hydrous carbonate mineral niveolanite with a complex crystal structure and composition ((Na0.90Ca0.09)Be(CO3)[(OH)0.92O0.09]·H2O) demonstrated that beryllium can be found in naturally occurring carbonates.18 In this compound, the Be2+ atoms are tetrahedrally coordinated. This raises the question if the very small ionic radius of beryllium in 4-fold coordination (r(Be2+) = 0.27 Å) is compatible with the formation of anhydrous Be(CO3).7
In the study presented here, we investigated the system BeO–CO2, in order to determine if a beryllium carbonate can be synthesized at elevated pressures and temperatures, in analogy to the successful synthesis of Al2(CO3)3 from an Al2O3 + CO2 mixture at ≈20 GPa.19 The experiments were carried out in laser-heated diamond anvil cells (LH-DACs) in a similar pressure range between 10 GPa and 20 GPa. First, BeO powder was compacted between a diamond and a glass plate. The experimental Raman spectrum of BeO is accurately reproduced by the DFT calculations in space group P63mc (see Fig. S1 in the ESI†), for ambient conditions.20 In a second step, the powder compact was placed on the culet of the lower diamond of the DAC (Fig. 1a). Additionally, we added a ruby chip for pressure determination.21 Afterward, CO2-I (dry ice) was cryogenically loaded into the DAC. The DAC was cooled down to ≈100 K and CO2 was directly condensed into the gasket hole from a gas jet until the gasket hole and the powder compact were completely covered. Then, the DAC was closed tightly (Fig. 1b).
 | ||
| Fig. 1 (a) Compacted BeO powder and ruby chip for pressure determination in the gasket hole of the DAC. (b) BeO + CO2-I (dry ice) mixture after cryogenic loading and closing of the DAC (10(1) GPa). (c) BeO + CO2 mixture after laser heating to ≈1500(200) K at 20(2) GPa. (d) Raman map of Be(CO3) (≈1195 cm−1) overlayed on a picture of the gasket hole. (e) Combined Raman maps of the CO2 phases present in the gasket hole after heating: phase III (≈280 cm−1), II (≈320 cm−1) and IV (≈110 cm−1). (f) Raman map of Be(CO3) (≈1195 cm−1) at 20(2) GPa. | ||
The BeO + CO2 mixture was compressed to target pressures between 10 GPa and 20 GPa in different experiments without intermediate heating. During the cold compression a phase transition from CO2-I (Pa![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) ) to CO2-III (Cmca) occurs in a broad (≈5 GPa) pressure interval around ≈12 GPa.22,23 In contrast, in earlier studies no phase transition had been found in BeO by Raman spectroscopy up to 55 GPa and by powder X-ray diffraction (PXRD) up to 137 GPa.24,25 At the target pressures of 10(1) GPa, 15(2) GPa and 20(2) GPa the BeO powder compact was laser-heated from one side in the CO2 atmosphere in different experiments up to temperatures of ≈1500 K. In this pressure range the direct and indirect heating of CO2-III causes the appearance of the high-temperature CO2 polymorphs CO2-II and CO2-IV.26,27 It should be noted, that CO2-II is assumed to be present only in a narrow temperature field ranging from ≈400 K to ≈600 K.28–30 Nevertheless, we clearly observed the presence of phase II in a narrow rim around the heated area where CO2-IV is present at 20(2) GPa (Fig. 1e), while CO2-III is still present in the unheated regions (Fig. 1e). At 20(2) GPa the Raman spectra of the CO2 phases III, II and IV measured in the same DAC are in agreement with our theoretical Raman spectra derived from the DFT calculations (Fig. 2a–c).
) to CO2-III (Cmca) occurs in a broad (≈5 GPa) pressure interval around ≈12 GPa.22,23 In contrast, in earlier studies no phase transition had been found in BeO by Raman spectroscopy up to 55 GPa and by powder X-ray diffraction (PXRD) up to 137 GPa.24,25 At the target pressures of 10(1) GPa, 15(2) GPa and 20(2) GPa the BeO powder compact was laser-heated from one side in the CO2 atmosphere in different experiments up to temperatures of ≈1500 K. In this pressure range the direct and indirect heating of CO2-III causes the appearance of the high-temperature CO2 polymorphs CO2-II and CO2-IV.26,27 It should be noted, that CO2-II is assumed to be present only in a narrow temperature field ranging from ≈400 K to ≈600 K.28–30 Nevertheless, we clearly observed the presence of phase II in a narrow rim around the heated area where CO2-IV is present at 20(2) GPa (Fig. 1e), while CO2-III is still present in the unheated regions (Fig. 1e). At 20(2) GPa the Raman spectra of the CO2 phases III, II and IV measured in the same DAC are in agreement with our theoretical Raman spectra derived from the DFT calculations (Fig. 2a–c).
