
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced H2 production assisted by anodic iodide oxidation using transparent tin oxide-based electrodes†
Shraddha
Paniya
 and
Kiran
Vankayala
*
and
Kiran
Vankayala
*
Functional Materials for Electrochemistry and Solar energy (FunMatES) group, Energy and Environmental Chemistry Lab, Department of Chemistry, Birla Institute of Technology and Science, Pilani, K K Birla Goa Campus, Goa, 403726, India. E-mail: kiranv@goa.bits-pilani.ac.in; kiran2cu@gmail.com
First published on 24th June 2024
Abstract
In this work, the direct use of transparent conducting oxides (TCOs) as cost-efficient anodes for the iodide oxidation reaction (IOR) is explored. Energy-saving hydrogen production assisted by the IOR is demonstrated using a hybrid water electrolysis system with FTO as the anode and Pt-wire as the cathode. The hybrid system delivers 10 mA cm−2 at a cell voltage as low as 1.15 V with the faradaic efficiency for H2 found to be ∼91%. This study may open avenues for developing novel systems that integrate the IOR with other high-value reduction reactions.
Hydrogen (H2)-based energy systems are promising for producing clean and sustainable energy owing to its high gravimetric heating value (141.9 mJ kg−1) and zero emissions on combustion.1,2 H2 production can be sustainable if generated through water electrolysis using renewable energy sources.1 However, the anodic oxygen evolution reaction (OER) is a bottleneck in water splitting owing to inherently high energy demand due to its multi-step multi-electron nature, which is known to limit the efficiency of water electrolysis.3 If the OER is replaced with thermodynamically more readily oxidizable molecules (whose oxidation potential is lower than the OER) such as alcohols,4 hydrazine,5 urea,6 benzylamine,7 hydroxymethylfurfural (HMF),8etc., then energy-saving H2 production can be realized.9 These hybrid water electrolysis systems are capable of generating H2 and high-value products concomitantly.9 The iodide oxidation reaction (IOR) to iodine (I2) is one such reaction whose thermodynamic potential is 0.54 V vs. NHE in acidic medium,10a which is ∼700 mV lower than that of the OER, and thus may lead to facile H2 production when hydrogen evolution reaction (HER) is integrated with the IOR.10a Recently, the electrocatalytic IOR has attracted significant attention for achieving energy-saving H2 production.10 In addition, the product of the IOR, for instance, I2 is a high-value chemical and widely used in the hygiene industry, pharmaceuticals, chemical synthesis, etc.10e,11 in comparison to less economic oxygen which is the product of the OER in traditional water electrolysis. Earlier studies on IOR have reported the use of materials which are either not cost-efficient or require stringent experimental conditions.10a,b,d,f The continuous quest for the development of novel and cost-efficient electrocatalysts prompted researchers to alleviate the dependency on noble-metal based materials in energy conversion devices. Transparent conducting oxide (TCO)-based materials, namely fluorine doped tin oxide (FTO), indium–tin oxide (ITO), and antimony-doped tin oxide (ATO), have been extensively used for various applications.12 TCOs are typically degenerately-doped wide band gap (3 eV) semiconductors possessing fascinating properties, such as high optical transparency (>80% at 550 nm for FTO) in the visible region of the electromagnetic spectrum, low electrical resistivity (∼10−4 Ω cm for FTO), etc.12,13 The majority of the studies use TCOs as conductive supports. For instance, FTO based electrodes have been used as transparent conductive substrates in solar cells, and organic light emitting diodes (OLEDs).12 Additionally, the TCOs find their existence in polymer electrolyte membrane (PEM) fuel cells, however as a conductive support for electrocatalysts.14 In other words, modified FTO/ITO based TCOs have been extensively studied for (photo)electrochemical applications. However, it is noteworthy that the direct use of TCOs as an electrocatalyst is scarce.15,16 The direct use of TCOs may attract attention due to the possibility of realizing practical applications, considering the commercial availability of large area FTO electrodes. Additionally, the direct use of transparent FTO provides scope to develop photoelectrochemical IOR systems that can be integrated with the HER or CO2 reduction reaction (CO2RR). Herein, the direct use of TCOs, namely FTO and ITO, in pristine form as an electrocatalyst for the IOR is studied. In addition to the high conductivity offered by TCOs, the appreciable stability of TCOs, especially of FTO in acidic environments,13,17 led us to choose TCOs in the present study. The ability of FTO for the IOR is studied and installed the same as anodes for the first time in hybrid water electrolysis that couples the HER and IOR.
