
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHexacoordinated tin complexes catalyse imine hydrogenation with H2†
Andrea
Žáková
a,
Pritha
Saha
a,
Alexandros
Paparakis
a,
Martin
Zábranský
 a,
Gabriela
Gastelu
b,
Jaroslav
Kukla
c,
Jorge G.
Uranga
b and
Martin
Hulla
*a
a,
Gabriela
Gastelu
b,
Jaroslav
Kukla
c,
Jorge G.
Uranga
b and
Martin
Hulla
*a
aDepartment of Inorganic Chemistry, Faculty of Science Charles, University Prague, 128 00, Czech Republic. E-mail: martin.hulla@natur.cuni.cz
bInstituto de Investigaciones en Físico-Química Córdoba Universidad Nacional de Córdoba (INFIQC-CONICET), Córdoba, 5000, Argentina
cInstitute of Environmental Studies, Faculty of Science Charles, University Prague, 128 00, Czech Republic
First published on 16th February 2024
Abstract
Frustrated Lewis pair (FLP) hydrogenation catalysts predominantly use alkyl- and aryl-substituted Lewis acids (LA) that offer a limited number of combinations of substituents, limiting our ability to tune their properties and, ultimately, their reactivity. Nevertheless, main-group complexes have numerous ligands available for such purposes, which could enable us to broaden the range of FLP catalysis. Supporting this hypothesis, we demonstrate here that hexacoordinated tin complexes with Schiff base ligands catalyse imine hydrogenation via activation of H2(g). As shown by hydrogen–deuterium scrambling, [Sn(tBu2Salen)(OTf)2] activated H2(g) at 25 °C and 10 bar of H2. After tuning the ligands, we found that [Sn(Salen)Cl2] was the most efficient imine hydrogenation catalyst despite having the lowest activity in H2(g) activation. Moreover, various imines were hydrogenated in yields up to 98% thereby opening up opportunities for developing novel FLP hydrogenation catalysts based on hexacoordinated LA of main-group elements.
Frustrated Lewis pairs (FLPs)1 combining bulky Lewis acids (LAs) with Lewis bases (LBs) catalyse imine hydrogenation via H2 activation (Fig. 1A).2–4 Tuning their electronic and steric properties improves the FLP hydrogenation activity, expands the substrate scope5,6 and, in some cases, imparts water tolerance.7–10 Notwithstanding these outcomes, tuning predominantly involves triaryl-substituted Lewis acids8,11,12 of boron, aluminium, gallium and indium13 with a narrow margin of manoeuvre, preventing us from further enhancing their reactivity. As a case in point, some authors argue that we cannot electronically tune the BAr3 LAs any further.7
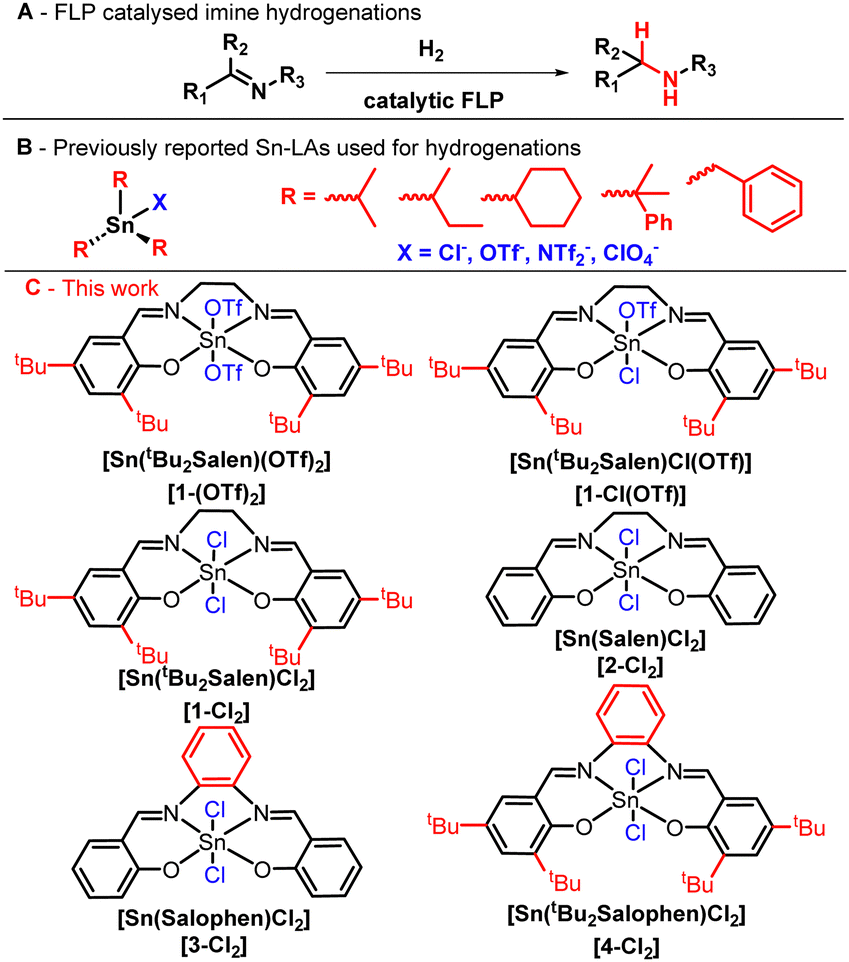 | ||
| Fig. 1 (A) Reaction scheme of FLP-catalysed imine hydrogenation, (B) previously reported Sn-based LAs used in FLP-catalysed hydrogenations, and (C) L4SnX2 catalysts developed for H2 activation and imine hydrogenations in this study. | ||