 | ||
| Fig. 2 Raman spectra of different CO2 phases measured in the same DAC at 20(2) GPa after laser heating (corresponding to the areas in Fig. 1e): (a) CO2-III, (b) CO2-II and (c) CO2-IV. (d) Raman spectra of Be(CO3) after the synthesis at elevated pressures and temperatures. Peaks of CO2-IV are marked by an asterisk (*). (e) Raman spectra of Be(CO3) after decompression of the DAC to ambient conditions. Experimental Raman spectra are shown in blue, DFT-based calculations are shown in black. The shifts of the calculated Raman spectra were rescaled by 2–6%. | ||
Besides the formation of CO2-II and CO2-IV, heating the BeO powder compact in the CO2 atmosphere at 20(2) GPa up to a maximum temperature of ≈1500(200) K causes the appearance of an unknown phase with a strong Raman mode at ≈1195 cm−1. A dominant Raman mode occurring at this wavenumber is characteristic for the C–O stretching mode in a [CO3]2−-group in alkali and alkaline earth metal carbonates such as Mg(CO3), Ca(CO3) or Sr(CO3).3,5,31 This was not observed at pressures <20 GPa. Heating the sample for 60 minutes resulted in an ambient temperature Raman spectrum of the unknown phase with very little contamination by other phases (Fig. 2d and e). We mapped the distribution of the unknown carbonate across the gasket hole in order to locate a suitable region for the subsequent single crystal X-ray diffraction experiments (Fig. 1d and f). The new phase is only present in the heated region where also the high-temperature phase CO2-IV is present, which only occurs at temperatures ≥600 K at this pressure (Fig. 1e).28–30
After the identification of an area where mainly the unknown phase is present by Raman spectroscopy, we employed X-ray diffraction imaging on a 60 × 60 μm2 grid with 3 μm steps at beamline ID27,32 European Synchrotron Radiation Facility (ESRF), using a spot size of 0.7 × 0.7 μm2 to locate a suitable spot in this region for the collection of single crystal diffraction data. Using the synchrotron single crystal X-ray diffraction data, we determined the crystal structure of the unknown phase and found that it is a “conventional” carbonate with Be(CO3) composition, i.e. neither a pyrocarbonate nor a sp3-carbonate.33,34 It crystallizes in the acentric, trigonal space group P3121 with Z = 3 (Fig. 3a). The refined structure model is in very good agreement with the results from our DFT-based full geometry optimizations (see Table S1 in the ESI†). Furthermore, the DFT calculated Raman spectrum nicely reproduces the experimental data in the whole frequency range (Fig. 2d).
