
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Modular synthesis of cyclic β-difluoroamines†
Natalie G.
Charlesworth
 a,
Dhanarajan
Arunprasath
a,
Mark A.
Graham
b,
Stephen P.
Argent
a,
Oleksandr P.
Datsenko
c,
Pavel K.
Mykhailiuk
cd and
Ross M.
Denton
*a
a,
Dhanarajan
Arunprasath
a,
Mark A.
Graham
b,
Stephen P.
Argent
a,
Oleksandr P.
Datsenko
c,
Pavel K.
Mykhailiuk
cd and
Ross M.
Denton
*a
aSchool of Chemistry, GlaxoSmithKline Carbon Neutral Laboratories for Sustainable Chemistry, University of Nottingham, 6 Triumph Road, Nottingham NG7 2GA, UK. E-mail: ross.denton@nottingham.ac.uk
bChemical Development, Pharmaceutical Technology & Development, Operations, AstraZeneca, Macclesfield SK10 2NA, UK
cEnamine Ltd, Winston Churchill Str. 78, 02094 Kyiv, Ukraine
dChemistry Department, Taras Shevchenko National University of Kyiv, Volodymyrska 64, 01601 Kyiv, Ukraine
First published on 10th June 2024
Abstract
Fluorine-containing saturated nitrogen heterocycles are very attractive structures in medicinal and biological chemistry because fluorine can be used to tune conformation as well as key properties such as basicity and bioavailability. At present cyclic fluorinated amines are accessed using hazardous reagents such as DAST or by lengthy synthesis routes. Here we report a modular two-step synthesis of cyclic β-fluoroalkyl amines using a photoredox-catalysed cyclisation/hydrogen atom transfer reaction of bromodifluoroethylamines.
Fluorinated organic molecules underpin a range of scientific fields1 and are used extensively in pharmaceutical and agrochemical research.2 For example, fluorination of amines lowers their basicity and can also modulate acute toxicity and lipophilicity.3–5 Given that saturated heterocyclic amines are very common in bioactive compounds, fluorine-containing analogues would be of significant value, particularly in the field of drug discovery.
Unfortunately, functionalised cyclic fluorinated amines are currently hard to access. They are generally synthesised through deoxydifluorination reactions using hazardous reagents (Fig. 1A) or lengthy Reformatsky-based sequences (Fig. 1B).6–9 To address this problem we envisaged a two-step synthesis of cyclic β-fluoroalkyl amines using a photoredox-catalysed cyclisation reaction of bromodifluoroethylamines, prepared through a modular three-component coupling (Fig. 1C).10 To the best of our knowledge, photoredox cyclisations11,12 of this type have not been previously described as such reactions could be complicated by competing redox-triggered C–H functionalization reactions adjacent to nitrogen.13 Related work has circumvented this problem by using redox inert sulfonamides14,15 or amides16–21 as cyclisation substrates, but subsequent deprotection or redox adjustments are then required to access synthetically useful products. Here we show that cyclisation of unprotected β-bromodifluoroalkyl amines prepared from readily available amines, aldehydes and carboxylic acids22 is indeed possible resulting in a concise synthesis of cyclic β-fluoroalkyl amines (Fig. 1C).