FLP reactivity has nevertheless been further modified by using group 15 and 14 LAs.14–21 In particular, R3SnX (R = alkyl or aryl, X = halogen, −OTf, −NTf2, and −ClO4) Lewis acids (Fig. 1B) in their cationic form are isolobal to group 13-based LAs,15 so they can act as their direct substitutes. In fact, tin-based FLPs effectively hydrogenate many functional groups and small molecules,14–16,22–25 but lack the diversity of metal complexes.
In addition to simple tetravalent alkyl- and aryl-substituted LAs, tin(IV) also forms hexacoordinated complexes with various ligands, including bipyridine,26 benzoylpyridine27 and Schiff bases.28 In particular, Schiff base ligands can stabilize various tin oxidation states, and their complexes are known LA catalysts.28,29 Moreover, they have been used in asymmetric catalysis,30,31 which is currently a prominent target of FLP chemistry.32,33 Despite the fact that a few main group complexes act as hydrogenation catalysts,34–36 we hypothesized that hexacoordinated tin complexes with Schiff base ligands may be applied as LAs in FLPs to catalyse imine hydrogenation.
In this study, we report that hexacoordinated complexes of tin with Schiff base ligands in the form L4SnX2, containing N and O donor atoms (Fig. 1C), activate H2 gas and act as hydrogenation catalysts for imine reduction. The presence of labile or hemi-labile axial ligands X−, such as triflate (OTf−) and chloride, promotes the formation of a vacant site on the LA metal centre, which is necessary for efficient catalysis.
Given the large positive polarization of tin(IV) and their labile triflate ligand(s), [Sn(tBu2Salen)(OTf)2] (1-(OTf)2) and [Sn(tBu2Salen)Cl(OTf)] (1-Cl(OTf)) showed high Lewis acidity, assessed using the Guttmann–Beckett (GB) method (AN = 83.6 and AN = 71.8, respectively), where 1-(OTf)2 presumably dissociates both triflate ligands and 1-Cl(OTf) dissociates the triflate but retains the chloride to form the Lewis acidic ions [Sn(tBu2Salen)]2+ and [Sn(tBu2Salen)Cl]+, respectively. Their Lewis acidity is similar to that of B(C6F5)3 (AN = 78.1). B(C6F5)3 is frequently used in FLP hydrogenations and is known to activate H2 with bases as weak as THF or dioxane.9,10,37,38 Calculation of hydride affinities (HIA) (Table 1, entries 1 and 2), following a protocol described by Greb et al., also indicates that HIA is much higher than B(C6F5)3 (HIA = 481 kJ mol−1).39 Accordingly, 1-(OTf)2 and 1-Cl(OTf) may activate H2 together with Lewis bases (LBs) comparable to FLPs based on B(C6F5)3 if a favourably oriented encounter complex forms.