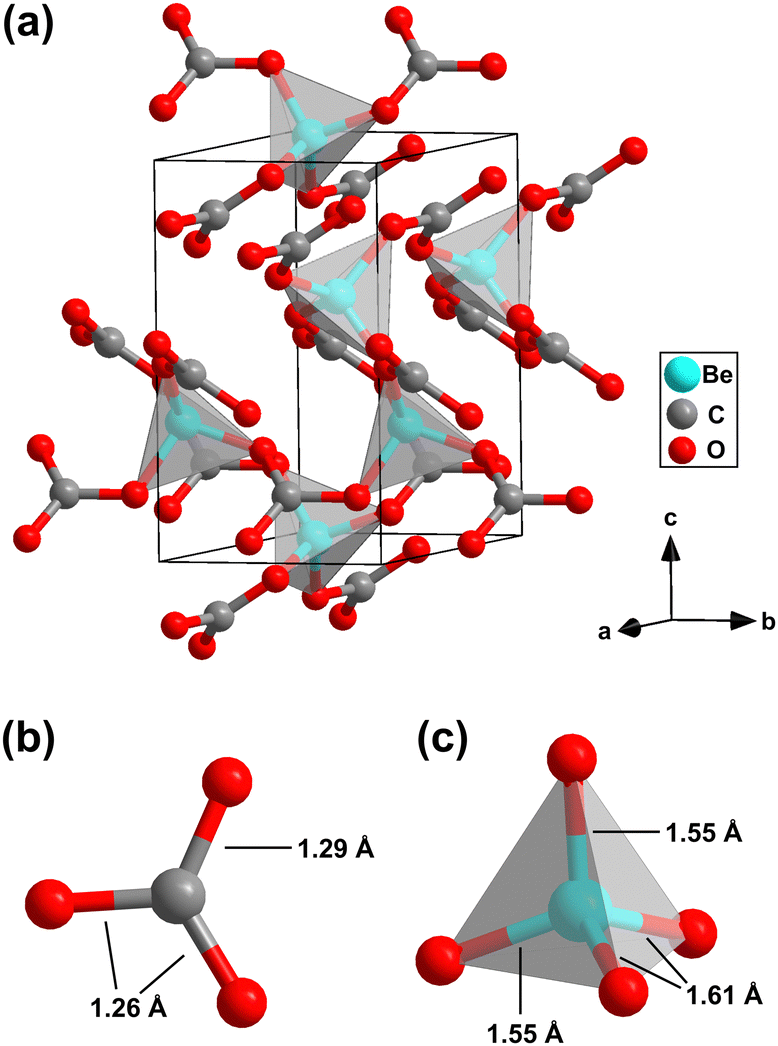 | ||
| Fig. 3 (a) Trigonal crystal structure (P3121, Z = 3) of Be(CO3) at 20(2) GPa obtained from synchrotron X-ray single crystal structure solution. (b) Geometry of the [CO3]2–groups and (c) of the BeO4-tetrahedra in Be(CO3). | ||
Be(CO3) is an anhydrous carbonate. Its crystal structure is characterized by the presence of isolated, trigonal-planar [CO3]2−-groups (Fig. 3b). The C–O bond distances within the [CO3]2−-group are nearly identical (1.26(1) Å and 1.29(1) Å). Hence, Be(CO3) completes the list of known crystal structures of alkaline earth metal carbonates with M2+(CO3) with M2+= Mg, Ca, Sr, Ba and Ra composition, by the lightest member.9,10,13,14 The second building block of Be(CO3) are almost ideally shaped BeO4-tetrahedra with Be–O bond distances between 1.55(1) Å and 1.61(1) Å (Fig. 3c). Due to the very small ionic radius, the Be atoms are only coordinated by four oxygen atoms, in contrast to eg. Mg or Ca in the corresponding carbonates at the same conditions which typically are coordinated by 6 or 9 oxygen atoms.3,5
After the synthesis of Be(CO3) at elevated pressures and temperatures we measured Raman spectroscopy during the decompression of the DAC. We did not observe any indication for a phase transition during pressure release. Furthermore, our Raman spectra clearly show that Be(CO3) can be recovered at ambient conditions, without storage under CO2 atmosphere. The experimental Raman spectrum at ambient pressure is unequivocally reproduced by the one from the DFT-based calculations in the trigonal space group P3121 (Fig. 2e), confirming that no pressure-induced phase transition had occurred. In addition, our calculations show that Be(CO3)-P3121 is more stable than a polymorph having aragonite structure type in the investigated pressure range.15 It should be noted, that the small amount of recovered material (≈0.00003 mm3) limits the possibility for further sample analysis, such as conventional PXRD.
The p,V relation of Be(CO3) obtained from the calculations between 0 GPa and 50 GPa is shown in Fig. S3 in the ESI.† The theoretical p,V data were fitted with an equation of state (Fig. S3 in the ESI†) to determine the bulk modulus. We obtained a bulk modulus of K0 = 95.5(8) GPa with Kp = 3.8(1). This is consistent with the bulk modulus obtained from the elastic stiffness tensor from our DFT-calculations (91(1) GPa). The bulk modulus of Be(CO3) is in the expected region for M2+(CO3) carbonates, which ranges from 67(2) GPa in Ca(CO3) (calcite) over 107(1) GPa in Mg(CO3) to 131(2) GPa in Ni(CO3).35 However, the bulk modulus of Be(CO3) does not fit to the proposed trend, where the bulk modulus increases with decreasing unit cell volume. In order to confirm the acentric space group symmetry we performed second harmonic generation (SHG) measurements in the DAC at 20(2) GPa and at ambient conditions. We measured strong SHG signals, ≈70 mV at 20 GPa and ≈60 mV at ambient conditions, from the sample (Fig. S4 in the ESI†). This is consistent with our DFT calculations, which reveal a relatively high SHG coefficient of deff = 1.34 pm V−1 at 20 GPa and of deff = 0.93 pm V−1 at ambient conditions.
