
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reversing the stereoselectivity of intramolecular [2+2] photocycloaddition utilizing cucurbit[8]uril as a molecular flask†
Xujun
Qiu
a,
Jasmin
Seibert
 a,
Olaf
Fuhr
bc,
Frank
Biedermann
*b and
Stefan
Bräse
*ad
a,
Olaf
Fuhr
bc,
Frank
Biedermann
*b and
Stefan
Bräse
*ad
aInstitute of Organic Chemistry (IOC), Karlsruhe Institute of Technology (KIT), Kaiserstraße 12, 76131 Karlsruhe, Germany. E-mail: braese@kit.edu
bInstitute of Nanotechnology (INT), Karlsruhe Institute of Technology (KIT), Kaiserstraße 12, 76131 Karlsruhe, Germany. E-mail: frank.biedermann@kit.edu
cKarlsruhe Nano Micro Facility (KNMFi), Karlsruhe Institute of Technology (KIT), Kaiserstraße 12, 76131 Karlsruhe, Germany
dInstitute of Biological and Chemical Systems-Functional Molecular Systems (IBCS-FMS), Karlsruhe Institute of Technology (KIT), Kaiserstraße 12, 76131 Karlsruhe, Germany
First published on 2nd February 2024
Abstract
Macrocyclic hosts, such as cucurbit[8]uril (CB8), can significantly influence the outcomes of chemical reactions involving encapsulated reactive guests. In this study, we demonstrate that CB8 completely reverses the stereoselectivity of intramolecular [2+2] photo-cycloaddition reactions. Notably, it was also found that CB8 can trigger the unreactive diene to be reactive.
Cyclobutanes, with their inherently strained scaffolds, are pivotal in numerous natural products and biological systems.1,2 The prevalent synthetic routes to cyclobutanes often employ inter/intramolecular [2+2] photo-cycloadditions using olefin precursors.3 Moreover, intramolecular [2+2] cycloadditions are commonly used to synthesize bioactive natural products.4 Yet, achieving precise control over the stereoselectivity of the photo [2+2] cycloaddition remains challenging.
Recent advancements in carrying out organic reactions within “molecular flasks”,5,6 specifically using supramolecular hosts, such as macrocycles,7–9 cages10–14 and capsules,15–17 have paved the way for novel reaction pathways. The reactivity and selectivity of reagents is controlled by the microenvironment, e.g., the confined space within the cavity of molecular flasks assists in preorganizing the substrates,18,19 which modulates the reaction rates or steers reactions in directions distinct from those in bulk solutions.
Cucurbit[n]urils (CBn) are characterized by their symmetrical, rigid, and hydrophobic cavities that can host organic molecules.20,21 Their nanocavities are excellent binders for hydrophobic molecules in aqueous environments, while the carbonyl-fringed portal regions provide an additional driving force for binding cationic guests. As a result, CBn have been employed as both reaction vessels and supramolecular catalysts22,23 to control or accelerate the outcome of organic reactions such as 1,3-dipolar Huisgen cycloaddition,24–26 Diels–Alder reaction, and photodimerization.27–29
In this study, we showcase how the outcome of intramolecular [2+2] photocycloaddition can be switched from an anti-head-to-tail to a syn-head-to-tail cycloaddition product using CB8 as a molecular flask. Furthermore, it was also found that CB8 can trigger the unreactive diene to be reactive, yielding a syn-head-to-head (1,2) product. As depicted in Scheme 1, diene 1 experiences intramolecular charge repulsion due to its two cationic methylpyridinium moieties. We started our investigation with the photolysis of 1 in water, using a standard photoreactor fitted with 14 UVA (365 nm) lamps. 1H NMR spectra (Fig. S4, ESI†) were recorded at fixed time intervals to monitor the reaction path.