| Entry | Lewis acid | ANa | Ionic formb | HIA (kJ mol−1)c |
|---|---|---|---|---|
| a Acceptor number. b Probable ionic fragments used for the calculation of hydride affinities assume dissociation of at least one ligand and all weakly coordinating anionic ligands. c Hydride ion affinities (HIA) were calculated at the DSD-PBEB86-D3BJ/def2-QZVP level. | ||||
| 1 | 1-OTf2 | 83.6 | [Sn(tBu2Salen)]2+ | 1170 |
| 2 | 1-Cl(OTf) | 71.8 | [Sn(tBu2Salen)Cl]+ | 660 |
| 3 | 1-Cl2 | 0 | [Sn(tBu2Salen)Cl]+ | 660 |
| 4 | 2-Cl2 | 0 | [Sn(Salen)Cl]+ | 688 |
| 5 | 3-Cl2 | 0 | [Sn(Salophen)Cl]+ | 701 |
| 6 | 4-Cl2 | 0 | [Sn(tBu2Salophen)Cl]+ | 682 |
As expected, 1-(OTf)2 and 1-Cl(OTf) activated H2 gas in the presence of THF, as shown by hydrogen–deuterium scrambling to H2 and D2 at 10 bar and at 25 and 60 °C, respectively (Fig. 2). Conversely, the dichloride complex [Sn(tBu2Salen)Cl2] (1-Cl2) does not possess a free binding site, at least at 25 °C, and in effect has the measured AN = 0 and the hydride affinity of 660 kJ mol−1 (Table 1, entry 3) remains masked by the Cl− ligands that must dissociate to reveal the cationic LA site. In line with Cl− coordination and the lack of binding site at low temperatures, 1-Cl2 failed to activate H2, even with additional base, 2,4,6-collidine, or DABCO, at temperatures between 25 and 60 °C. These results indicate that H2 activation requires a free binding site via a labile ligand.
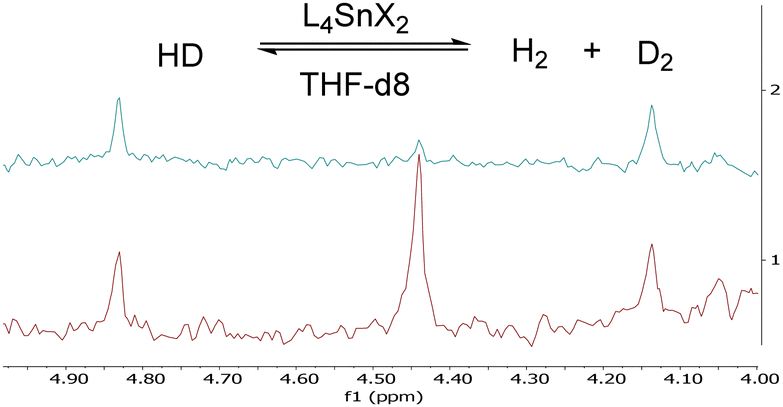 | ||
| Fig. 2 2D NMR spectra of HD scrambling by 1-(OTf)2 in THF-d8 at 25 °C and 10 bar of HD. The blue trace is T = 0 h, and the red trace is T = 19 h. The peak at 4.45 ppm corresponds to D2 gas, suggesting HD scrambling. Comparable spectra are obtained with 1-Cl(OTf) (ESI†). | ||
Under the reaction conditions used for H2 activation, however, neither of the complexes 1-(OTf)2 and 1-Cl(OTf)2 displayed any hydrogenation activity, suggesting that hydride transfer could be hindering the desired catalytic reactivity in line with the high HIA of 1170 and 660 kJ mol−1 respectively and associated poor hydride donor ability. Catalytic hydrogenation of the FLP model substrate, N-tert-butyl-1-phenylmethanimine, was observed only at 180 °C and 50 bar of H2 in sulfolane with 1-OTf2, 1-Cl(OTf) and even 1-Cl2, in 10, 49 and 29% yield, respectively (Table 2, entries 1–3). These findings suggest that chloride becomes a sufficient leaving group for H2 activation at high temperatures, albeit less so than triflate.
