
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mixing and matching N,N- and N,O-chelates in anionic Mg(I) compounds: synthesis and reactivity with RN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) CNR and CO†
CNR and CO†
Andrea
O'Reilly
 a,
Matthew D.
Haynes
b,
Zoë R.
Turner
b,
Claire L.
McMullin
*c,
Sjoerd
Harder
d,
Dermot
O'Hare
*b,
J. Robin
Fulton
*a and
Martyn P.
Coles
*a
a,
Matthew D.
Haynes
b,
Zoë R.
Turner
b,
Claire L.
McMullin
*c,
Sjoerd
Harder
d,
Dermot
O'Hare
*b,
J. Robin
Fulton
*a and
Martyn P.
Coles
*a
aSchool of Chemical and Physical Sciences, Victoria University of Wellington, PO Box 600, Wellington 6012, New Zealand. E-mail: martyn.coles@vuw.ac.nz; j.robin.fulton@vuw.ac.nz
bChemistry Research Laboratory, Department of Chemistry, University of Oxford, Mansfield Road, Oxford OX1 3TA, UK. E-mail: dermot.ohare@chem.ox.ac.uk
cDepartment of Chemistry, University of Bath, Bath, BA2 7AY, UK. E-mail: cm2025@bath.ac.uk
dInorganic and Organometallic Chemistry, Friedrich-Alexander-Universität, Erlangen-Nürnberg, Egerlandstraße 1, 91058 Erlangen, Germany
First published on 20th June 2024
Abstract
Reduction of [Mg(NON)]2 ([NON]2− = [O(SiMe2NDipp)2]2−, Dipp = 2,6-iPr2C6H3) affords Mg(I) species containing NON- and NNO-ligands ([NNO]2− = [N(Dipp)SiMe2N(Dipp)SiMe2O]2−). The products of reactions with iPrN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CNiPr and CO are consistent with the presence of reducing Mg(I) centres. Extraction with THF affords [K(THF)2]2[(NNO)Mg–Mg(NNO)] with a structurally characterised Mg–Mg bond that was examined using density functional theory.
CNiPr and CO are consistent with the presence of reducing Mg(I) centres. Extraction with THF affords [K(THF)2]2[(NNO)Mg–Mg(NNO)] with a structurally characterised Mg–Mg bond that was examined using density functional theory.
Since the initial report of (BDI)Mg–Mg(BDI) in 2007 (I, Fig. 1; BDI = [HC{C(Me)NDipp}2]−),1 Mg(I) reagents have demonstrated their capacity to act as soluble, electron precise reducing agents.2 The discovery of these compounds has undoubtedly fuelled the recent development and application of low valent group 2 (alkaline earth, Ae) metal complexes.3 Indeed, over 35 examples of neutral Mg(I) species (L)Mg–Mg(L) (L = monoanionic ligand) have now been structurally characterised. In contrast, examples in which the low valent magnesium centres are present in an anionic [(L′)Mg–Mg(L′)]2− unit (L′ = dianionic ligand) are a recent addition to this important family of compounds (Fig. 1).
 | ||
| Fig. 1 Examples of neutral (I) and anionic (II, III and IV) Mg(I) compounds. | ||
The first anionic Mg(I) compounds II were synthesised in a one-pot reaction of the neutral diimine or diamine with MgCl2 in the presence of excess potassium.4 The products have a coplanar [(L′)Mg–Mg(L′)]2− unit with THF-solvated potassium cations above and below the MgN2C2 metallacycles. In 2021, Hill and co-workers isolated the dimeric Na/Mg complex III by reducing the neutral diamido magnesium precursor with 5% Na/NaCl.5 The structure showed non-solvated sodium cations with Na⋯π(arene) interactions to ligand substituents. The [(L′)Mg–Mg(L′)]2− unit is twisted, with a long Mg–Mg bond. The most recent addition to this family IV, derives from the steric modulation of a rigid [xanth-EtNONAr]2− backbone ([xanth-EtNONAr]2− = 4,5-Ar2-2,7-Et2-9,9-Me2-xanthene). Previous work had shown that when Ar = 2,4,6-Cy3C6H2, the dinitrogen complex [K{Mg(xanth-EtNONAr)}]2(μ-N2) was isolated,6 presumed to be due to the ligand bulk preventing Mg–Mg bond formation and the resulting Mg(I) radicals reducing N2. Reducing the size of the Ar substituents to 2,4,6-iPr3C6H2 allowed Mg–Mg bond formation in IV.7 The solid-state structure of IV adopts a folded structure with the potassium atoms coordinated via η4-OCN2 and η6-aryl interactions to the ligands.