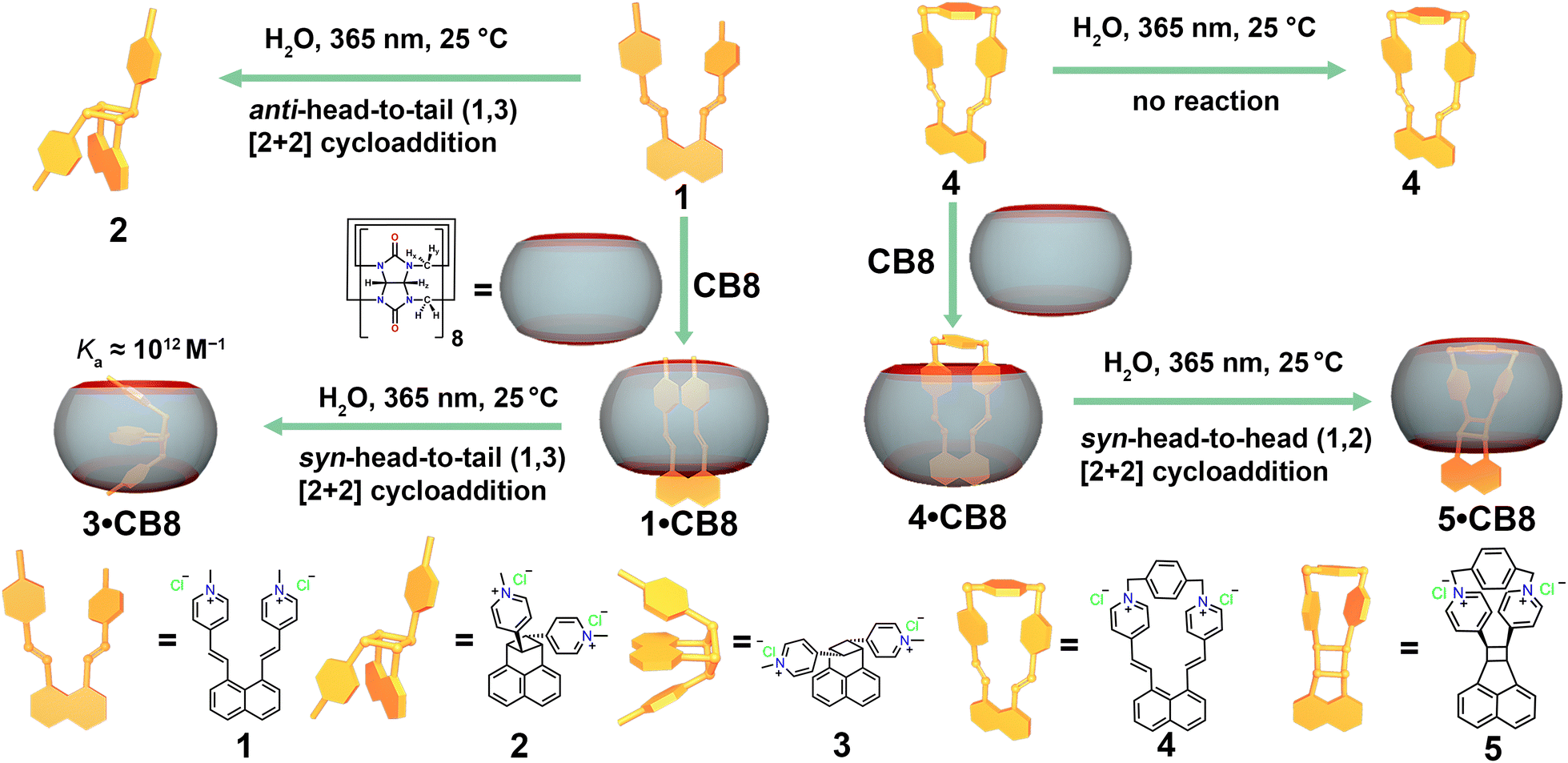 | ||
| Scheme 1 Schematic illustration of modulating the photolysis reaction outcome of 1 utilizing cucurbit[8]uril as a molecular flask. | ||
As irradiation progressed, the 1H peaks from 1 gradually disappeared, while new signals corresponding to a single intramolecular cycloaddition product emerged. After approximately 18 minutes of UV exposure, no further significant changes were observed in the NMR spectra, indicating the completion of the photolysis. Structural assignment of the product peaks revealed that UV irradiation of 1 in water proceeds via photoisomerization and [2+2] cycloaddition, yielding an anti-head-to-tail (1,3) product where the charge repulsion between the pyridinium moieties is minimized (refer to Fig. S7–S15 in the ESI† for a detailed structural assignment of the reaction product.)
