
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Asymmetric synthesis of enantioenriched α-allyl esters through Pd(BINAPHANE)-catalysed allylation of disubstituted ketenes†
Ahmad A.
Ibrahim
b,
Stephen C. J.
O’Reilly
a,
Margot
Bottarel
a and
Nessan J.
Kerrigan
 *a
*a
aSchool of Chemical Sciences and Life Sciences Institute, Dublin City University, Glasnevin, Dublin 9, Ireland. E-mail: nessan.kerrigan@dcu.ie
bDepartment of Chemistry, Oakland University, 2200 N. Squirrel Rd, Rochester, MI 48309, USA
First published on 26th February 2024
Abstract
Pd2dba3·CHCl3 (2.5 mol%)-BINAPHANE (5 mol%) was used to promote the first catalytic enantioselective allylation of disubstituted ketenes to give α-allyl esters. The ester products were formed in good to excellent yields (61–93% yield for 13 examples, 16 examples in all), with moderate to good enantioselectivity (68–80% ee for 7 examples).
One of the most important methods for the asymmetric synthesis of carbonyl compounds possessing an α-stereogenic centre is the transition metal-catalysed asymmetric allylic alkylation of enolates or related nucleophiles (AAA reaction).1 Stabilised ‘soft’ enolates have been found to be most successful as nucleophiles and the reaction has generally performed best with pronucleophiles such as cyclic β-ketoesters, rather than simple acyclic substrates (esters, acids, ketones, aldehydes, ketenes).1 A new catalytic allylation method for such substrates would provide a powerful tool for C–C bond formation. Recently, Carreira's group and Dong's group independently demonstrated the stereodivergent synthesis of α,β-substituted chiral aldehydes through dual catalysis involving chiral Ir(I) complex/chiral amine or chiral Rh(I) complex/chiral amine-catalysed reaction of racemic aldehydes with allylic alcohols and internal alkynes respectively.2,3 Zhang's group and List's group also independently showed that in situ-generated enamines could be used as an alternative to enolates for enantioselective allylations of ketones and aldehydes.4 Snaddon and co-workers reported the α-allylation of aryl acetic acid esters to form α-allylated esters bearing a tertiary chiral centre using cooperative Pd(0)/isothiourea catalysis, while Hartwig reported a chiral Ir(I)/isothiourea-catalysed stereodivergent variant of the latter reaction to form α,β-substituted chiral esters.5,6 In 2017, Ding and co-workers communicated the asymmetric allylic alkylation of ketoesters to give products bearing vicinal tertiary and quaternary chiral centres.7 However the latter method was restricted to cyclic β-ketoesters as substrates.
Previously, in 1986, Watanabe's group had reported the non-enantioselective α-allylation of disubstituted ketenes to afford dienes or allylated esters, often obtained as mixtures of products or in low yields.8 Recently our group, as part of a program of studies on the development of new reactions of ketenes, reported the Pd(0)-catalysed stereospecific reaction of enantioenriched vinyl cyclopropanes with ketenes to provide access to enantioenriched tetrahydrofurans.9 However, there has been no report of a direct catalytic enantioselective allylation of ketene-derived enolates to give α-allylated esters bearing an α-quaternary stereogenic centre.10 In this communication we describe our initial results toward that goal.