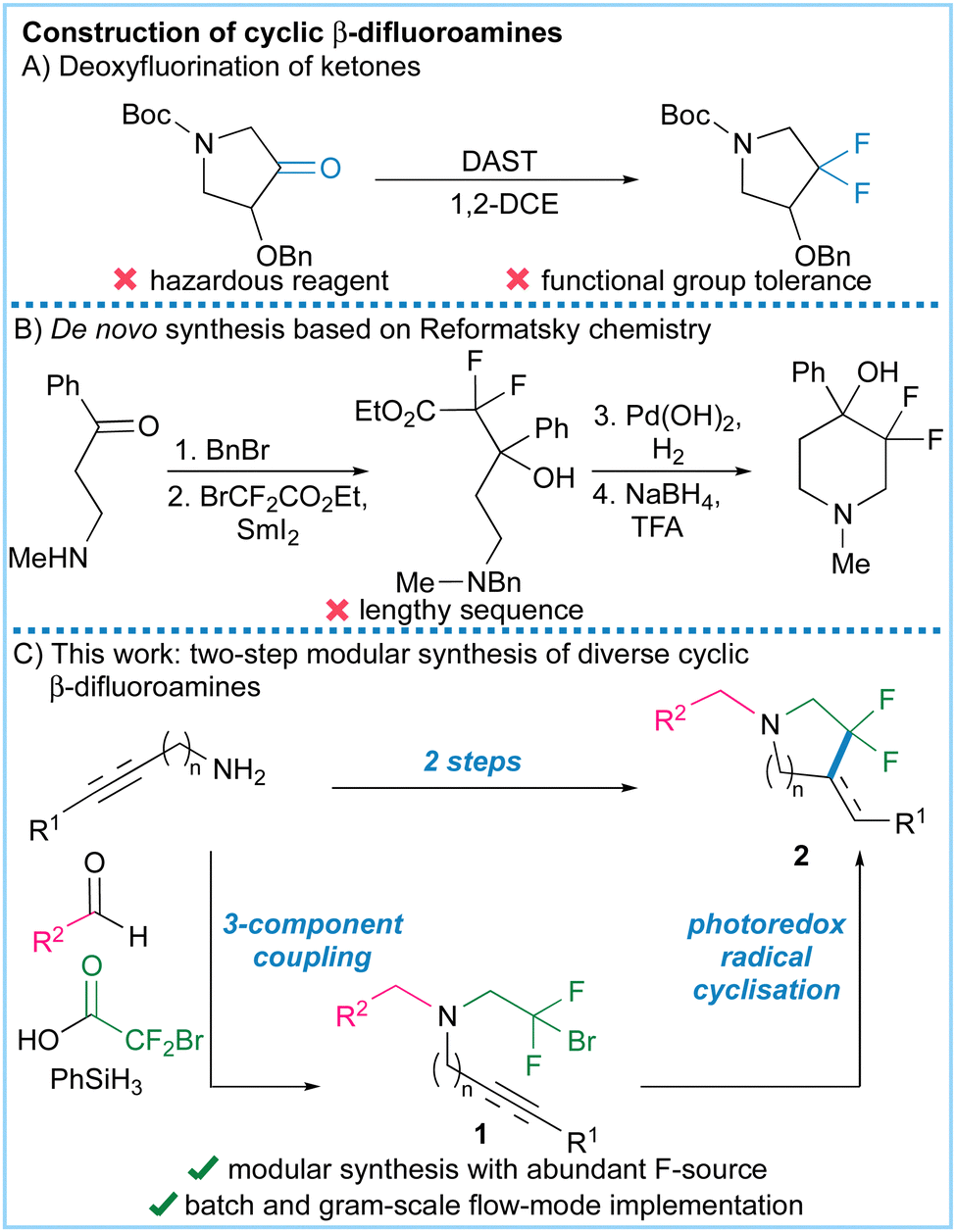 | ||
| Fig. 1 Methods for the construction of difluorinated amines. (A) Deoxyfluorination of ketones. (B) Reformatsky chemistry using ethyl bromodifluoroacetate. (C) This work – a two-step modular amine synthesis using primary amines, aldehydes and bromodifluoroacetic acid as the fluorine source. | ||
We began our studies by screening conditions using model alkynyl amine substrate 3, which was prepared by a three- component coupling using bromodifluoroacetic acid as the source of the bromodifluoromethyl group (Table 1).10 We opted to use the strongly reducing Ir(ppy)3 catalyst (−1.73 V vs. SCE) as a starting point and, gratifyingly, the desired cyclised product 4 was detected and no products arising from oxidation of the substrate or product were obtained (Table 1, entry 1). We then surveyed additional hydrogen atom sources and, while the Hantzsch ester (entry 2) and tris(trimethylsilyl)silane (TTMSS) (entry 3) were found to be poor, formic and acetic acid were superior (entries 4 and 5).