The as-obtained commercial TCOs such as FTO and ITO coated glass substrates after thorough cleaning were used as electrodes in the present study. The basic characterization studies were carried out to ensure the characteristics of the FTO and ITO electrodes. Fig. S1 (ESI†) shows the XRD pattern of FTO, which matches well with the cassiterite phase of SnO2 (JCPDS file no. #00-41-1445). The XRD pattern of ITO consisted of reflections corresponding to SnO2 (JCPDS file no. #00-41-1445) and In2O3 (JCPDS file no. #00-006-0416), as expected.18 The Raman spectra of FTO and ITO show characteristic Raman bands at 115, 435, 556, and 772 cm−1 which are assigned for the B1g, Eg, Eu and B2g modes, respectively for cassiterite SnO2 (Fig. S2, ESI†).19 Fig. S3 (ESI†) shows the morphology of the FTO and ITO coated glass substrates obtained from field emission scanning electron microscopy (FESEM) images.
Electrochemical activity of FTO and ITO coated glass substrates were studied in Ar-saturated 0.1 M HClO4 at 25 °C in a standard three-electrode system. Pre-cleaned FTO/ITO coated glass was directly used as the working electrode (WE), and a saturated calomel electrode (SCE) and graphite rod acted as the reference electrode (RE) and counter electrode (CE), respectively. Fig. 1a represents the iR-corrected (80%) linear sweep voltammograms (LSVs) depicting OER and IOR activity when the measurements were carried out in the absence and presence of NaI, respectively. It is noted that the potential required to observe OER currents is very high indicating that FTO is not an efficient OER electrode. However, in the presence of NaI, the voltammograms show a clear and dominant oxidation of iodide at low potentials, signifying the fact that FTO can oxidize I− to I2. It is observed that the FTO electrode requires a potential of ∼1.04 V vs. RHE to reach the benchmark current density of 10 mA cm−2 for the IOR. Table S1 (ESI†) presents the comparison of the performance of FTO for the IOR with some of the recently reported electrocatalysts. Furthermore, it is observed that the currents increase with increase in the concentration of NaI, confirming that the currents originated in the presence of NaI are due to IOR at the FTO surface (Fig. 1b). The LSV measurements with lower concentrations of I− shown in Fig. S4 (ESI†) indicate that FTO is capable of oxidizing even low concentrations of I−. It should be noted that by replacing the OER by IOR, the potential required to reach 1 mA cm−2 can be significantly lowered by ∼1.65 V. The data clearly suggest that the IOR can be a potential alternative to the OER to realize energy-saving H2 production. Furthermore, when the electrode is pre-treated at 3.3 V vs. RHE (outside the stability limits of FTO)13,17 for 10 min prior to IOR measurements, the IOR activity substantially decreased as the chosen potential is known to leach out some of the Sn-ions from the FTO.13 As shown in Fig. S5 (ESI†), the potential required to reach 10 mA cm−2 is increased from 1.09 V vs. RHE for pristine FTO to 1.51 V vs. RHE for pre-treated FTO, suggesting the role of Sn-ions in dictating IOR activity. Additionally, the ability of FTO towards IOR activity is further tested by carrying out measurements in alkaline (0.1 M NaOH) solutions of NaI (Fig. 1c), as the product of the IOR depends on pH.10b For instance, the product of the IOR at low pH is predominantly iodine (2I− → I2 + 2e−; pH ∼ 1),10b while it is iodate when the IOR is performed in high pH (I− + 6OH−→ IO3− + 3H2O + 6e−; pH ∼ 13)10b conditions. As indicated with digital pictures in Fig. 1c, the generation of yellow colour product formed at low pH conditions (in HClO4 medium) confirms the formation of iodine (in the form of I3−), while it is colourless at high pH conditions (in NaOH medium) as the product formed is IO3−, which is colourless. It should be noted that the IOR in alkaline medium requires higher potentials than that in acidic medium due to the complex 6-electron process.10b,11 Therefore, further studies in the present study are not carried out in an alkaline medium. The IOR studies performed using ITO coated glass are given in Fig. S6 (ESI†). The data indicate that FTO and ITO show comparable IOR activity, with ITO exhibiting relatively higher current densities as compared to FTO. Electrochemical impedance spectroscopy (EIS) data for the OER and IOR recorded using FTO and ITO are given in Fig. S6(b) (ESI†). Among FTO & ITO, the radius of the semicircle for the IOR is noted to be smaller for ITO indicating the lower resistance for charge transfer at the electrode/electrolyte interface than that of FTO. The Nyquist plot of the OER for both FTO and ITO suggests a higher charge transfer resistance than IOR, reconfirming the facile oxidation of iodide (0.54 V vs. NHE) than water (1.23 V vs. NHE) on FTO and ITO electrodes. Though ITO performed slightly better than FTO for the IOR, further studies were carried out with FTO due to its relatively higher stability than ITO in acidic environments.13,17 The stability aspects will be discussed in detail in the subsequent section.