We began our studies by reinvestigating the work of Watanabe's group using Pd(PPh3)4 as catalyst to promote the reaction of ethylphenylketene with allyl carbonate or allyl acetate in THF. Interestingly, in our hands only α-allylated carbonyl products (e.g.3aa and 3b) were formed, with no diene 4, in contrast to Watanabe's observations in THF (Scheme 1).8
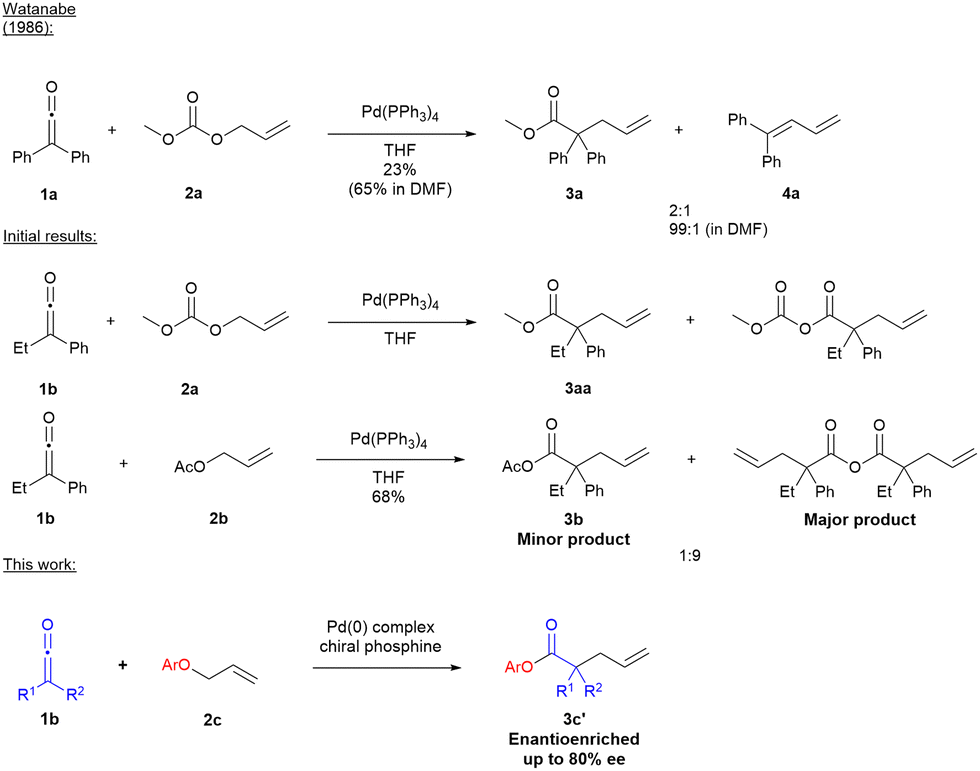 | ||
| Scheme 1 Precedent and initial results. | ||
We proposed that an aryl oxide (leaving group/counterion) generated during Pd(II)-π-allyl 5 formation would act as a better nucleophile than acetate or carbonate and add to the ketene to form an ester enolate 6 in stereoselective fashion (Scheme 2). We anticipated that ketene dimerisation would be minimised under the reaction conditions where the putative ester enolate would be stabilised by the Pd(II)-π-allyl species (perhaps through inner sphere coordination of enolate to Pd).9 The ester enolate 6 would then undergo C-allylation by the associated Pd(II)-π-allyl species to provide the desired allyl ester product 3 (as the linear regioisomer) along with simultaneous Pd(0) regeneration (Scheme 2). The use of chiral phosphine ligands on Pd would be expected to control enantioselectivity in formation of the new α-quaternary stereogenic centre in 3.
 | ||
| Scheme 2 Proposed reaction mechanism. | ||
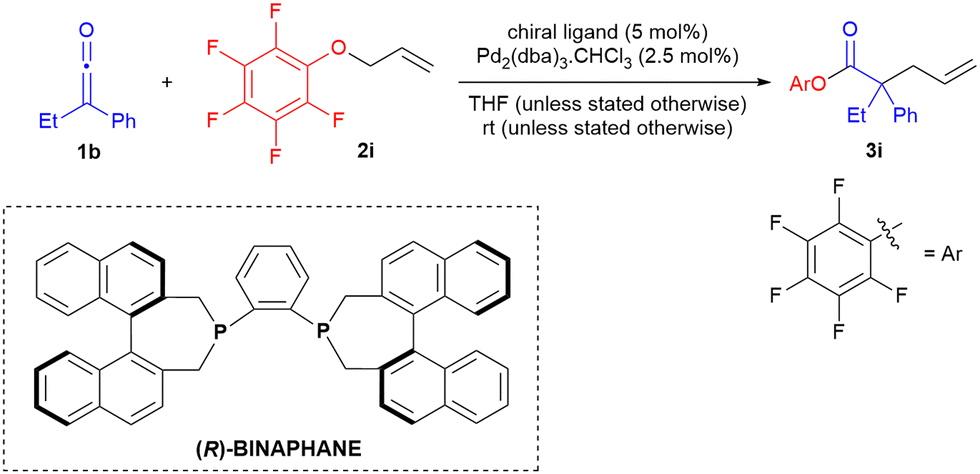
Inspired by Lectka's work on α-halogenation of ketenes where an aryl oxide was used in a rebound mechanism to regenerate nucleophilic catalyst for the catalytic cycle, we examined a number of allyl aryl ethers bearing electron withdrawing substituents on the aryl moiety.11 We determined that allyl pentafluorophenoxide gave best results in terms of yield of 3 and ee when Pd2(dba)3·CHCl3-BINAPHANE was used as catalyst (Table 1, entry 6). To our delight, ketene dimerisation was not found to proceed to any great extent. The lack of ketene dimer implied that phosphonium enolate formation, formed through addition of dissociated BINAPHANE ligand to ketene, was not a significant reaction pathway (we have previously demonstrated that BINAPHANE is an excellent catalyst for ketene dimerisation and related cycloadditions).12

With the desired allyl ester product being favoured, we examined, in parallel, other chiral phosphines in combination with Pd2(dba)3·CHCl3 in order to attempt to enhance enantioselectivity in the reaction. However, surprisingly, BINAPHANE (and BINAPINE) was found to be virtually unique in providing enantioselectivity of >30% ee, as 26 other chiral ligands in our study were found to provide ee generally <10% (Table 2, see ESI† for full details). Axial chirality associated with the phosphepine skeleton appeared to be essential for enantioselectivity, with the only ligands providing ee of ≥20% being