|
|
|||
|---|---|---|---|
| Entry | Base (equiv.) | H-source (equiv.) | Product yielda/% |
| Reactions conducted on a 0.05 mmol scale.a Yield determined by 19F NMR spectroscopy using trifluorotoluene as an internal standard. | |||
| 1 | Et3N (10) | None | 56 |
| 2 | Et3N (10) | Hantzsch ester (1.5) | 7 |
| 3 | Et3N (10) | TTMSS (1.5) | 12 |
| 4 | Et3N (10) | HCOOH (1.5) | 71 |
| 5 | Et3N (10) | HCOOH (5.0) | 77 |
| 6 | Et3N (10) | AcOH (5.0) | 88 |
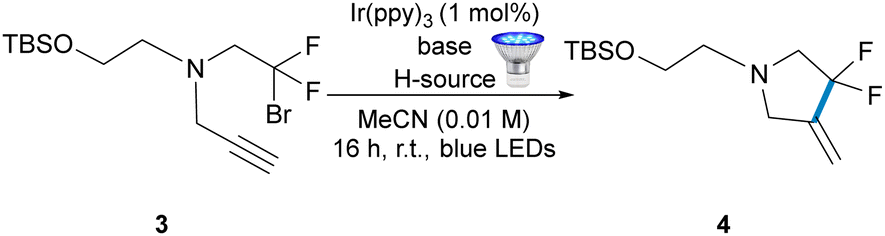
Optimal conditions (entry 6) were obtained using ten equivalents of triethylamine in combination with five equivalents of acetic acid. Increasing the concentration proved to be detrimental, as did the use of other photoredox catalysts (Ir(ppy)2(dtbbpy), Ru(bpy)3Cl2, EosinY and 4CzIPN) as detailed in the ESI.†
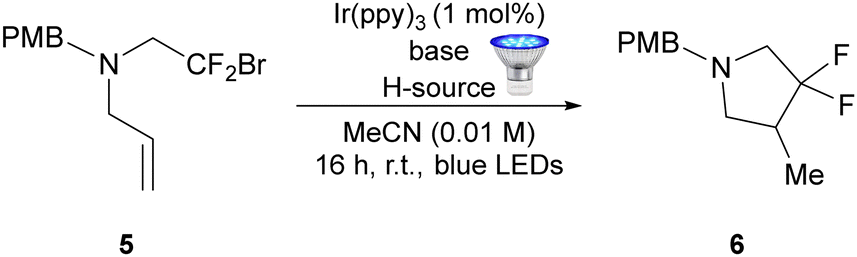
We next conducted a further screen of conditions for the cyclisation of alkenyl amine 5 (Table 2). In this instance, the use of DIPEA as the base resulted in a moderately improved yield (44% vs. 38%, entries 1 and 2) for the transformation vs. triethylamine. Interestingly, for these substrates the addition of acetic acid was detrimental and only 10% of the product was obtained in this case (entry 3). Fortunately, TTMSS proved to be an excellent hydrogen atom donor for this substrate class (entry 4), with the desired compound 6 being obtained in a 97% yield.
Having optimised the reaction conditions for the cyclisation of alkyne- and alkene-containing tertiary amines, we explored the scope of both photoredox cyclisation reactions (Fig. 2A and B). In all cases the yields were determined using 19F NMR spectroscopy and after chromatographic purification (values in parentheses). Key findings were as follows.