The unit cell volumes of “conventional” chemically simple carbonates characterized by the presence of isolated [CO3]2–groups strongly depend on the ionic radius of the cation. For carbonates with calcite-type crystal structures and 6-fold coordination (M2+(CO3)) the unit cell volume decreases systematically from Ca(CO3) (r(Ca2+) = 1.00 Å) to Ni(CO3) (r(Ni2+) = 0.69 Å) (Fig. 4).7–9,11 The recently synthesized sp2-carbonate Al2(CO3)3 does not crystallize in calcite-type crystal structure, but the Al3+-cations (r(Al3+) = 0.535 Å) are also coordinated by 6 oxygens. The normalized unit cell volume of this compound fits in the systematic trend of the calcite-type carbonates (Fig. 4).7,19 In contrast to the other M2+(CO3) carbonates the ionic radius of beryllium is significantly smaller. It is 0.45 Å in 6-fold coordination and decreases to 0.27 Å in 4-fold coordination.7 In Be(CO3) the coordination number is four and the unit cell volume at ambient conditions fits perfectly in the systematic decrease of the unit cell volume of the M2+(CO3) carbonates with decreasing ionic radii (Fig. 4).
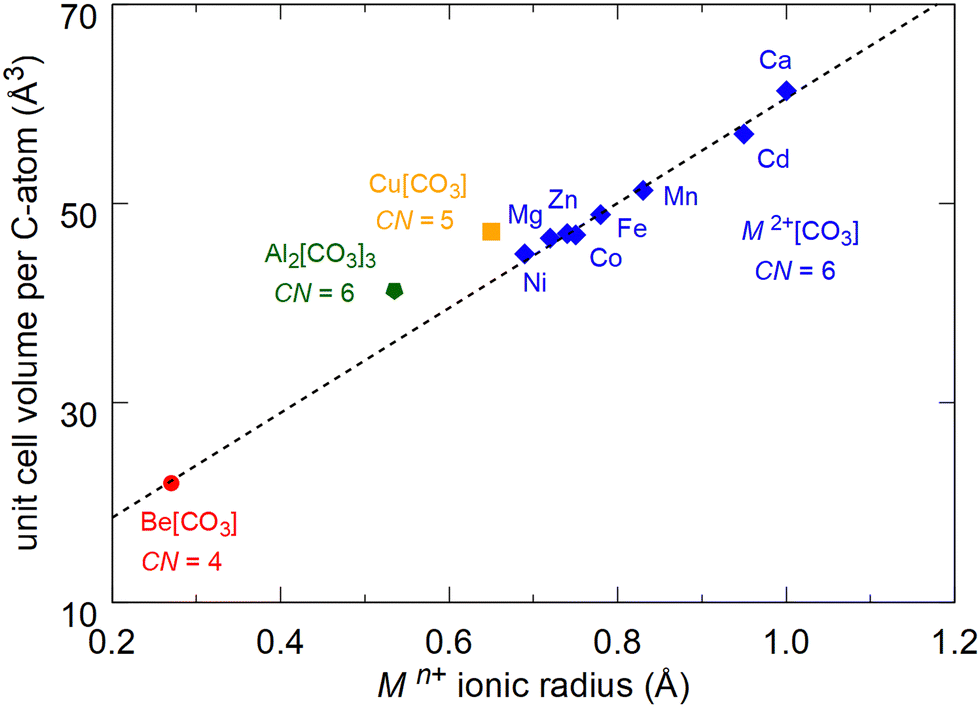 | ||
| Fig. 4 Unit cell volumes per carbon atom for carbonates with Mn+(CO3) composition as a function of the ionic radius.7 Calcite-type carbonates (CN = 6) are shown in blue, Cu(CO3) (CN = 5) in Cm space group symmetry is shown in orange and Al2(CO3)3 (CN = 6) in Fdd2 space group symmetry is shown in green.8,9,11,12,19 The DFT-calculated volume of Be(CO3) (CN = 4) in P3121 space group symmetry is shown in red. The dashed line is a linear fit to the unit cell volumes of the carbonates with M2+(CO3) composition. | ||
In conclusion, we have synthesized the alkaline earth metal carbonate Be(CO3) by a reaction of BeO and CO2 at 20(2) GPa and 1500(200) K. The structure was determined by synchrotron based single crystal diffraction and characterized by Raman spectroscopy and DFT calculations. The acentric space group symmetry (P3121) was confirmed by SHG measurements. We found that Be(CO3) can be recovered to ambient conditions. The synthesis and crystal structure determination of Be(CO3) significantly enlarged the chemistry of carbonates, as it was the last missing member of the family of alkaline earth metal carbonates. In Be(CO3) the beryllium cations are coordinated by four oxygen atoms. This arrangement was not observed in other carbonates up to now. Be(CO3) crystallizes in a new structure type.