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Solvent | Temperature (°C) | Yield (%) |
| Reaction conditions: N-tert-butyl-1-phenylmethanimine (1 mmol), reaction solvent (4 mL), catalyst (0.05 mmol), H2 (50 bar), 17 h. All reactions were performed in triplicate, quantifying the reaction product by 1H NMR with CH2Br2 as the internal standard and confirming the structure by ESI-MS. | ||||
| 1 | 1-(OTf)2 | Sulfolane | 180 | 10 |
| 2 | 1-Cl(OTf) | Sulfolane | 180 | 49 |
| 3 | 1-Cl2 | Sulfolane | 180 | 29 |
| 4 | 2-Cl2 | Sulfolane | 180 | 53 |
| 5 | 3-Cl2 | Sulfolane | 180 | 42 |
| 6 | 4-Cl2 | Sulfolane | 180 | 28 |
| 7 | 1-(OTf)2 | Toluene | 180 | 30 |
| 8 | 1-Cl(OTf) | Toluene | 180 | 86 |
| 9 | 1-Cl2 | Toluene | 180 | 84 |
| 10 | 2-Cl2 | Toluene | 180 | 98 |
| 11 | 1-Cl2 | Toluene | 150 | NR |
| 12 | 2-Cl2 | Toluene | 150 | 27 |
| 13 | 2-Cl2 | Collidine | 180 | 85 |
Replacing the bulky -tBu groups with -H on 1-Cl2 to form [Sn(Salen)Cl2] (2-Cl2) improved the product yield from 29 to 53% (Table 2, entries 3 and 4), implicating steric hindrance in the low hydrogenation activity of the complexes. This hypothesis was tested with [Sn(Salophen)Cl2] (3-Cl2) and [Sn(tBu2Salophen)Cl2] (4-Cl2), which yielded the desired product in 42 and 28% yield, respectively (Table 2, entries 5 and 6), thus confirming that –tBu groups on complexes 1-Cl2, and 4-Cl2 hinder substrate access to the metal centre. Moreover, the –tBu-substituted complexes 1-Cl2 and 4-Cl2 had similar yields of the target product, at 29 and 28%, respectively (Table 2, entries 3 and 6) and the HIA of the best catalyst 2-Cl2 without the –tBu groups and the worst 4-Cl2 with –tBu groups are almost identical (Table 1, entries 4 and 6), further demonstrating that steric hindrance of –tBu groups is the limiting factor of the activity of chloride complexes. Attempts to calculate an optimized FLP structure were only successful for 2-Cl2 (Fig. 4B) as the –tBu groups on 1-Cl2 and 4-Cl2 prevented FLP formation.