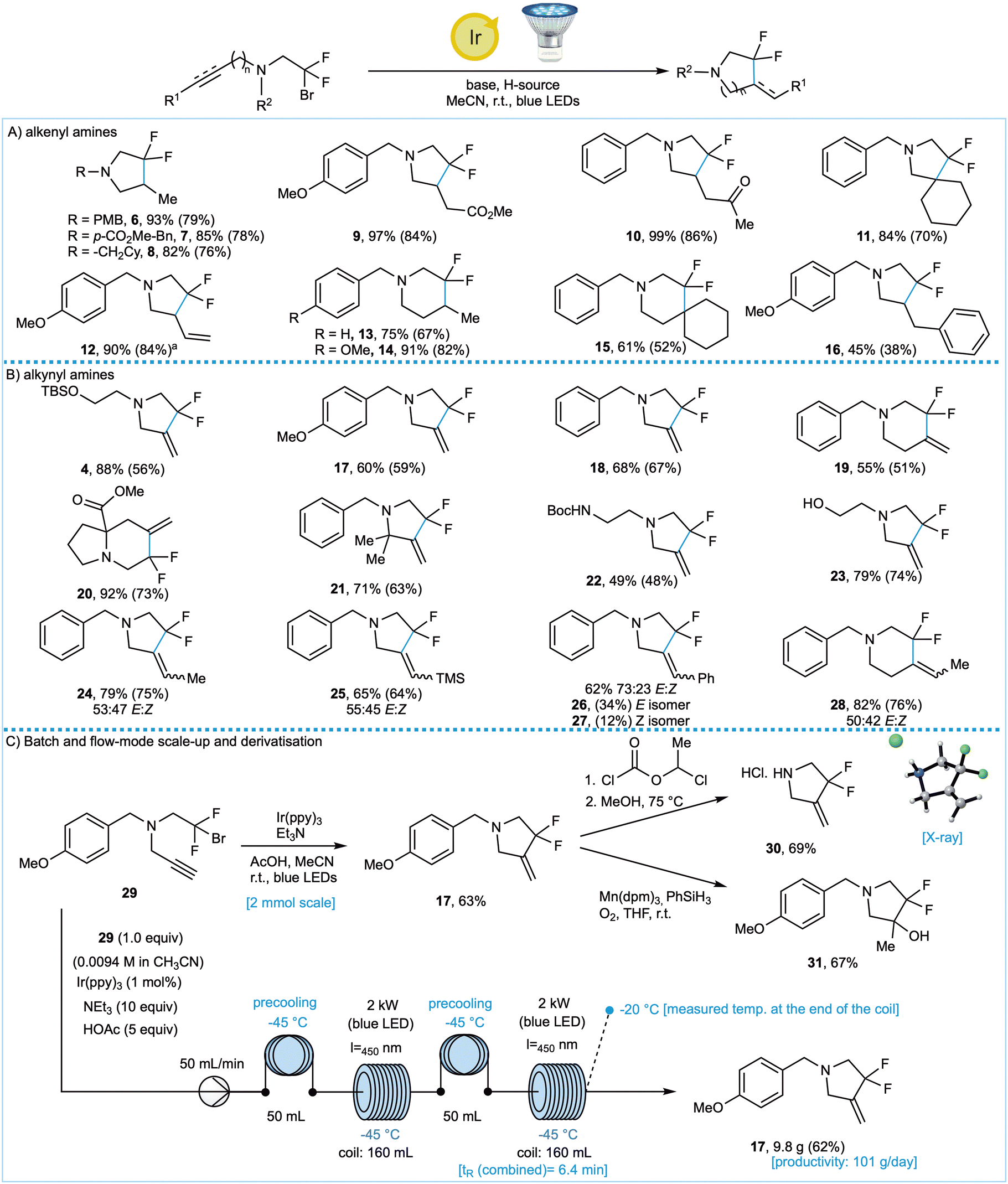 | ||
Fig. 2 Photoredox radical cyclisation of alkenyl and alkynyl amines. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Reaction conditions: (A) alkenyl amine cyclisation – Ir(ppy)3 (1 mol%), DIPEA (10 equiv.), TTMSS (5 equiv.), MeCN (0.01 M), r.t., blue LED irradiation; (B) alkynyl amine cyclisation – Ir(ppy)3 (1 mol%), Et3N (10 equiv.), acetic acid (5 equiv.), MeCN (0.01 M), r.t., blue LED irradiation; PMB deprotection – 1-chloroethyl chloroformate (1.1 equiv.), 1,2-DCE (0.4 M), 90 °C, 2 h, then MeOH (1.0 M), 75 °C, 1 h; Mukaiyama hydration – Mn(dpm)3 (5 mol%), PhSiH3 (2 equiv.), O2 (1 atm), THF (0.2 M), r.t., 24 h. Yields were determined by NMR spectroscopy vs. internal standard (1,3-benzodioxole or trifluorotoluene); yields of isolated material after chromatography are given in parentheses. aDIPEA (20 equiv.), TTMSS (2 equiv.). (C) Batch and flow-mode scale-up reactions. Reaction conditions: (A) alkenyl amine cyclisation – Ir(ppy)3 (1 mol%), DIPEA (10 equiv.), TTMSS (5 equiv.), MeCN (0.01 M), r.t., blue LED irradiation; (B) alkynyl amine cyclisation – Ir(ppy)3 (1 mol%), Et3N (10 equiv.), acetic acid (5 equiv.), MeCN (0.01 M), r.t., blue LED irradiation; PMB deprotection – 1-chloroethyl chloroformate (1.1 equiv.), 1,2-DCE (0.4 M), 90 °C, 2 h, then MeOH (1.0 M), 75 °C, 1 h; Mukaiyama hydration – Mn(dpm)3 (5 mol%), PhSiH3 (2 equiv.), O2 (1 atm), THF (0.2 M), r.t., 24 h. Yields were determined by NMR spectroscopy vs. internal standard (1,3-benzodioxole or trifluorotoluene); yields of isolated material after chromatography are given in parentheses. aDIPEA (20 equiv.), TTMSS (2 equiv.). (C) Batch and flow-mode scale-up reactions. | ||
Acyclic alkenes were successfully utilised as radical traps, with particularly high yields observed for products 9 and 10 where the cyclic radical intermediates are stabilised. This, however, was not crucial as products 6, 13 and 14 derived from terminal alkenes substrates were obtained in good yield. The use of an endocyclic alkene afforded spirocyclic amine 11 in a good, isolated yield of 74%. Silyl-protected alcohols (4) are tolerated, as are free hydroxyl groups (23), carbamates (22), esters (9 and 20) and ketones (10). These products would be challenging to access using conventional nucleophilic fluorinating reagents such as DAST. Conformational restriction in the proline-derived substrate also promoted the desired cyclisation to give fluoroindolizidine 20. Cyclisation of internal alkynes afforded products 24–28 with E:Z ratios close to 50:50.