We gratefully acknowledge funding from the DFG (WI1232, BA4020 and FOR2125/CarboPaT). EB and MB acknowledge the support of the DFG Emmy-Noether Program (projects BY101/2-1 and BY112/2-1). BW is grateful for support by the BIOVIA Science Ambassador program. We acknowledge the European Synchrotron Radiation Facility (ESRF) for provision of synchrotron radiation facilities under proposal number CH-7022. Parts of this research were carried out at beamline ID27 (doi: 10.15151/ESRF-ES-1550913042).
Data availability
Experimental and computational details together with the crystallographic data associated with this article are available in the ESI.† Experimental and theoretical structural models are deposited at the Cambridge Crystallographic Data Centre (2361893, 2361963, 2361964†).Conflicts of interest
There are no conflicts to declare.References
- Carbonates: Mineralogy and Chemistry, ed. R. J. Reeder and De Gruyter, Berlin, Boston, 1983 Search PubMed.
- B. Winkler, J. Zemann and V. Milman, Acta Crystallogr., Sect. B: Struct. Sci., 2000, 56, 648–653 CrossRef CAS PubMed.
- L. Bayarjargal, C.-J. Fruhner, N. Schrodt and B. Winkler, Phys. Earth Planet. Inter., 2018, 281, 31–45 CrossRef CAS.
- J. Binck, S. Chariton, M. Stekiel, L. Bayarjargal, W. Morgenroth, V. Milman, L. Dubrovinsky and B. Winkler, Phys. Earth Planet. Inter., 2020, 299, 106403 CrossRef CAS.
- J. Binck, L. Bayarjargal, S. S. Lobanov, W. Morgenroth, R. Luchitskaia, C. J. Pickard, V. Milman, K. Refson, D. B. Jochym, P. Byrne and B. Winkler, Phys. Rev. Mater., 2020, 4, 055001 CrossRef CAS.
- V. Cerantola, E. Bykova, I. Kupenko, M. Merlini, L. Ismailova, C. McCammon, M. Bykov, A. I. Chumakov, S. Petitgirard, I. Kantor, V. Svitlyk, J. Jacobs, M. Hanfland, M. Mezouar, C. Prescher, R. Rüffer, V. B. Prakapenka and L. Dubrovinsky, Nat. Commun., 2017, 8, 15960 CrossRef PubMed.
- R. D. Shannon, Acta Crystallogr., 1976, A32, 751–767 CrossRef CAS.
- L.-G. Liu and C.-C. Lin, Am. Mineral., 1997, 82, 643–646 CAS.
- H. Effenberger, K. Mereiter and J. Zemann, Z. Kristallogr., 1981, 156, 233–243 CAS.
- S. M. Antao and I. Hassan, Can. Mineral., 2009, 47, 1245–1255 CrossRef CAS.
- D. L. Graf, Am. Mineral., 1961, 46, 1283–1316 CAS.
- H. Seidel, K. Viswanathan, W. Johannes and H. Ehrhardt, Z. Anorg. Allg. Chem., 1974, 410, 138–148 CrossRef CAS.
- F. Weigel and A. Trinkl, Radiochim. Acta, 1973, 19, 199–202 CrossRef CAS.