 | ||
| Fig. 1 iR-corrected (a) linear sweep voltammograms obtained using FTO with (red) and without (black) 1 M NaI in 0.1 M HClO4, (b) variation of the voltammograms with concentration of NaI, (c) voltammograms recorded when 1 M NaI dissolved in different electrolytes. The scan rate used was 5 mV s−1. | ||
To gain insight into the reaction kinetics at the FTO/electrolyte interface and the electron transfer, in situ EIS measurements were performed in 0.1 M HClO4 with and without 1 M NaI at different DC potentials (Fig. S7a and b, ESI†). As can be seen from the Bode phase plots of the OER in Fig. S7a (ESI†), FTO showed no frequency peak between 1.65 V and 1.9 V vs. RHE in 0.1 M HClO4 without NaI. The commencement of a peak in the low-frequency range started at ∼1.95 V vs. RHE, which can be attributed to the adsorption of oxygen active species on the surface and the corresponding faradaic reaction (OER),20 complementing the LSV results. After the addition of 1 M NaI (Fig. S7b, ESI†), the peak in the low-frequency range started to appear from 0.65 V vs. RHE indicating that iodide ions adsorb on the FTO surface in a more favourable manner. This accounts for the lowering of the potential by ∼1300 mV.
Motivated by the preliminary electrochemical measurements, the product quantification studies were performed using an H-type two-compartment cell (Fig. S8, ESI†). A commercial Pt-wire was chosen as the CE due to its high HER activity in acidic electrolytes. The use of a divided cell eliminates the unwanted counter reactions to the IOR (reduction of product formed in the IOR) at the CE. The H-cell setup was tested by recording voltammograms prior to quantification studies. The data obtained in the H-type cell (Fig. 2a) are similar to the LSVs recorded in the 3-electrode cell given in Fig. 1a. The products formed at the CE (H2) and WE (I2) when the system was held at a potential of 1.08 V vs. RHE were quantified using gas chromatography (GC) and UV-visible spectroscopy, respectively. Fig. 2b represents typical GC traces recorded for the gaseous products collected from the CE compartment, confirming the formation of H2. Similarly, Fig. 2c shows typical absorption spectra of the liquid product (with a dilution factor of 8×) collected from the WE compartment at different intervals of time. The data clearly show the appearance of a peak corresponding to I3− (at 350 nm) that grows with time, confirming that the oxidation of I− leads to the formation of I2 (since I2 in the presence of I−, exists as I3−). The concentration of iodine in the form of I3− was estimated using a calibration plot obtained by recording absorption spectra of various concentrations of standard iodine (dissolved in I−) (Fig. S9 (ESI†)). The faradaic efficiency (FE) is found to be ∼95% for the gaseous product (H2) while the FE value for the liquid product (I2(I3−)) is ∼98% (Fig. 2d). To confirm the formation of I2 during the IOR process, the electrolyte obtained after long-term chronoamperometry tests carried out at 1.1 V vs. RHE for 14 h, was collected and subjected to extraction of I2 in chloroform, as shown in Fig. S10a (ESI†). The absorption spectrum of the extract (Fig. S10b, ESI†) shows a peak at 511 nm which is similar to that of the standard I2, confirming the