The reactivity of anionic Mg(I) compounds with a range of small molecules (H2, CO, N2O, THF),5,7,8 and organic functional groups (alkynes, nitriles, carbodiimides, ketones, polyaromatic compounds)9 has been explored, confirming the reductive capability of these species. We report herein the synthesis of a magnesium compound supported by the NON-ligand ([NON]2− = [O(SiMe2NDipp)2]2−), its reduction to Mg(I) and the reactivity of the low-valent species with iPrNCNiPr and CO.
Prior to this work, Mg(NON)(THF)2 was the only reported group 2 metal NON-compound, isolated as a product of over reduction and ligand transfer during the synthesis of a Bi(II) radical.10 In this work the non-solvated compound was targeted as a precursor to reduced Mg(I) species, using an alkane elimination route between MgBu2 and (NON)H2 (Scheme 1). The reaction proceeded smoothly in hexane or toluene, affording colourless crystals of [Mg(NON)]2 (1).
 | ||
| Scheme 1 Synthesis and thermal displacement plot (30% probability; H-atoms omitted; peripheral carbons as spheres) of 1 (′ = −x, 1 − y, −z;). Selected bond lengths (Å) and angles (°): Mg–N1 1.9414(10), Mg–O 2.0605(9), Mg–N2′ 1.9542(10), Mg⋯Mg′ 4.102(1); N1–Mg–O 77.54(4), N1–Mg–N2′ 143.74(5), O–Mg–N2′ 138.48(4). | ||
Compound 1 is sparingly soluble in hydrocarbon solvents, preventing the acquisition of meaningful spectroscopic data at room temperature.11 The crystal structure revealed a dimer located on an inversion centre (Scheme 1). The bidentate ligand adopts a κN1,O-μ-N2-coordination mode in which the oxygen atom is in a four-membered N–Si–O–Mg metallacycle and the pendant N2 atom bonds to the symmetry related Mg. We note that the Mg⋯Mg separation of 4.102(1) Å is too great to support a Mg–Mg bond in this dimeric arrangement, necessitating a reorganisation of the ligand to permit formation of the target [(L′)Mg–Mg(L′)]2− unit.
The reduction of 1 by KC8 in Et2O reproducibly forms a new product A (Scheme 2). 1H NMR analysis indicated two distinct sets of ligand resonances in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio that are consistent with a symmetric and a non-symmetric environment at magnesium. This is most clear from the three SiMe2 singlets that appear at δH 0.38 (12H), 0.04 (6H) and −0.04 (6H). The symmetric environment at magnesium is consistent with a Mg(NON) group with a κN,N′- or κN,O,N′-coordination mode of the NON-ligand.10 Based on NMR spectra of a related aluminium system, we attribute the non-symmetric ligand environment to a previously reported intramolecular 1,3-silyl retro-Brook rearrangement of the NON-ligand to form an N,O-chelated Mg(NNO) group ([NNO]2− = [N(Dipp)SiMe2N(Dipp)SiMe2O]2−).12 We are unable to crystallise A and cannot therefore assign it a precise chemical structure. Furthermore, analytical data does not allow us to discriminate between a discrete heteroleptic [(NON)Mg–Mg(NNO)]2− unit or a mixture that also includes homoleptic [(NON)Mg–Mg(NON)]2− and [(NNO)Mg–Mg(NNO)]2− species. It is possible that a dynamic equilibrium exists, hindering crystallisation of a single species. However, onward reactivity confirms the presence of both K[Mg(NON)] and K[Mg(NNO)] groups in A.