phosphepines (Table 2 entries 8–10). Decreasing temperature of the Pd-BINAPHANE catalysed reaction did not have any benefit on enantioselectivity, with reaction efficiency greatly reduced at −78 °C (Table 2, entry 12). THF proved to be the optimal reaction solvent, as employment of CH2Cl2 and toluene led to slightly lower yield and ee, and in DMF the reaction was found not to favour allylated ester at all, highlighting the sensitivity of the ketene allylation to reaction conditions. Interestingly, Watanabe's group had found DMF to be a good solvent for the Pd(PPh3)4-catalysed allylation of diphenylketene with allyl phenyl ether.8 When we investigated allyl phenyl ether as substrate, under the conditions of Table 2 entry 8, a complex mixture resulted.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Chiral ligand | Solvent | Temp. | Yielda [%] | eeb [%] |
| a Isolated yield after flash column chromatography through silica gel or iatrobeads. b ee for 3i determined by chiral HPLC analysis (OD-H) or by derivatisation with (S)-α-methylbenzylamine and GC-MS analysis (ratio of diastereomers to determine ee). | |||||
| 1 | (R)-BINAP | THF | rt | 98 | 4 |
| 2 | (S,S)-DACH naphthyl-Trost | THF | rt | 93 | 5 |
| 3 | (R,R)-ANDEN phenyl-Trost | THF | −25 °C | 73 | 5 |
| 4 | (1R,1′R,2S,2′S)-DUANPHOS | THF | rt | 80 | 3 |
| 5 | (R,R)-DIPAMP | THF | rt | 71 | 8 |
| 6 | (R)-Cl-MeO-BIPHEP | THF | rt | 96 | 7 |
| 7 | (R)-PHANEPHOS | THF | rt | 88 | 7 |
| 8 | (R)-BINAPHANE | THF | rt | 90 | 34 |
| 9 | (S)-f-BINAPHANE | THF | rt | 79 | −27 |
| 10 | (S)-BINAPINE | THF | rt | 86 | −34 |
| 11 | (R)-BINAPHANE | THF | −25 °C | 75 | 37 |
| 12 | (R)-BINAPHANE | THF | −78 °C | 13 | −35 |
| 13 | (R)-BINAPHANE | CH2Cl2 | rt | 72 | 32 |
| 14 | (R)-BINAPHANE | Toluene | rt | 70 | 32 |
| 15 | (R)-BINAPHANE | DMF | rt | <1 | nd |
With the optimal catalytic system in hand we then proceeded to explore the substrate scope of the reaction, examining variations of the ketene structure (R1, R2 = aryl or alkyl), and of the allyl ether substitution pattern (Table 3). Most reactions proceeded, after purification, to afford the highly non-polar allyl ester products in good to excellent yields (61–93% for 13 examples) and purity, with some examples contaminated by non-polar byproducts/impurities, such as branched regioisomer (3′, Scheme 2).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | R4 | Yielda [%] | eeb [%] | 3 |
a Isolated yield after flash column chromatography through silica gel or neutral silica (iatrobeads).
b ee for 3 determined by chiral HPLC analysis (OD-H, AD-H and AD) or by derivatisation with (S)-α-methylbenzylamine and GC-MS analysis (ratio of diastereomers to determine ee).
c Major regioisomer = linear isomer for all examples (rs ≥8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1).