formation of I2 as the product of the IOR in acidic medium. Furthermore, the robustness of the FTO electrode is evaluated by performing long-term (24 h) tests at 10 mA cm−2. As shown in Fig. S11 (ESI†), the FTO electrode exhibited stable electrochemical performance. The FTO electrode after long term testing was characterised using XRD, XPS and Raman spectroscopy to decipher the stability of the electrode. The XRD patterns (Fig. S12, ESI†) and Raman spectra (Fig. S13, ESI†) of FTO before and after the long term stability test show negligible changes indicating the robustness of the FTO substrate. Additionally, the surface morphology of the FTO electrodes remained intact after the long-term test as evidenced by FESEM images shown in Fig. S14 (ESI†). Notably, the energy dispersive X-ray spectrum (EDS) clearly shows the presence of I-species suggesting the adsorption of I-species on the FTO surface. The XPS (Fig. S15, ESI†) data of the FTO electrode after the stability test show characteristic peaks corresponding to Sn 3d and O 1s that do not show any significant variation. The presence of I-species is noted from the I 3d XPS data of the post catalysis sample (Fig. S15d, ESI†), complementing the EDS data. As mentioned above, the characterization studies of the post catalysis ITO electrode reveal relatively poorer stability than FTO (More details in ESI;† Fig. S16). It should be noted that spontaneous oxidation of I− under acidic conditions (4H+ + 6I− + O2 → 2H2O + 2I3−) can be a competitive reaction which may generate iodine (in the form of I3−). To understand this, a control experiment was carried out at 25 °C, similar to the conditions used in electrochemical experiments; however in the absence of applied voltage. As shown in Fig. 2e, the formation of iodine or I3− is not very evident in voltage-free conditions as compared to that obtained via electrochemical oxidation carried out at 1.17 V vs. RHE. It is observed that the amount of triiodide produced by electrochemical oxidation is 63.4 mM while it is 23 μM in control experiments after 22 h (see ESI† Fig. S17 and S18). This clearly suggests the electrocatalytic effect of the FTO electrode in oxidizing iodide to iodine. Additionally, the IOR in a neutral medium (0.1 M Na2SO4) was also studied and the product formed during IOR was quantified (see Fig. S19, ESI†).
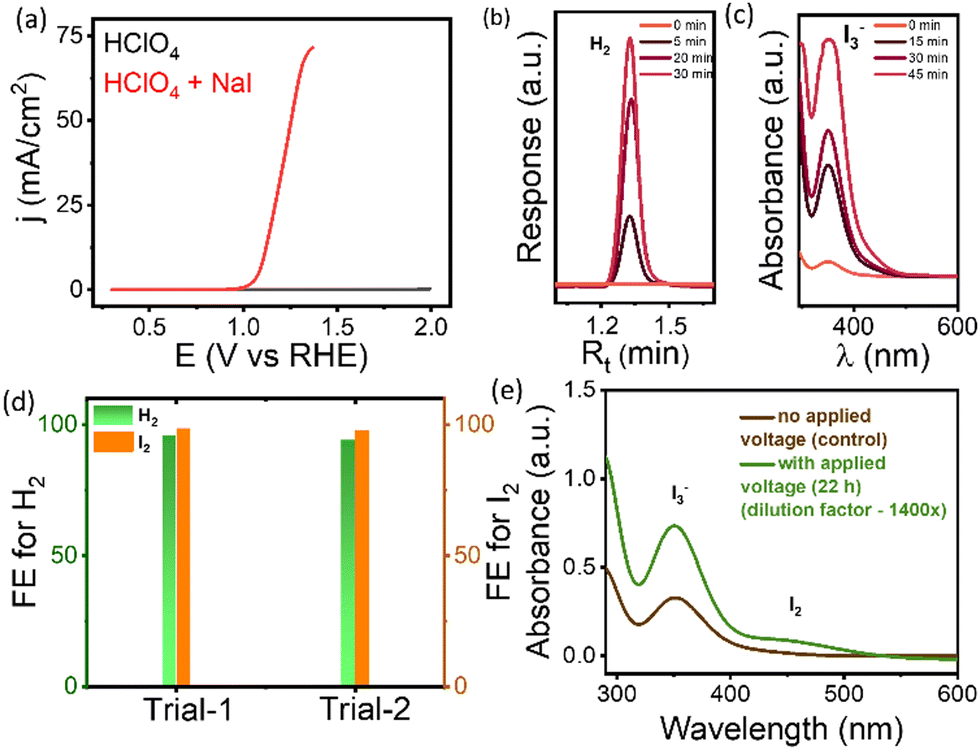 | ||
| Fig. 2 (a) iR-corrected linear sweep voltammetry curves obtained in a H-cell using FTO as WE and Pt-wire as CE in the presence and absence of 1 M NaI in 0.1 M HClO4, at a scan rate of 5 mV s−1, (b) and (c) representative gas chromatograms and UV-Vis absorbance spectra, respectively, recorded during the course of potentiostatic measurements at 1.08 V vs. RHE, (d) faradaic efficiency of products H2 and I2 determined in two different trials, and (e) UV-vis absorbance spectra of the aliquot of the product obtained with and without applied voltage. | ||