Intrigued by the potential impact of a confined environment on the photoreaction process, we explored whether the photoisomerization of molecule 1 could be inhibited within the cavity of a molecular host, and whether this altered the outcome of the [2+2] cycloaddition. We selected the water-soluble molecular host CB8 as our choice, given its spacious and hydrophobic cavity, which could accommodate both vinylpyridinium residues of 1 simultaneously. To test this binding hypothesis, we first examined the host–guest properties between 1 and CB8 using 1H NMR. As depicted in Fig. 2a, when an aqueous solution of 1 and CB8 was mixed in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio in D2O, the NMR signals a–d, corresponding to the vinylpyridinium residues, showed significant upfield shifts, indicating that these are bound inside the shielding CB8 cavity. Conversely, the peaks corresponding to the naphthyl moieties (e–g) displayed slight downfield shifts, suggesting that these moieties are located outside or near the rim of the CB8 portal.30 Notably, the signals Hx and Hy from CB8 split into four sets, reflecting the deformation of CB8 when molecule 1 is accommodated within its cavity. The formation of the 1:1 complex of 1·CB8 was further confirmed by ESI-MS experiments (Fig. S16, ESI†), where a peak with a mass-to-charge ratio (m/z) of 846.2923 for [1·CB8-2Cl]2+ was observed.
:1 ratio in D2O, the NMR signals a–d, corresponding to the vinylpyridinium residues, showed significant upfield shifts, indicating that these are bound inside the shielding CB8 cavity. Conversely, the peaks corresponding to the naphthyl moieties (e–g) displayed slight downfield shifts, suggesting that these moieties are located outside or near the rim of the CB8 portal.30 Notably, the signals Hx and Hy from CB8 split into four sets, reflecting the deformation of CB8 when molecule 1 is accommodated within its cavity. The formation of the 1:1 complex of 1·CB8 was further confirmed by ESI-MS experiments (Fig. S16, ESI†), where a peak with a mass-to-charge ratio (m/z) of 846.2923 for [1·CB8-2Cl]2+ was observed.