Substituting sulfolane for toluene improved the catalytic performance of 1-OTf2, 1-Cl(OTf) and 1-Cl2 reaching 30, 86 and 84% yields of the desired amine, respectively (Table 2, entries 7–9), thus approximately doubling and trebling those obtained in sulfolane (Table 2, entries 1, 2 and 3). Among other solvent effects, this can be attributed to the enhanced solubility of H2 gas in toluene40 that can promote tin hydride formation via the Le Chatelier principle. As a result, 1-Cl(OTf) and 1-Cl2 showed similar activities. The failure to further improve the yield of 1-(OTf)2 was attributed to the instability of this catalyst under these reaction conditions. 2-Cl2 also demonstrated improved activity in toluene and the desired product was obtained in 98% yield (Table 2, entry 10). The use of 2,4,6-collidine as the solvent, which can also act as an FLP base and enhance H2 activation, decreased the yield from 98 to 85% (Table 2, entries 10 and 13). Lowering the temperature to 150 °C, in toluene, also decreased the yield and confirmed 2-Cl2 as the best catalyst (Table 2, entries 11 and 12). Based on these results, we established the optimal reaction conditions, which were 180 °C, toluene, 50 bar of H2 and 17 hours over the catalyst 2-Cl2 (5 mol%).
Under optimal reaction conditions, 2-Cl2 was used as the catalyst to assess the substrate scope of the reaction (Fig. 3). The model substrate N-tert-butyl-1-phenylmethanimine was converted into the corresponding amine (1) in 98% yield. Introduction of functional group(s) onto the benzene ring such as 4-chloride-, 4-trifluoromethyl- or 4,5-dimethoxy- decreased the reaction yields to 61 (2), 42 (3) and 50% (5) respectively or halted the reaction in the case of 4-nitro- (4), and 2-hydroxo- (6 and 7). As shown by mass spectrometry, nitro- and hydroxy-substitution completely inhibited imine reduction possibly due to the slow but preferential –NO2 group reduction to –NH2 and to the relative Brønsted acidity of phenol(s), respectively. Phenols can protonate the hydride formed in the reaction and hence reverse H2 activation. Simultaneous reduction of an alkene was also observed and N-(tert-butyl)-N-cinnamylamine (13) was obtained in only 10% yield, while the rest was further hydrogenated to remove the alkene moiety.
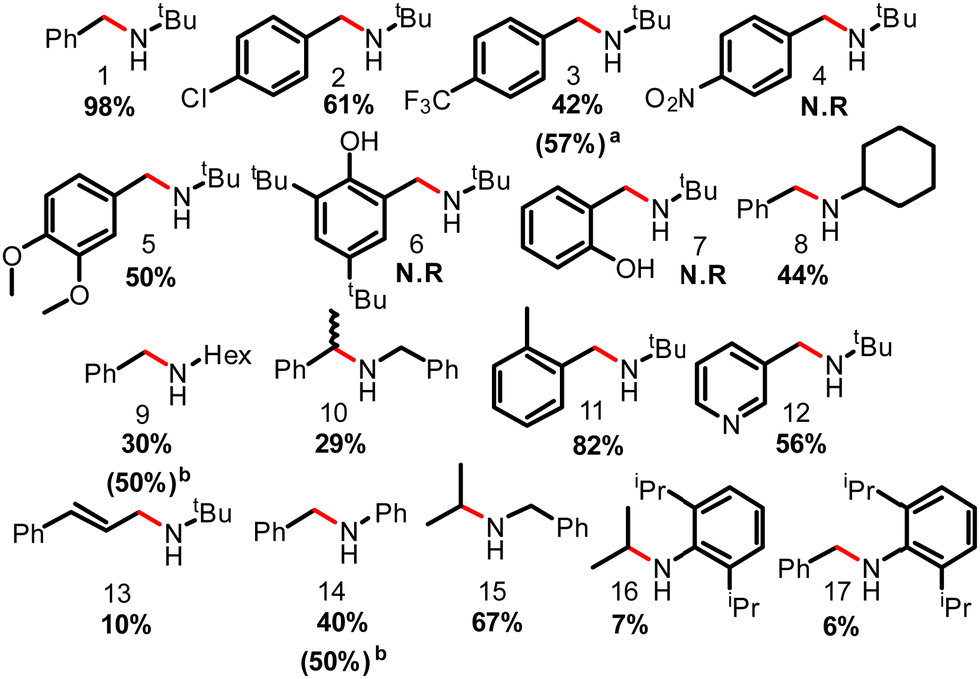 | ||
| Fig. 3 Substrate scope of the imine hydrogenation reaction with H2 over 2-Cl2. Reaction conditions: imine (1 mmol), toluene (4 mL) and LA (5 mol%), H2 (50 bars), 180 °C, 17 h. All yields were determined by 1H NMR with CH2Br2 as the internal standard, and all structures were confirmed by ESI-MS. (a) 70 bar (b) Extended reaction time to 48 h. | ||
 | ||
| Fig. 4 (A) X-ray structure of 2-Cl2 and (B) calculated FLP structure at the DSD-PBEB86-D3BJ/def2-QZVP level. | ||
Substitution of the –tBu group for –Hex, Cy, –Ph, –Bn or other unfunctionalized aliphatic or aromatic hydrocarbon substituents resulted in the corresponding amine formation in 6 to 82% yield (8 to 17). Noteworthy is the tolerance of ortho-methyl substitution, which yielded the corresponding amine 11 in 82% yield, whereas ortho-diisopropyl inhibited the reactivity and the corresponding amine 17 was obtained in 6% yield.