:1 ratio that are consistent with a symmetric and a non-symmetric environment at magnesium. This is most clear from the three SiMe2 singlets that appear at δH 0.38 (12H), 0.04 (6H) and −0.04 (6H). The symmetric environment at magnesium is consistent with a Mg(NON) group with a κN,N′- or κN,O,N′-coordination mode of the NON-ligand.10 Based on NMR spectra of a related aluminium system, we attribute the non-symmetric ligand environment to a previously reported intramolecular 1,3-silyl retro-Brook rearrangement of the NON-ligand to form an N,O-chelated Mg(NNO) group ([NNO]2− = [N(Dipp)SiMe2N(Dipp)SiMe2O]2−).12 We are unable to crystallise A and cannot therefore assign it a precise chemical structure. Furthermore, analytical data does not allow us to discriminate between a discrete heteroleptic [(NON)Mg–Mg(NNO)]2− unit or a mixture that also includes homoleptic [(NON)Mg–Mg(NON)]2− and [(NNO)Mg–Mg(NNO)]2− species. It is possible that a dynamic equilibrium exists, hindering crystallisation of a single species. However, onward reactivity confirms the presence of both K[Mg(NON)] and K[Mg(NNO)] groups in A.
 | ||
| Scheme 2 Formation of A and reaction with iPrNCNiPr, CO and THF to afford 2, 3·Et2O and 4·THF (isolated yields reported). | ||
Evidence for A reacting as a discrete heteroleptic [(NON)Mg–Mg(NNO)]2− unit is inferred from its reaction with diisopropylcarbodiimide to form 2 (Scheme 2). Previous studies with I and II-a showed that addition of RNCNR (R = Cy, tBu) afforded the magnesioamidinate species from the reductive insertion of carbodiimide into a Mg–Mg bond.1,9c The proposed mechanism involves initial N-coordination of the carbodiimide to one Mg centre, followed by migration of the second Mg to the sp2 carbon of the carbodiimide.1,13
NMR spectra of 2 retain a 1:1 ratio of NON- and NNO-ligand resonances, with additional signals for magnetically equivalent NiPr groups, and a high frequency resonance at δC 226.1 for the CN2 carbon atom. The crystal structure reveals both Mg(NON) and Mg(NNO) groups bridged by a [C(NiPr)2]2− unit (Fig. 2). The C–N distances in the amidinate unit (1.344(3)–1.358(3) Å) indicate delocalisation in the CN2 fragment, confirming a two-electron reduction of the carbodiimide. The regiochemistry in 2 is consistent with N-coordination of the carbodiimide at the less sterically encumbered Mg(NNO) centre, followed by migration of the Mg(NON) fragment to the carbon atom.12a
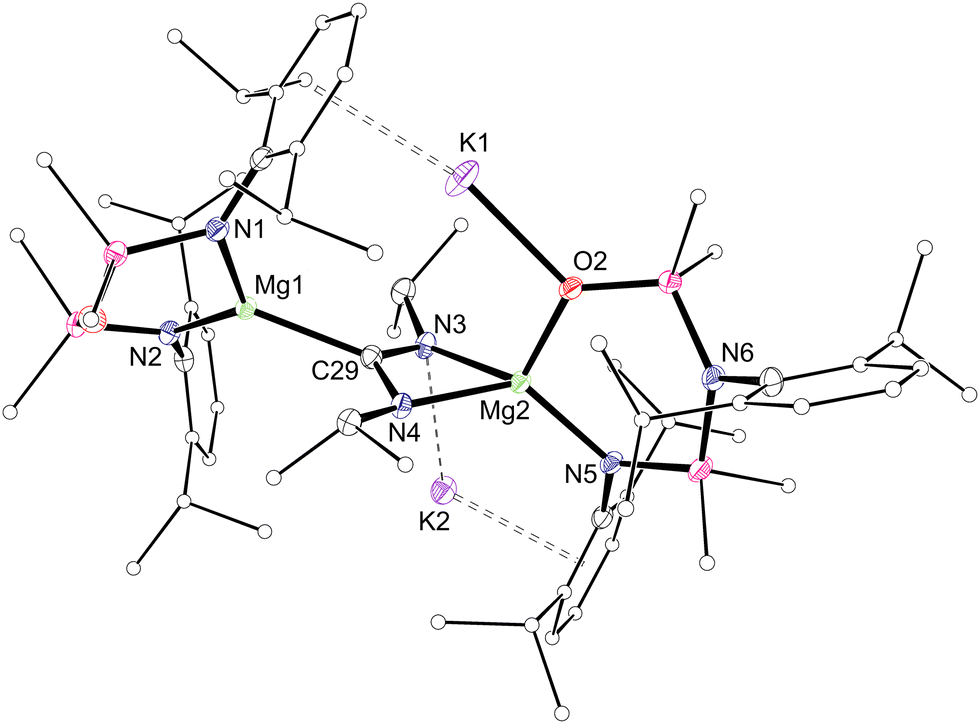 | ||
| Fig. 2 Thermal displacement plot (30% probability; H-atoms omitted; peripheral carbons as spheres) of one of the independent molecules of 2. Selected bond lengths (Å) and angles (°): Mg1–N1 2.0594(19), Mg1–N2 2.0415(19), Mg2–N5 2.0513(19), Mg2–O2 1.9122(16), C29–N3 1.358(3), C29–N4 1.344(3), Mg1–C29 2.232(2), Mg2–N3 2.1737(19), Mg2–N4 2.069(2); N1–Mg1–N2 123.18(8), N5–Mg2–O2 106.09(8), N3–Mg2–N4 63.51(7). | ||