Further, hybrid water electrolysis (IOR||HER) measurements were performed in an H-type electrochemical cell as mentioned above, using optimized NaI concentrations (Fig. S20, ESI†) with FTO as the anode and Pt-wire as cathode (FTO||Pt-wire) (Fig. 3a). The polarization curves obtained at a scan rate of 5 mV s−1 are shown in Fig. 3b. The data suggest that to achieve a current density of 10 mA cm−2, a cell voltage of 1.15 V is required in hybrid water electrolysis, which is comparable to several hybrid water electrolysis systems recently reported in the literature (Table S2, ESI†). Additionally, the energy-saving efficiency at 1 mA cm−2 is found to be 56% when the IOR is integrated with the HER. Furthermore, the H2 generated in the hybrid electrolyzer was also quantified by holding the cell at a cell voltage of 1.15 V. A FE of around 91% was observed for H2. It should be noted that H2 production is not observed in traditional water electrolysis setups under the conditions (1.15 V) used in hybrid water electrolysis (Fig. S21, ESI†). Fig. 3c shows the long-term stability of FTO in the two electrode–electrolyzer setup when the system is at 10 mA cm−2 and the cell voltage-time data suggest that even after 20 h, the FTO shows only a little change in the cell voltage of 12.87 mV h−1 as compared to the initial value, reaffirming the stability of FTO for the IOR. It is worth noting that the pristine FTO as an anode for the IOR reported in the present study is capable of delivering efficiencies comparable to several of the reported systems that use anodes either noble metal-based or that require tedious experimental conditions. This study may open up new avenues for the further exploration of the use of FTO-based electrodes for energy-saving H2 production assisted by an IOR process.
 | ||
| Fig. 3 (a) Schematic illustration of a hybrid water electrolysis cell consisting of Pt-wire as the cathode and FTO as the anode, (b) linear sweep voltammetry curves obtained for hybrid water electrolysis (red) and traditional water electrolysis (black), (c) variation of the cell voltage with time monitored when the hybrid water electrolysis system is held at 10 mA cm−2. Inset of (c) represents the snapshot depicting the formation of iodine at the anode and H2 at the cathode captured during the electrolysis. | ||
In summary, the use of TCOs, especially FTO, as an efficient electrode for the IOR is demonstrated. The IOR is proven to be a potential alternative to the OER. Impressively, FTO electrodes exhibited high activity for the IOR with a FE value of 98%. The integration of the IOR with the HER in hybrid water electrolysis enables the realization of efficient H2 production at less input voltage, when FTO is used as the anode and Pt is used as the cathode. The hybrid water electrolysis cell (FTO||Pt-wire) delivered 10 mA cm−2 at a cell voltage of only 1.15 V with 91% FE for H2 production.
SP has carried out the experimental work and the data analysis. KV conceived the idea, procured funding, and monitored the execution and data analysis of the project. Shivam Nikam, undergraduate student is acknowledged for initial assistance. The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.
KV acknowledges BITS Pilani for the partial support under the CDRF scheme (SC/07/23/108). SP acknowledges BITS Pilani, K K Birla Goa campus for the research fellowship. We are grateful to the Central Sophisticated Instrumentation Facility (CSIF), BITS Pilani, K K Birla Goa campus for FESEM, XRD and Raman facilities and the Central Instrumentation Facility, BITS Pilani, Pilani campus for the XPS facility.