Nevertheless, further reaction trends are difficult to establish and analysis of side reactions, reaction mass balance and catalyst stability indicate that the desired reaction yield is a balance between the rate of substrate hydrogenation, its decomposition and catalyst deactivation, which decomposes to an inactive mixture of metallic tin, tin oxides and ligand fragments under the reaction conditions.
In toluene, the reaction substrate also acts as the Lewis base to activate H2 because the tin(IV) complexes lack this ability on their own. Furthermore, a ligand must also dissociate from the coordinatively saturated complexes (Fig. 4A) to generate a Lewis acidic site, and generate an active FLP catalyst (Fig. 4B) as shown in our H2 activation studies (Fig. 2). Based on these observations and on previous literature,4,41–44 we propose a catalytic cycle for hydrogenation over hexacoordinate tin(IV) complexes (Scheme 1).
 | ||
| Scheme 1 Catalytic cycle proposed for imine hydrogenation with H2 catalysed by tin(IV)-Schiff base complexes. | ||
In the initial phase (Scheme 1, I), a ligand X− (Cl− or OTf−) dissociates from the complex to reveal an LA site. Interaction with the imine then forms the active FLP catalyst (II) and splits H2 yielding L4SnHX and [imineH][X] (III). The protonation activates the imine towards hydride transfer from the metal centre to the iminium double bond (IV).43 The produced amine reversibly binds the tin(IV) complex, which slows down the reaction as demonstrated by the addition of the product (1 mmol) to the reaction, which decreased the conversion of the starting imine from 85% to 30% in a 6-hour reaction. Eventual dissociation of the produced amine then regenerates the active LA with a free binding site (V).
In conclusion, hexacoordinated tin(IV) complexes with Schiff base and labile or hemi-labile axial ligands can be used as LA components of FLPs for H2 activation and imine hydrogenation. H2 activation takes place with bases as weak as THF at 25 °C and can be reversible, as shown by HD scrambling. Nevertheless, imine reduction only occurs at temperatures ≥ 150 °C and optimally at 180 °C, suggesting that hydride transfer hinders the reaction. The best catalyst is [Sn(Salen)Cl2] (2-Cl2) even though dichloride complexes have the lowest ability to activate H2 among all complexes tested in this study. In effect, various imines are hydrogenated in yields ranging from 6 to 98% in toluene depending on substrate substitution and functionalization. The high variability of these complexes opens up opportunities for developing hydrogenation catalysts based on hexacoordinated compounds of main-group elements.
The authors thank the Czech Science Foundation (GAČR 21-27431M) and Charles University Research Centre program No. UNCE/24/SCI/010 for funding the study. We also thank Carlos V. Melo for editing the manuscript. The research used computational resources of the Centro de Computación de Alto Desempeño – CCAD of Universidad Nacional de Cordoba – UNC (https://ccad.unc.edu.ar/), part of Sistema Nacional de Computación de Alto Desempeño – SNCAD of the Ministry of Science, Technology and Innovation, Argentina.