Having established the scope, we sought to further explore the practicality of the reaction. In particular, we were interested in scaling up the reaction and conducting deprotection/derivatisation sequences in order to generate novel building blocks.
Pleasingly, with no detriment to the yield, we were able to achieve a ten-fold scale-up of the batch reaction for the synthesis of 17 from 0.25 mmol to 2 mmol (Fig. 2C). Accounting for the presence of a PMB group in some of our substrates, we developed a cleavage protocol using 1-chloroethyl chloroformate, which gave 30 in a 69% isolated yield.
Our two-step protocol represents a very efficient method to access this pyrrolidine analogue and the calculated pKaH and logP of 30 (pKaH = 15.1, logP = 1.20) vs. pyrrolidine (pKaH = 19.5, logP = 0.77)23 indicate the impact fluorine has on key properties which can, in principle, modulate biological activity and metabolic stability. Next, the alkene in product 17 was transformed into tertiary alcohol 31 in 67% yield, via Mukaiyama hydration conditions, thereby demonstrating the versatility of the alkene handle and providing a new method to access the difluorohydrin motif previously generated through Reformatsky chemistry (Fig. 1B). In order to obtain still larger quantities of product 17, we developed a flow-chemistry platform (Fig. 2C) which allowed us to prepare over 9 g of 17 with a productivity of 101 g day−1. In principle this method can be applied to access multigram quantities of any of the products described above.
Finally, we investigated the mechanism of the cyclisation reactions (Fig. 3). Given that Ir(ppy)3 has a reduction potential of −1.73 V vs. SCE and taking into account mechanistic investigations into cyclisations of un-activated alkyl iodides,24 an oxidative quenching cycle is likely to be operating. We propose that irradiation of the catalyst forms the excited Ir(III)* complex which then reduces the bromodifluoroalkyl starting material 1 by single electron transfer to give radical 32 and the corresponding Ir(IV) species. Intramolecular radical addition is favoured under our dilute conditions to give cyclic radical 33, which is then able to participate in hydrogen atom transfer with multiple donors (vide infra).