d When Pd(PPh3)4 (5 mol%) was employed: 51% yield for (±)-3i; 75% for (±)-3l; 71% for (±)-3q; 87% for (±)-3s; 77% for (±)-3w.
e Pd(PPh3)4 (5 mol%) used as catalyst. :1).
d When Pd(PPh3)4 (5 mol%) was employed: 51% yield for (±)-3i; 75% for (±)-3l; 71% for (±)-3q; 87% for (±)-3s; 77% for (±)-3w.
e Pd(PPh3)4 (5 mol%) used as catalyst.
|
|||||||
| 1 | Et | Ph | H | H | 90d | 34 | 3i |
| 2 | Et | Ph | H | Ph | 82 | 70 | 3j |
| 3 | Et | Ph | H | 4-NO2C6H4 | 89 | 75 | 3k |
| 4d | Me | Ph | H | H | 65 | 34 | 3l |
| 5 | Me | Ph | H | Ph | 43 | 70 | 3m |
| 6 | Me | Ph | H | 4-NO2C6H4 | 85 | 80 | 3n |
| 7 | Me | Ph | H | 2-NO2C6H4 | <10 | nd | 3o |
| 8 | Me | Ph | H | CO2Me | 75 | 79 | 3p |
| 9d | Me | Ph | CO2Et | H | 68 | 44 | 3q |
| 10e | Me | Ph | Me | H | 72 | — | 3r |
| 11d | i-Bu | Ph | H | H | 54 | 41 | 3s |
| 12 | i-Bu | Ph | H | Ph | 92 | 68 | 3t |
| 13 | Me | 2-MeC6H4 | H | Ph | 77 | 14 | 3u |
| 14 | Me | 4-MeO-Naphthyl | H | Ph | 77 | 69 | 3v |
| 15d | Me | i-Pr | H | 4-NO2C6H4 | 93 | 3 | 3w |
| 16e | Me | i-Pr | H | CO2Me | 39 | — | 3x |
| 17e | Ph | Ph | H | H | 61 | — | 3y |
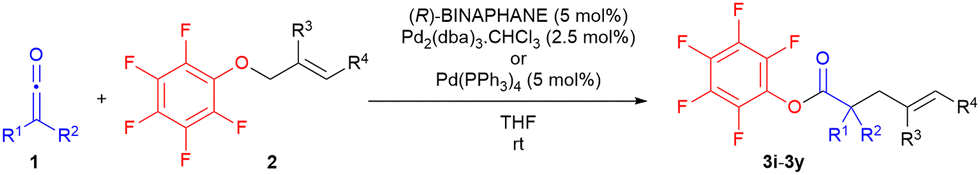
Significantly, it was noted that good to excellent regioselectivity (rs) was displayed in all cases (rs 8:1–>32:1), favouring formation of the linear, less substituted product rather than the branched, more highly substituted product, e.g. rs 24:1 for 3j; rs >32:1 for 3m; rs 10:1 for 3t; rs 32:1 for 3x. The regioselectivity was interpreted in terms of preferential nucleophilic addition of ester enolate to the less sterically hindered end of the Pd(II)-π-allyl intermediate in 6 (Scheme 2). This outcome is in agreement with previously observed trends in most regioselectivity studies of Pd(0)-catalysed allylic alkylations of enolate species.1
Gratifyingly, we generally found that with aryl-substituted allyl ethers (R4 = aryl) an increase in enantioselectivity to a good level (67–80% ee) was observed. A range of ketenes (seven in all) such as methylphenylketene, ethylphenylketene, i-butylphenylketene, methyl-i-propylketene, diphenylketene and alkylarylketenes bearing electron donating groups on the aryl ring performed well from a reactivity standpoint, albeit with quite varying effects on enantioselectivity (3–80% ee).13,14 An ortho-donating group, in particular, was found to be detrimental to enantioselectivity, with an ee of 14% (versus 70% ee for no ortho-substituent, Table 3 entry 13 vs. entry 5) being obtained, and this was ascribed to the increase in steric bulk associated with the ortho-substituent leading to a mixture of enolate isomers from reversible addition of ArO− to the ketene. The greatest influence on enantioselectivity was noted for the presence of an aryl substituent (R4 = Ph) on the β-allylic carbon of the allyl aryl ether (Table 3 entry 2 vs. entry 1 or entry 4 vs. entry 5), with an increase in ee of >30% compared to the unsubstituted case (R4 = H). The presence of a para-electron withdrawing group (e.g. NO2) on the R4 aryl group led to a further 5–10% increase in enantioselectivity (e.g.Table 3, entry 6 vs. entry 5). On the other hand, an ortho-electron withdrawing group on the R4 aryl substituent effectively shut down the reaction (Table 3, entry 7).