Conflicts of interest
There are no conflicts to declare.Notes and references
- G. C. Welch, R. R. San Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124–1126 CrossRef CAS PubMed.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46–76 CrossRef CAS PubMed.
- D. W. Stephan, Acc. Chem. Res., 2015, 48, 306–316 CrossRef CAS PubMed.
- D. J. Scott, M. J. Fuchter and A. E. Ashley, Chem. Soc. Rev., 2017, 46, 5689–5700 RSC.
- D. W. Stephan, Science, 2016, 354, aaf7229–aaf7229 CrossRef PubMed.
- J. Lam, K. M. Szkop, E. Mosaferi and D. W. Stephan, Chem. Soc. Rev., 2019, 48, 3592–3612 RSC.
- É. Dorkó, B. Kótai, I. Pápai, T. Soós, M. Szabó and A. Domján, Angew. Chem., Int. Ed., 2017, 56, 9512–9516 CrossRef PubMed.
- D. J. Scott, T. R. Simmons, E. J. Lawrence, G. G. Wildgoose, M. J. Fuchter and A. E. Ashley, ACS Catal., 2015, 5, 5540–5544 CrossRef CAS PubMed.
- T. Mahdi and D. W. Stephan, J. Am. Chem. Soc., 2014, 136, 15809–15812 CrossRef CAS PubMed.
- D. J. Scott, M. J. Fuchter and A. E. Ashley, J. Am. Chem. Soc., 2014, 136, 15813–15816 CrossRef CAS PubMed.
- V. B. Saptal, G. Juneja and B. M. Bhanage, New J. Chem., 2018, 42, 15847–15851 RSC.
- V. Fasano and M. J. Ingleson, Chem. – Eur. J., 2017, 23, 2217–2224 CrossRef CAS PubMed.
- M. Xu, J. Possart, A. E. Waked, J. Roy, W. Uhl and D. W. Stephan, Philos. Trans. Royal Soc. A, 2017, 375, 20170014 CrossRef PubMed.
- A. Paparakis, R. C. Turnell-Ritson, J. S. Sapsford, A. E. Ashley and M. Hulla, Catal. Sci. Technol., 2023, 13, 637–644 RSC.
- D. J. Scott, N. A. Phillips, J. S. Sapsford, A. C. Deacy, M. J. Fuchter and A. E. Ashley, Angew. Chem., Int. Ed., 2016, 55, 14738–14742 CrossRef CAS PubMed.
- J. Sapsford, D. Scott, N. Allcock, M. Fuchter, A. Ashley and C. Tighe, Adv. Synth. Catal., 2018, 360, 1066–1071 CrossRef CAS PubMed.
- P. Sarkar, S. Das and S. K. Pati, Chem. – Asian J., 2022, 17, e202200148 CrossRef CAS PubMed.
- T. Thorwart, D. Hartmann and L. Greb, Chem. – Eur. J., 2022, 28, e202202273 CrossRef CAS PubMed.
- T. A. Kinder, R. Pior, S. Blomeyer, B. Neumann, H. G. Stammler and N. W. Mitzel, Chem. – Eur. J., 2019, 25, 5899–5903 CrossRef CAS PubMed.
- P. Holtkamp, F. Friedrich, E. Stratmann, A. Mix, B. Neumann, H. G. Stammler and N. W. Mitzel, Angew. Chem., Int. Ed., 2019, 58, 5114–5118 CrossRef CAS PubMed.
- J. M. Bayne and D. W. Stephan, Chem. Soc. Rev., 2016, 45, 765–774 RSC.
- R. T. Cooper, J. S. Sapsford, R. C. Turnell-Ritson, D. H. Hyon, A. J. P. White and A. E. Ashley, Philos. Trans. Royal Soc. A, 2017, 375, 20170008 CrossRef PubMed.
- G. R. Whittell, E. I. Balmond, A. P. M. Robertson, S. K. Patra, M. F. Haddow and I. Manners, Eur. J. Inorg. Chem., 2010, 3967–3975 CrossRef CAS.