Data availability
Data are available upon request to the corresponding author.Conflicts of interest
There are no conflicts to declare.Notes and references
- I. Staffell, D. Scamman, A. Velazquez Abad, P. Balcombe, P. E. Dodds, P. Ekins, N. Shah and K. R. Ward, Energy Environ. Sci., 2019, 12, 463–491 RSC
.
-
(a) J. H. Kim, D. Hansora, P. Sharma, J. W. Jang and J. S. Lee, Chem. Soc. Rev., 2019, 48, 1908–1971 RSC
- B. You and Y. Sun, Acc. Chem. Res., 2018, 51, 1571–1580 CrossRef CAS PubMed
- J. Li, L. Li, X. Ma, X. Han, C. Xing, X. Qi, R. He, J. Arbiol, H. Pan, J. Zhao, J. Deng, Y. Zhang, Y. Yang and A. Cabot, Adv. Sci., 2023, 10, 2300841 CrossRef CAS PubMed
-
(a) L. Zhu, J. Huang, G. Meng, T. Wu, C. Chen, H. Tian, Y. Chen, F. Kong, Z. Chang, X. Cui and J. Shi, Nat. Commun., 2023, 14, 1997 CrossRef CAS PubMed
- Y. Wang, L. Chen, H. Zhang, M. Humayun, J. Duan, X. Xu, Y. Fu, M. Boudoudina and C. Wang, Green Chem., 2023, 25, 8181–8195 RSC
- I. Mondal, P. V. Menezes, K. Laun, T. Diemant, M. Al-Shakran, I. Zebger, T. Jacob, M. Driess and P. W. Menezes, ACS Nano, 2023, 17, 14043–14052 CrossRef CAS PubMed
- T. Wei, W. Liu, S. Zhang, Q. Liu, J. Luo and X. Liu, Chem. Commun., 2023, 59, 442–445 RSC
-
(a) E. A. Moges, C.-Y. Chang, M.-C. Tsai, W.-N. Su and B. J. Hwang, EES Catal., 2023, 1, 413–433 RSC
-
(a) D. B. Adam, M. C. Tsai, Y. A. Awoke, W. H. Huang, Y. W. Yang, C. W. Pao, W. N. Su and B. J. Hwang, ACS Sustainable Chem. Eng., 2021, 9, 8803–8812 CrossRef CAS
- L. Yao, Y. P. Liu, H. H. Cho, M. Xia, A. Sekar, B. P. Darwich, R. A. Wells, J. H. Yum, D. Ren, M. Gratzel, N. Guijarro and K. Sivula, Energy Environ. Sci., 2021, 14, 3141–3151 RSC
- P. P. Edwards, A. Porch, M. O. Jones, D. V. Morgan and R. M. Perks, Dalton Trans., 2004, 2995–3002 RSC
- S. Geiger, O. Kasian, A. M. Mingers, K. J. J. Mayrhofer and S. Cherevko, Sci. Rep., 2017, 7, 4595 CrossRef PubMed
- J. M. Kim, Y. J. Lee, S. Kim, K. H. Chae, K. R. Yoon, K. A. Lee, A. Byeon, Y. S. Kang, H. Y. Park, M. K. Cho, H. C. Ham and J. Y. Kim, Nano Energy, 2019, 65, 104008 CrossRef CAS
- A. Herman, J. L. Mathias and R. Neumann, ACS Catal., 2022, 12, 4149–4155 CrossRef CAS
- G. Gobel, A. Talke and F. Lisdat, Electroanalysis, 2018, 30, 225–229 CrossRef
- J. D. Benck, B. A. Pinaud, Y. Gorlin and T. F. Jaramillo, PLoS One, 2014, 9, e107942 CrossRef PubMed
- L. Cojocaru, C. Olivier, T. Toupance, E. Sellier and L. Hirsch, J. Mater. Chem. A, 2013, 1, 13789 RSC
- A. Korjenic and K. S. Raja, J. Electrochem. Soc., 2019, 166, C169–C184 CrossRef CAS
- S. Bai, L. Chen, J. Bai, C. Lv, S. Xu, D. Zhang, H. Meng, C. Guo, H. Yang and C. Shang, Inorg. Chem. Front., 2023, 10, 4695–4701 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc01717j |
| This journal is © The Royal Society of Chemistry 2024 |