We further explored the binding affinity between 1 and CB8 through UV titration, gradually introducing CB8 into a solution of 1. As depicted in Fig. 1b, the addition of CB8 led to a decrease in absorbance, with the two maximum absorbance peaks at 281 nm and 396 nm shifting to isosbestic points 293 nm and 421 nm, respectively. We plotted the changes in absorbance at 281 nm against the concentration of CB8, as shown in Fig. 1c. Least-square fitting of this data with a direct binding assay (DBA) model yielded a binding constant of (2.53 ± 0.11) × 106 M−1, indicating a strong interaction between 1 and CB8.
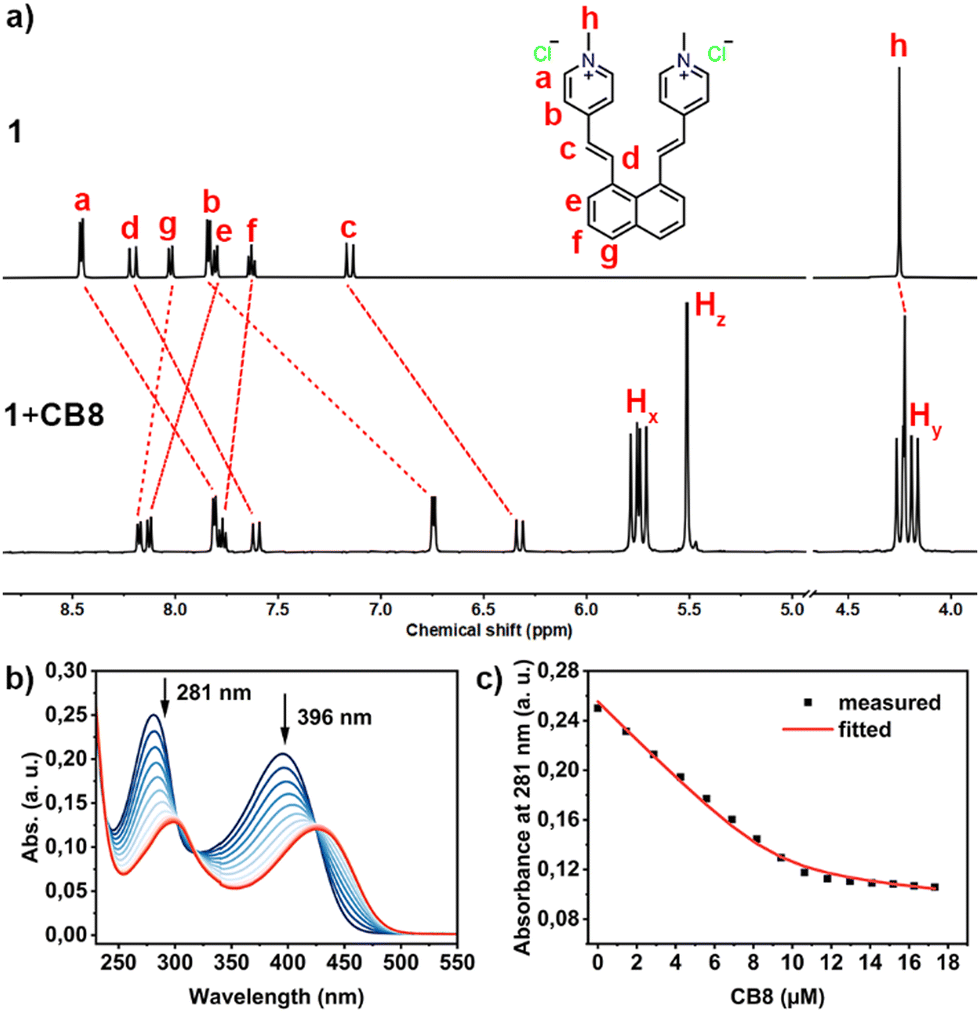 | ||
| Fig. 1 (a) Partial 1H NMR spectra of 1 (0.5 mM) and 1 + CB8 (1:1, 0.5 mM) in D2O; (b) UV-vis titration spectra of 1 (1 = 10 μM) upon addition of CB8 (CB8 = 0–17.3 μM) in an aqueous solution at 298 K; (c) least-square curve fitting of the UV absorbance changes at 281 nm against the concentration of CB8 for determining the binding constant between 1 and CB8. | ||
Furthermore, we performed the photoreaction of 1 encapsulated in the CB8 cavity under the same photoreaction conditions as for 1 in water for comparison. Upon irradiation, the 1H NMR signals were observed to be completely different from the initial state, suggesting the formation of new species (Fig. S19, ESI†). After around 21 min, no more significant variation in the NMR suggests the end of the reaction.
A similar phenomenon in UV absorbance and fluorescence emission change was observed (Fig. S20 and S21, ESI†). As shown in Fig. 2b, the absorbance was observed to decrease dramatically within the first 3 min, in which the maximum absorption peak at 421 nm was eventually barely visible after being irradiated for 21 min. The fluorescence intensity was identical upon UV irradiation, which decreased rapidly to no emission at the end, Fig. 2c. The formation of [2+2] cycloaddition product 3 in the CB8 cavity was also evidenced by ESI-MS (Fig. S24, ESI†), in which a strong signal peak with a mass-to-charge ratio (m/z) of 846.2927 that corresponded to [3·CB8-2Cl]2+ was observed, which suggested no mass change of 3 in the CB8 cavity compared to starting reagent 1.