The absolute stereochemistry of 3l was determined to be (R) by comparison of specific rotation value for the derived alcohol 7 with the literature value reported by Kanai and co-workers.5d The absolute configuration for all other examples were assigned to be (R) by analogy (Scheme 3).
 | ||
| Scheme 3 Synthetic utility of ester products: elaboration to chiral alcohol. | ||
In conclusion, we have developed a Pd(BINAPHANE)-catalysed enantioselective synthesis of allyl aryl esters, bearing an α-quaternary centre, from allyl aryl ethers and disubstituted ketenes in good to excellent yields (up to 93%) and with moderate to good enantioselectivity (up to 80% ee). Future studies will seek to improve enantioselectivity through cooperative chiral Lewis base-Pd(L*) catalysis and to explore the scope of the reaction with respect to in situ generated ketenes.5a,13e
Support has been provided by the RSC Research Enablement Grant (E22-5472593338) to N.J.K. Open access funding provided by IReL.
Conflicts of interest
There are no conflicts to declare.Notes and references
- General Pd-catalysed allylic alkylation references: (a) O. Pamies, J. Margalef, S. Canellas, J. James, E. Judge, P. J. Guiry, C. Moberg, J.-E. Backvall, A. Pfaltz, M. A. Pericas and M. Dieguez, Chem. Rev., 2021, 121, 4373 CrossRef CAS PubMed; (b) A. Y. Hong and B. M. Stoltz, Eur. J. Org. Chem., 2013, 2745 CrossRef CAS PubMed; (c) J. D. Weaver, A. Recio, III, A. J. Grenning and J. A. Tunge, Chem. Rev., 2011, 111, 1846 CrossRef CAS PubMed; (d) B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921 CrossRef CAS PubMed; (e) B. M. Trost and D. L. Van Vranken, Chem. Rev., 1996, 96, 395 CrossRef CAS PubMed.
- S. Krautwald, D. Sarlah, M. A. Schafroth and E. M. Carreira, Science, 2013, 340, 1065 CrossRef CAS PubMed.
- F. A. Cruz and V. M. Dong, J. Am. Chem. Soc., 2017, 139, 1029 CrossRef CAS PubMed.
- (a) X. Zhao, D. Liu, H. Guo, Y. Liu and W. Zhang, J. Am. Chem. Soc., 2011, 133, 19354 CrossRef CAS PubMed; (b) G. Jiang and B. List, Angew. Chem., Int. Ed., 2011, 50, 9471 CrossRef CAS PubMed.
- Catalytic enantioselective allylation of esters and acids: (a) K. J. Schwarz, J. L. Amos, J. C. Klein, D. T. Do and T. N. Snaddon, J. Am. Chem. Soc., 2016, 138, 5214 CrossRef CAS PubMed; (b) H.-C. Lin, G. J. Knox, C. M. Pearson, C. Yang, V. Carta and T. N. Snaddon, Angew. Chem., Int. Ed., 2022, 61, e202201753 CrossRef CAS PubMed; (c) R. Visse, M.-A. Mollemann and M. Braun, Eur. J. Org. Chem., 2019, 4604 CrossRef CAS; (d) T. Fujita, T. Yamamoto, Y. Morita, H. Chen, Y. Shimizu and M. Kanai, J. Am. Chem. Soc., 2018, 140, 5899 CrossRef CAS PubMed; (e) M. Braun, P. Meletis and R. Visse, Adv. Synth. Catal., 2011, 353, 3380 CrossRef CAS.
- X. Jiang, J. J. Beiger and J. F. Hartwig, J. Am. Chem. Soc., 2017, 139, 87 CrossRef CAS PubMed.
- J. Liu, Z. Han, X. Wang, F. Meng, Z. Wang and K. Ding, Angew. Chem., Int. Ed., 2017, 56, 5050 CrossRef CAS PubMed.
- (a) T.-a Mitsudo, M. Kadokura and Y. Watanabe, J. Chem. Soc., Chem. Commun., 1986, 1539 RSC; (b) T.-a Mitsudo, M. Kadokura and Y. Watanabe, J. Org. Chem., 1987, 52, 1695 CrossRef CAS.