Compounds III, IV and the aforementioned dimagnesium(μ-N2) complex reductively dimerise CO to afford ethynediolate complexes.5–7 The reaction of A with 1 bar CO proceeded with an immediate colour change from yellow to red-orange, affording a low yield of crystals of 3·Et2O (Scheme 2). The 1H NMR spectrum showed only symmetric ligand environments, consistent with Mg(NON) fragments. The 13C{1H} NMR spectrum showed a signal at δC 76.6, downfield of the [C2O2]2− resonances noted in previous ethynediolate products (δC 50.2/55.3).
X-ray diffraction analysis of 3·Et2O confirmed formation of the homoleptic ethynediolate complex, K2[{Mg(NON)(Et2O)}2(μ-C2O2)] containing Et2O solvated Mg(NON) groups (Fig. 3). Previous studies showing that the isomerisation of Al(NON) to Al(NNO) is exergonic12a suggest that the formation of Mg(NNO) groups during the synthesis of A is non-reversible. The absence of Mg(NNO) groups in 3·Et2O therefore suggests that its formation involves the preferential reaction of CO with the Mg(NON) components of A, and may indicate the presence of symmetrical [(NON)Mg–Mg(NON)]2− units. The structural parameters of the ‘Mg(OC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CO)Mg’ unit closely resemble the previous examples,5,6,14 with a short C–C bond of 1.211(8) Å and a slight trans-bent geometry (O2–C29–C29′ = 164.6(6)°).
CO)Mg’ unit closely resemble the previous examples,5,6,14 with a short C–C bond of 1.211(8) Å and a slight trans-bent geometry (O2–C29–C29′ = 164.6(6)°).
 | ||
| Fig. 3 Thermal displacement plot (30% probability; H-atoms omitted; peripheral carbons as spheres) of one of the independent molecules of 3·Et2O. Selected bond lengths (Å) and angles (°): Mg1–N1 2.044(4), Mg1–N2 2.049(3), Mg1–O2 1.936(3), O2–C29 1.309(5), C29–C29′ 1.211(8); N1–Mg1–N2 121.24(17), O2–C29–C29′ 164.6(6). | ||
Extraction of A with THF afforded a mixture of products including K2[NON], and the new compound 4·THF that was isolated by fractional crystallisation in 37% yield. The 1H NMR spectrum of 4·THF shows splitting according to a compound containing only NNO-ligands. This is confirmed by X-ray diffraction, revealing 4·THF as the anionic Mg(I) species, [(NNO)Mg–Mg(NNO)]2− charge balanced with two [K(THF)2]+ cations (Fig. 4). This result indicates either an analogous redistribution of the [Mg(L′)]− fragments from a mixture present in A, or isomerisation of the Mg(NON) group to the more stable Mg(NNO) isomer during the extraction with THF. There is no residual electron density in the region expected for hydride ligands, confirming formation of the anionic Mg(I) compound (Fig. S24, ESI†). The Mg–Mg bond length (2.9393(11) Å) is in the range reported for (solvent free) (L)Mg–Mg(L) compounds (2.7907(9)–3.0513(8) Å),15 and is shorter than found in the anionic Mg(I) complexes II (3.2077(10) Å/3.2124(11) Å)5 and III (3.1369(15) Å).14 We attribute the short bond to the reduction in steric stress imparted by the NNO-isomer, and the reduction in electron density at Mg that results from the high electronegativity of the O-ligand.