 | ||
| Fig. 2 (a) 1H NMR spectra of 1·CB8 (1:1, 0.5 mM), 3·CB8 (1:1, 0.5 mM), and 3 (0.5 mM), in the aromatic region in D2O; (b) UV-vis spectra of 1·CB8 (1:1, 0.5 mM, diluted to 20 μM for UV-vis measurements) upon irradiation at 298 K; (c) emission spectra of 1·CB8 (1:1, 0.5 mM, and diluted to 20 μM for UV-vis measurements) upon irradiation at 298 K (excited at 410 nm). | ||
Upon addition of the strongly CB8-binding competitive guest, memantine hydrochloride, to the 3·CB8 complex, unbound 3 was obtained, as shown in Fig. 2a (for full spectra see Fig. S25, ESI†). The symmetric signals in Fig. 2a (Fig. S25, ESI†) indicate a syn-head-to-tail (1,3) photo cycloaddition product 3, which is different from the similar structure reported in the literature.31 Notably, the binding constant of 3 between CB8 was determined to be (2.04 ± 0.29) × 1012 M−1 using a competitive binding assay (CBA), where memantine was utilized as a competitive guest (Fig. S42, ESI†).32 The dramatically increased binding constant of 3 compared with starting molecule 1 could result from the rigidified structure of 3 having a better complemental effect with the CB8 void. Further studies were conducted to understand the mechanistic pathway of the reaction. The rate of product formation was found to be significantly influenced by the concentration of CB8, suggesting a pivotal role of the macrocycle in the reaction kinetics. Control experiments, conducted in the absence of CB8 or in the presence of smaller macrocycle CB7 that cannot fully encapsulate 1 in its cavity due to sterical reasons, further corroborated its role in steering the reaction towards the syn-head-to-tail (1,3) cycloaddition product. In the meantime, controlled experiments in the dark under thermal conditions were performed for 1 and 1·CB8, and no reaction traces were observed for both cases. To gain more insights into the stereo structures of the different possible products, we performed theoretical calculations and predicted the NMR composition (Table S1, ESI†), which further supported our conclusions.
We then wondered whether the photo cycloaddition process could still take place if two olefin branches were covalently locked. Therefore, molecule 4, in which the pyridine moieties were bridged by 1,4-bis(bromomethyl)benzene, was designed, synthesized, and fully characterized (Fig. S27–S29, ESI†). As shown in Fig. 3a and b, the crystal structure suggests that molecule 4 bears a rigid structure and the olefin groups are separated distinctly. Thus, photolysis of molecule 4 in aqueous media results in no change. In the same fashion, we then studied the host–guest interaction between 4 and CB8. Indeed, molecule 4 can be encapsulated within the CB8 cavity, as indicated by the ESI-MS (Fig. S30, ESI†), and NMR spectra (Fig. S31, ESI†). The NMR spectra of 4 and 4·CB8 suggest that CB8 selectively encapsulates a naphthyl moiety, while the bridged pyridinium locates at the carbonyl portal of the CB8, which is comparable to the literature.33 The binding constant of 4 and CB8 was obtained using UV-vis titration, which was fitted as (2.52 ± 0.15) × 106 M−1 (Fig. S33, ESI†).
 | ||
| Fig. 3 (a) Molecular structure of 4 (front view, water molecules are omitted for clarity); (b) crystal packing of 4 (water molecules are omitted for clarity); (c) 1H NMR spectra of 4, 4·CB8 (1:1, 0.5 mM), 5·CB8 (1:1, 0.5 mM), and 5 (0.5 mM) in D2O; for full spectra see Fig. S27, S31, S38 and S40 (ESI†). | ||
Surprisingly, upon the irradiation of 4·CB8 under the same reaction conditions as photolysis of 1 and 1·CB8, new species were observed in the NMR spectra (Fig. 3c and Fig. S34, ESI†). Absorption (Fig. S35, ESI†) and emission spectra (Fig. S36, ESI†) also revealed the occurrence of the photoreaction. The mass spectra (Fig. S37, ESI†) of the 5·CB8 complex suggest no mass change of the photoreaction product. The NMR spectra (Fig. 3c and Fig. S38, ESI†) indicate that a dissymmetric product is formed, and the identical signal at 5.18–5.27 ppm suggests that the syn-head-to-head (1,2) cycloaddition product is generated, which is comparable to the similar structure.31 To obtain unbound product 5 out of the CB8 cavity, memantine hydrochloride was introduced to the reaction mixture (Fig. 3c and Fig. S40, ESI†).
In conclusion, we presented a strategy to selectively tune the photoreaction pathway utilizing CB8 as a molecular flask. The photoreaction of molecule 1 in bulk water was assisted with the photoisomerization and [2+2] cycloaddition process, resulting in an anti-head-to-tail (1,3) photoproduct 2. However, with the template from CB8, the photoreaction selectively formed syn-head-to-tail (1,3) cycloaddition product 3. Notably, the binding constant between 3 and CB8 was estimated to be as high as (2.04 ± 0.29) × 1012 M−1. When molecule 4 was intramolecularly locked, photoreaction was prevented upon irradiation. On the contrary, when the photoreaction was performed inside the CB8 cavity, a syn-head-to-head (1,2) product was obtained.