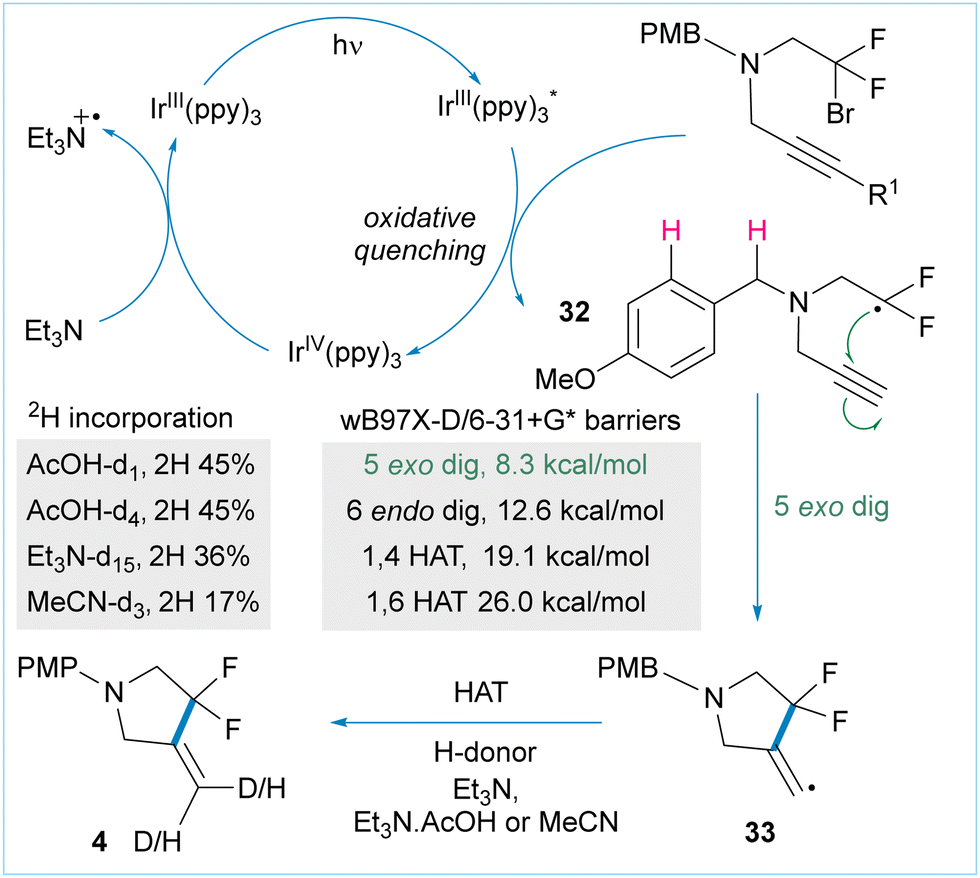 | ||
| Fig. 3 Proposed catalytic cycle based on control experiments, deuterium quenching and quantum chemical calculations. | ||
We examined other possible reactions of radical 32 using QM calculations (see ESI†) and large barriers were obtained for the potentially competitive 1,4-HAT and 1,6-HAT pathways as well as the 6-endo cyclisation. The exo-cyclisation has a barrier of 8.3 kcal mol−1 (Fig. 3) which is consistent with related processes.25 Possible C–H functionalisation products arising from oxidation of either the amine substrate or product were absent. This is due to the negative inductive effect of the β-difluoro group which substantially alters the redox potential of both compounds, thereby preventing C–H functionalisation adjacent to nitrogen.
Next, we carried out a series of deuterium-incorporation studies in which all possible H atom sources were systematically exchanged with their 2H counterparts. This revealed that Et3N, AcOH and MeCN were all competent hydrogen atom donors (Fig. 2 and ESI†) for the HAT reaction of intermediate 33.
A series of control experiments (see ESI†) confirmed that the reaction does not proceed in the absence of light, nor in the absence of tertiary amine base.
The reaction does proceed (albeit in lower yield – 56% for 4) in the absence of acetic acid which suggests that triethylamine can act as an electron and hydrogen atom donor in this process.
In summary, we have demonstrated a mild and efficient visible-light-mediated intramolecular cyclisation reaction of bromodifluoroalkyl amines. In two-steps, complex fluorinated nitrogen-containing heterocycles are generated using a carboxylic acid as the source of fluorine.
R. M. D., N. G. C. and M. A. G. designed the experiments. N. G. C. carried out the experiments and collected the data. R. M. D. carried out the computational calculations. R. M. D. and N. G. C. analysed the data. R. M. D. and N. G. C. drafted the manuscript. S. A. P. determined the crystal structure of compound 30. P. M. and O. P. carried out flow chemistry. All the authors approved the final version of the manuscript for submission.