- J. S. Sapsford, D. Csókás, R. C. Turnell-Ritson, L. A. Parkin, A. D. Crawford, I. Pápai and A. E. Ashley, ACS Catal., 2021, 11, 9143–9150 CrossRef CAS.
- A. Paparakis and M. Hulla, ChemCatChem, 2023, 15, e202300510 CrossRef CAS.
- Y. M. Ahmed and G. G. Mohamed, Inorg. Chem. Commun., 2022, 144, 109864 CrossRef CAS.
- A. Pérez-Rebolledo, G. M. de Lima, N. L. Speziali, O. E. Piro, E. E. Castellano, J. D. Ardisson and H. Beraldo, J. Organomet. Chem., 2006, 691, 3919–3930 CrossRef.
- H. Jing, S. K. Edulji, J. M. Gibbs, C. L. Stern, H. Zhou and S. B. T. Nguyen, Inorg. Chem., 2004, 43, 4315–4327 CrossRef CAS PubMed.
- A. M. Abu-Dief and I. M. A. Mohamed, J. Basic Appl. Sci., 2015, 4, 119–133 Search PubMed.
- M. Palucki, P. J. Pospisil, W. Zhang and E. N. Jacobsen, J. Am. Chem. Soc., 1994, 116, 9333–9334 CrossRef CAS.
- S. De, A. Jain and P. Barman, ChemistrySelect, 2022, 7, e202104334 CrossRef CAS.
- W. Meng, X. Feng and H. Du, Acc. Chem. Res., 2018, 51, 191–201 CrossRef CAS PubMed.
- W. Meng, X. Feng and H. Du, Chin. J. Chem., 2020, 38, 625–634 CrossRef CAS.
- Y. Liang, J. Luo, Y. Diskin-Posner and D. Milstein, J. Am. Chem. Soc., 2023, 145, 9164–9175 CrossRef CAS PubMed.
- H. Elsen, C. Färber, G. Ballmann and S. Harder, Angew. Chem., Int. Ed., 2018, 57, 7156–7160 CrossRef CAS PubMed.
- A. Friedrich, J. Eyselein, H. Elsen, J. Langer, J. Pahl, M. Wiesinger and S. Harder, Chem. – Eur. J., 2021, 27, 7756–7763 CrossRef CAS PubMed.
- D. J. Scott, M. J. Fuchter and A. E. Ashley, Angew. Chem., Int. Ed., 2014, 53, 10218–10222 CrossRef CAS PubMed.
- L. J. Hounjet, C. Bannwarth, C. N. Garon, C. B. Caputo, S. Grimme and D. W. Stephan, Angew. Chem., Int. Ed., 2013, 52, 7492–7495 CrossRef CAS PubMed.
- E. Philipp and L. Greb, ChemPhysChem., 2021, 22, 935–943 CrossRef PubMed.
- J. J. Simnick, H. M. Sebastian, H.-M. Lin and K.-C. Chao, J. Chem. Eng. Data, 1978, 23, 339 CrossRef CAS.
- P. Pérez, D. Yepes, P. Jaque, E. Chamorro, L. R. Domingo, R. S. Rojas and A. Toro-Labbé, Phys. Chem. Chem., 2015, 17, 10715–10725 RSC.
- S. Grimme, H. Kruse, L. Goerigk, G. Erker, S. Grimme, H. Kruse, L. Goerigk and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 1402–1405 CrossRef CAS PubMed.
- J. Paradies, Eur. J. Org. Chem., 2019, 283–294 CrossRef CAS.
- P. A. Chase, T. Jurca and D. W. Stephan, Chem. Commun., 2008, 1701–1703 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Catalyst synthesis and analysis; NMRs, MS, IRs, sXRD, and Lewis acidity measurements. CCDC 2330171 and 2333521. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc05878f |
| This journal is © The Royal Society of Chemistry 2024 |