- (a) M. Mondal, M. Panda, V. McKee and N. J. Kerrigan, J. Org. Chem., 2019, 84, 11983 CrossRef CAS PubMed; (b) M. Mondal, M. Panda, N. W. Davis, V. McKee and N. J. Kerrigan, Chem. Commun., 2019, 55, 13558 RSC; (c) M. Mondal, S. Mitra, D. J. Twardy, M. Panda, K. A. Wheeler and N. J. Kerrigan, Chem. – Eur. J., 2022, 28, e202104391 CrossRef CAS PubMed; (d) M. Mondal, S. Chen, N. Othman, K. A. Wheeler and N. J. Kerrigan, J. Org. Chem., 2015, 80, 5789 CrossRef CAS PubMed; (e) S. Chen, A. A. Ibrahim, M. Mondal, A. J. Magee, A. J. Cruz, K. A. Wheeler and N. J. Kerrigan, Org. Lett., 2015, 17, 3248 CrossRef CAS PubMed.
- (a) A. D. Allen and T. T. Tidwell, Arkivoc, part (i), 415, 2016; (b) S. Chen, E. C. Salo and N. J. Kerrigan, in Science of Synthesis Reference Library, Asymmetric Organocatalysis, Lewis Base and Acid Catalysts, ed. B. List, Thieme, Stuttgart, 2012, vol. 1, ch. 1.1.10, p. 455 Search PubMed; (c) D. H. Paull, A. Weatherwax and T. Lectka, Tetrahedron, 2009, 65, 6771 CrossRef CAS PubMed; (d) T. T. Tidwell, Ketenes, Wiley, New York, 2nd edn, 2006 Search PubMed; (e) S. Takeuchi, N. Miyoshi and Y. Ohgo, Chem. Lett., 1992, 551 CrossRef CAS; (f) M. M. Li, Y. Wei, J. Liu, H. W. Chen, L. Q. Lu and W. J. Xiao, J. Am. Chem. Soc., 2017, 139(41), 14707 CrossRef CAS PubMed.
- (a) H. Wack, A. E. Taggi, A. M. Hafez, W. J. Drury and T. Lectka, J. Am. Chem. Soc., 2001, 123, 1531 CrossRef CAS PubMed; (b) J. Erb, D. H. Paull, L. Belding, T. Dudding and T. Lectka, J. Am. Chem. Soc., 2011, 133, 7536 CrossRef CAS PubMed.
- (a) A. A. Ibrahim, P.-H. Wei, G. D. Harzmann, D. Nalla, M. Mondal, K. A. Wheeler and N. J. Kerrigan, Tetrahedron, 2021, 78, 131838 CrossRef CAS; (b) S. Chen, M. Mondal, A. A. Ibrahim, K. A. Wheeler and N. J. Kerrigan, J. Org. Chem., 2014, 79, 4920 CrossRef CAS PubMed.
- Preparation of ketenes: (a) B. L. Hodous and G. C. Fu, J. Am. Chem. Soc., 2002, 124, 10006 CrossRef CAS PubMed; (b) S. L. Wiskur and G. C. Fu, J. Am. Chem. Soc., 2005, 127, 6176 CrossRef CAS PubMed; (c) A. D. Allen, L. M. Baigrie, L. Gong and T. T. Tidwell, Can. J. Chem., 1991, 69, 138 CrossRef CAS; (d) J. E. Wilson and G. C. Fu, Angew. Chem., Int. Ed., 2004, 43, 6358 CrossRef CAS PubMed; (e) S. Chen, A. A. Ibrahim, N. J. Peraino, D. Nalla, M. Mondal, M. Van Raaphorst and N. J. Kerrigan, J. Org. Chem., 2016, 81, 7824 CrossRef CAS PubMed; (f) J. J. Douglas, G. Churchill, A. M. Z. Slawin, D. J. Fox and A. D. Smith, Chem. – Eur. J., 2015, 21, 16354 CrossRef CAS PubMed; (g) M. Panda, M. Mondal, S. Chen, A. A. Ibrahim, D. J. Twardy and N. J. Kerrigan, Eur. J. Org. Chem., 2020, 5752 CrossRef CAS.
- Ketenes bearing electron-withdrawing groups (e.g. 4-CF3) on the aryl substituent were found to provide good reactivity (up to 64% yield), but enantioselectivity was not determined due to a lack of resolution on a variety of chiral HPLC columns .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc00057a |
| This journal is © The Royal Society of Chemistry 2024 |