We thank AstraZeneca for financial support (CASE studentship to N. G. C.) and Dr Kevin Butler, Dr Mattia Silvi and Dr Liam Ball for valuable discussions.
Data availability
The experimental and computational data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- C. Ni and J. Hu, Chem. Soc. Rev., 2016, 45, 5441–5454 RSC.
- N. A. Meanwell, J. Med. Chem., 2018, 61, 5822–5880 CrossRef CAS PubMed.
- D. O’Hagan, J. Fluorine Chem., 2010, 131, 1071–1081 CrossRef.
- E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315–8359 CrossRef CAS PubMed.
- B. Jeffries, Z. Wang, H. R. Felstead, J. Y. Le Questel, J. S. Scott, E. Chiarparin, J. Graton and B. Linclau, J. Med. Chem., 2020, 63, 1002–1031 CrossRef CAS PubMed.
- GENENTECH INC., US Pat., US2014/044401, 2014.
- M. K. Nielsen, C. R. Ugaz, W. Li and A. G. Doyle, J. Am. Chem. Soc., 2015, 137, 9571–9574 CrossRef CAS PubMed.
- L. Mohammadkhani and M. M. Heravi, J. Fluorine Chem., 2019, 225, 11–20 CrossRef CAS.
- A. B. Beeler, R. S. V. S. Gadepalli, S. Steyn, N. Castagnoli and J. M. Rimoldi, Bioorg. Med. Chem., 2003, 11, 5229–5234 CrossRef CAS PubMed.
- K. G. Andrews, R. Faizova and R. M. Denton, Nat. Commun., 2017, 8, 15913 CrossRef PubMed.
- M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed.
- T. Koike and M. Akita, Inorg. Chem. Front., 2014, 1, 562–576 RSC.
- L. Shi and W. Xia, Chem. Soc. Rev., 2012, 41, 7687–7697 RSC.
- J. J. Devery, J. D. Nguyen, C. Dai and C. R. J. Stephenson, ACS Catal., 2016, 6, 5962–5967 CrossRef CAS.
- J. D. Nguyen, E. M. D’Amato, J. M. R. Narayanam and C. R. J. Stephenson, Nat. Chem., 2012, 4, 854–859 CrossRef CAS PubMed.
- E. Fava, M. Nakajima, M. B. Tabak and M. Rueping, Green Chem., 2016, 18, 4531–4535 RSC.
- T. Sato, Y. Wada, M. Nishimoto, H. Ishibashi and M. Ikeda, J. Chem. Soc., Perkin Trans. 1, 1989, 879–886 RSC.
- K. Sun, S. Wang, R. Feng, Y. Zhang, X. Wang, Z. Zhang and B. Zhang, Org. Lett., 2019, 21, 2052–2055 CrossRef CAS PubMed.
- X. Zhuang, X. Shi, R. Zhu, B. Sun, W. Su and C. Jin, Org. Chem. Front., 2021, 8, 736–742 RSC.
- F. Zhu, Z. Li and X. F. Wu, Org. Lett., 2023, 25, 8535–8539 CrossRef CAS PubMed.
- G. Wang, C. Shen and K. Dong, Org. Lett., 2023, 25, 2878–2882 CrossRef CAS PubMed.
- M. Morgenthaler, E. Schweizer, A. Hoffmann-Röder, F. Benini, R. E. Martin, G. Jaeschke, B. Wagner, H. Fischer, S. Bendels, D. Zimmerli, J. Schneider, F. Diederich, M. Kansy and K. Müller, ChemMedChem, 2007, 2, 1100–1115 CrossRef CAS PubMed.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 42717 CrossRef PubMed.
- J. D. Nguyen, E. M. D’Amato, J. M. R. Narayanam and C. R. J. Stephenson, Nat. Chem., 2012, 4, 854–859 CrossRef CAS PubMed.
- G. V. M. Sharma, D. H. Chary, N. Chandramouli, F. Achrainer, S. Patrudu and H. Zipse, Org. Biomol. Chem., 2011, 9, 4079–4084 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Details of experimental procedures and characterisation of compounds. CCDC 2083564. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc00640b |
| This journal is © The Royal Society of Chemistry 2024 |