 | ||
| Fig. 4 Thermal displacement plot (30% probability; H-atoms omitted; peripheral carbons as spheres) of 4·THF (′ = −x, −y, 2 − z). Selected bond lengths (Å) and angles (°): Mg–Mg′ 2.9393(11), Mg–N1 2.0493(15), Mg–O1 1.9203(13), O1–K 2.5859(13); N1–Mg–O1 106.37(6), N1–Mg–Mg′ 131.70(5), O1–Mg–Mg′ 121.91(5), Mg–O1–K 109.97(6). | ||
DFT analysis of I and IV revealed a non-nuclear attractor (NNA) at the centre point of the Mg–Mg bond.7,16 This feature was not prominent in the core of III,5 which the authors suggest is best described as a [Mg2Na2]4+ unit. Investigation of 4·THF by DFT (see ESI† for details) show that the HOMO corresponds to the Mg–Mg σ-bond with equal contribution from both metals and a high s-character of ∼93% (Fig. 5a). Natural bond order (NBO) calculations show a Mg–Mg Wiberg bond index (WBI) of 0.724, with NPA charges qMg = +1.00 and qK = +0.88. These values compare well with those reported for III (WBI = 0.656, qMg = +1.03 and qNa = +0.84),5 and IV (WBI = 0.694, qMg = +0.97 and qK = +0.90).6 An NNA of 0.72 electrons is present in the middle of the Mg–Mg bond in 4·THF (Fig. 5b), similar to that noted in I (0.8 electrons).16
 | ||
| Fig. 5 (a) HOMO of 4·THF. (b) Laplacian distribution for 4·THF, showing non-nuclear attractor (NNA215) at the centre of the Mg–Mg bond. Both calculated at the BP86/6-311++G**//BP86/BS1 level of theory. | ||
In conclusion, we have demonstrated that the reduction of the [Mg(NON)]2 dimer 1 affords product A, formulated as containing K[Mg(NON)] and K[Mg(NNO)] units. Although the composition of A has not been explicitly identified and an equilibrium of different species may be present, analytical data and reactivity studies confirm the presence of reducing Mg(NON) and Mg(NNO) groups. Isolation and structural characterisation of [K(THF)2]2[(NNO)Mg–Mg(NNO)] (4⋅THF) prompted analysis by DFT, showing a non-nuclear attractor of 0.72 electrons at the centre of the Mg–Mg bond. Access to both K[Mg(NON)] and K[Mg(NNO)] groups from A may expand the scope of reactivity compared with a system in which only one ligand-type was available. This flexibility imparted by the opportunity to ‘mix and match’ [Mg(L′)]− fragments according to the reaction may be important in stabilising the reduction products and will form part of our ongoing investigations.
A. O’. R., M. P. C. and J. R. F. acknowledge funding from the Royal Society Te Apārangi (MFP-VUW2020). M. D. H. acknowledges funding from the EPSRC Centre for Doctoral Training in Inorganic Chemistry for Future Manufacturing (OxICFM), EP/S023828/1, and the RSC for a Researcher Development and Travel Grant. C. L. M. acknowledges the RSC Research Enablement Grant (E21-8355738114). The authors gratefully acknowledge the University of Bath's Research Computing Group (https://doi.org/10.15125/b6cd-s854) for their support in this work.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- S. P. Green, C. Jones and A. Stasch, Science, 2007, 318, 1754–1757 CrossRef CAS PubMed.
- (a) C. Jones, Nat. Rev. Chem., 2017, 1, 0059 CrossRef CAS; (b) A. Stasch and C. Jones, Dalton Trans., 2011, 40, 5659–5672 RSC.
- (a) J. T. Boronski, Dalton Trans., 2024, 53, 33–39 RSC; (b) L. A. Freeman, J. E. Walley and R. J. Gilliard, Nat. Synth., 2022, 1, 439–448 CrossRef; (c) K. M. Fromm, Coord. Chem. Rev., 2020, 408, 213193 CrossRef CAS.
- (a) Y. Liu, S. Li, X.-J. Yang, P. Yang and B. Wu, J. Am. Chem. Soc., 2009, 131, 4210–4211 CrossRef CAS PubMed; (b) M. Ma, H. Wang, J. Wang, L. Shen, Y. Zhao, W.-H. Xu, B. Wu and X.-J. Yang, Dalton Trans., 2019, 48, 2295–2299 RSC.