- A. V. Matyskin, B. Ebin, S. Allard, N. Torapava, L. Eriksson, I. Persson, P. L. Brown and C. Ekberg, Inorg. Chem., 2023, 62, 12038–12049 CrossRef CAS.
- Y. Duan and D. C. Sorescu, J. Chem. Phys., 2010, 133, 074508 CrossRef.
- H. Effenberger and J. Zemann, Z. Kristallogr., 1979, 150, 133–138 CrossRef CAS.
- Encyclopedia of the Alkaline Earth Compounds, ed. R. C. Ropp, Elsevier B.V., Oxford, Amsterdam, 2013 Search PubMed.
- I. V. Pekov, N. V. Zubkova, N. V. Chukanov, A. A. Agakhanov, D. I. Belakovskiy, L. Horváth, Y. E. Filinchuk, E. R. Gobechiya, D. Y. Pushcharovsky and M. K. Rabadanov, Can. Mineral., 2008, 46, 1343–1354 CrossRef CAS.
- L. Bayarjargal, D. Spahr, V. Milman, J. Marquardt, N. Giordano and B. Winkler, Inorg. Chem., 2023, 62, 13910–13918 CrossRef CAS PubMed.
- R. M. Hazen and L. W. Finger, J. Appl. Phys., 1986, 59, 3728–3733 CrossRef CAS.
- H. K. Mao, J. Xu and P. M. Bell, J. Geophys. Res., 1986, 91, 4673–4676 CrossRef CAS.
- K. Aoki, H. Yamawaki, M. Sakashita, Y. Gotoh and K. Takemura, Science, 1994, 263, 356–358 CrossRef CAS PubMed.
- H. Olijnyk and A. P. Jephcoat, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 879–888 CrossRef CAS.
- A. P. Jephcoat, R. J. Hemley and H. K. Mao, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 4727–4734 CrossRef CAS PubMed.
- Y. Mori, T. Ikai and K. Takarabe, Photon factory Activity Report Part B, 2003, 20, 2015.
- C. S. Yoo, H. Kohlmann, H. Cynn, M. F. Nicol, V. Iota and T. LeBihan, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 104103 CrossRef.
- F. Datchi, B. Mallick, A. Salamat and S. Ninet, Phys. Rev. Lett., 2012, 108, 125701 CrossRef PubMed.
- V. Iota and C.-S. Yoo, Phys. Rev. Lett., 2001, 66, 5922 CrossRef PubMed.
- V. Iota, C.-S. Yoo, J.-H. Klepeis, Z. Jenei, W. Evans and H. Cynn, Nat. Mater., 2007, 6, 34–38 CrossRef CAS PubMed.
- D. Scelta, K. F. Dziubek, M. Ende, R. Miletich, M. Mezouar, G. Garbarino and R. Bini, Phys. Rev. Lett., 2021, 126, 065701 CrossRef CAS PubMed.
- N. Biedermann, S. Speziale, B. Winkler, H. J. Reichmann, M. Koch-Müller and G. Heide, Phys. Chem. Miner., 2017, 44, 335–343 CrossRef CAS.
- M. Mezouar, G. Garbarino, W. Bauchau, S. Morgenroth, K. Martel, S. Petitdemange, P. Got, C. Clavel, A. Moyne, H.-P. Van Der Kleij, A. Pakhomova, B. Wehinger, M. Gerin, T. Poreba, A. Rosa, G. Forestier, A. Weck, F. Datchi, M. Wilke, S. Jahn, D. Andrault, L. Libon, L. Pennacchioni and D. Laniel, High Pressure Res., 2024, 44, 171–198 CrossRef CAS.
- D. Spahr, J. König, L. Bayarjargal, V. Milman, A. Perlov, H.-P. Liermann and B. Winkler, J. Am. Chem. Soc., 2022, 144, 2899–2904 CrossRef CAS PubMed.
- J. König, D. Spahr, L. Bayarjargal, P. N. Gavryushkin, V. Milman, H.-P. Liermann and B. Winkler, Earth. Space. Chem., 2022, 6, 73–80 CrossRef.
- J. Zhang and R. J. Reeder, Am. Mineral., 1999, 84, 861–870 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational details, crystallographic data, Raman measurements and DFT calculations. CCDC 2361893, 2361963 and 2361964. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc03462g |
| This journal is © The Royal Society of Chemistry 2024 |