- H.-Y. Liu, R. J. Schwamm, S. E. Neale, M. S. Hill, C. L. McMullin and M. F. Mahon, J. Am. Chem. Soc., 2021, 143, 17851–17856 CrossRef CAS PubMed.
- R. Mondal, M. J. Evans, T. Rajeshkumar, L. Maron and C. Jones, Angew. Chem., Int. Ed., 2023, 62, e202308347 CrossRef CAS PubMed.
- R. Mondal, M. J. Evans, D. T. Nguyen, T. Rajeshkumar, L. Maron and C. Jones, Chem. Commun., 2024, 60, 1016–1019 RSC.
- (a) H.-Y. Liu, S. E. Neale, M. S. Hill, M. F. Mahon, C. L. McMullin and B. L. Morrison, Chem. Commun., 2023, 59, 3846–3849 RSC; (b) H.-Y. Liu, S. E. Neale, M. S. Hill, M. F. Mahon, C. L. McMullin and E. Richards, Angew. Chem., Int. Ed., 2023, 62, e202213670 CrossRef CAS PubMed.
- (a) L. Yang, Y. Zhang, Y. Zhao and X.-J. Yang, Polyhedron, 2023, 244, 116632 CrossRef CAS; (b) J. Wang, J. Wang, L. Shen, Y. Zhao, B. Wu and X.-J. Yang, Organometallics, 2019, 38, 2674–2682 CrossRef CAS; (c) L. Yang, Y. Qu, J. Wang, W. Xu, Y. Zhao and X.-J. Yang, Chem. – Eur. J., 2023, 29, e202301266 CrossRef CAS PubMed; (d) H.-Y. Liu, S. E. Neale, M. S. Hill, M. F. Mahon, C. L. McMullin and E. Richards, Organometallics, 2024, 43, 879–888 CrossRef CAS PubMed.
- R. J. Schwamm, J. R. Harmer, M. Lein, C. M. Fitchett, S. Granville and M. P. Coles, Angew. Chem., Int. Ed., 2015, 54, 10630–10633 CrossRef CAS PubMed.
- 1H and 13C{1H} NMR spectra recorded at 373 K show a symmetric ligand environment (Fig. S3 and S4, ESI†). The addition of THF to aid solubilisation forms a partially solvated dimer. Further details and related studies with heavier group 2 NON-compounds will be the subject of a forthcoming publication.
- (a) A. O’Reilly, M. G. Gardiner, C. L. McMullin, J. R. Fulton and M. P. Coles, Chem. Commun., 2024, 60, 881–884 RSC; (b) F. Haftbaradaran, G. Mund, R. J. Batchelor, J. F. Britten and D. B. Leznoff, Dalton Trans., 2005, 2343–2345 RSC; (c) F. Haftbaradaran, A. M. Kuchison, M. J. Katz, G. Schatte and D. B. Leznoff, Inorg. Chem., 2008, 47, 812–822 CrossRef CAS PubMed.
- L. Xiao, W. Chen, L. Shen, L. Liu, Y. Xue, Y. Zhao and X.-J. Yang, Chem. Commun., 2020, 56, 6352–6355 RSC.
- R. Mondal, K. Yuvaraj, T. Rajeshkumar, L. Maron and C. Jones, Chem. Commun., 2022, 58, 12665–12668 RSC.
- (a) X. Cao, J. Li, A. Zhu, F. Su, W. Yao, F. Xue and M. Ma, Org. Chem. Front., 2020, 7, 3625–3632 RSC; (b) T. X. Gentner, B. Rösch, G. Ballmann, J. Langer, H. Elsen and S. Harder, Angew. Chem., Int. Ed., 2019, 58, 607–611 CrossRef CAS PubMed.
- J. A. Platts, J. Overgaard, C. Jones, B. B. Iversen and A. Stasch, J. Phys. Chem. A, 2011, 115, 194–200 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and characterization data; full details of computational experiments. CCDC 2357068 (1), 2357069 (2), 2357070 (3·Et2O) and 2357071 (4·THF). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc02594f |
| This journal is © The Royal Society of Chemistry 2024 |