The authors acknowledge the support of the China Scholarship Council (CSC grant: 202010190002) and the Deutsche Forschungsgemeinschaft (DFG) under Germany's Excellence Strategy-3DMM2O-EXC-2082/1-390761711. F. B. acknowledges the Deutsche Forschungsgemeinschaft (DFG grant BI-1805/2-1) for financial support.
Conflicts of interest
There are no conflicts to declare.References
- J. C. J. M. D. S. Menezes and M. F. Diederich, Eur. J. Med. Chem., 2019, 182, 111637 CrossRef CAS PubMed.
- T. Bach and J. P. Hehn, Angew. Chem., Int. Ed., 2011, 50, 1000–1045 CrossRef CAS PubMed.
- S. Poplata, A. Tröster, Y.-Q. Zou and T. Bach, Chem. Rev., 2016, 116, 9748–9815 CrossRef CAS PubMed.
- M. D. Kärkäs, J. A. Porco, Jr. and C. R. J. Stephenson, Chem. Rev., 2016, 116, 9683–9747 CrossRef PubMed.
- M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418–3438 CrossRef CAS PubMed.
- B. Breiner, J. K. Clegg and J. R. Nitschke, Chem. Sci., 2011, 2, 51–56 RSC.
- P. Zhang, J. Meijide Suárez, T. Driant, E. Derat, Y. Zhang, M. Ménand, S. Roland and M. Sollogoub, Angew. Chem., Int. Ed., 2017, 56, 10821–10825 CrossRef CAS PubMed.
- S. Roland, J. M. Suarez and M. Sollogoub, Chem. – Eur. J., 2018, 24, 12464–12473 CrossRef CAS PubMed.
- B. Tang, J. Zhao, J.-F. Xu and X. Zhang, Chem. – Eur. J., 2020, 26, 15446–15460 CrossRef CAS PubMed.
- C. Zhao, F. D. Toste, K. N. Raymond and R. G. Bergman, J. Am. Chem. Soc., 2014, 136, 14409–14412 CrossRef CAS PubMed.
- B. Mondal and P. S. Mukherjee, J. Am. Chem. Soc., 2018, 140, 12592–12601 CrossRef CAS PubMed.
- S. S. Nurttila, W. Brenner, J. Mosquera, K. M. van Vliet, J. R. Nitschke and J. N. H. Reek, Chem. – Eur. J., 2019, 25, 609–620 CrossRef CAS PubMed.
- J. Guo, Y.-W. Xu, K. Li, L.-M. Xiao, S. Chen, K. Wu, X.-D. Chen, Y.-Z. Fan, J.-M. Liu and C.-Y. Su, Angew. Chem., Int. Ed., 2017, 56, 3852–3856 CrossRef CAS PubMed.
- H. Takezawa, T. Kanda, H. Nanjo and M. Fujita, J. Am. Chem. Soc., 2019, 141, 5112–5115 CrossRef CAS PubMed.
- Q. Zhang, L. Catti and K. Tiefenbacher, Acc. Chem. Res., 2018, 51, 2107–2114 CrossRef CAS PubMed.
- A. Cavarzan, A. Scarso, P. Sgarbossa, G. Strukul and J. N. H. Reek, J. Am. Chem. Soc., 2011, 133, 2848–2851 CrossRef CAS PubMed.
- J. Kang and J. Rebek, Nature, 1997, 385, 50–52 CrossRef CAS PubMed.
- Y. Yu, J.-M. Yang and J. Rebek, Chem, 2020, 6, 1265–1274 CAS.
- J. L. Bolliger, in Effects of Nanoconfinement on Catalysis, ed. R. Poli, Springer International Publishing, Cham, 2017, pp. 17–48 DOI:10.1007/978-3-319-50207-6_2.
- K. I. Assaf and W. M. Nau, Chem. Soc. Rev., 2015, 44, 394–418 RSC.
- S. J. Barrow, S. Kasera, M. J. Rowland, J. del Barrio and O. A. Scherman, Chem. Rev., 2015, 115, 12320–12406 CrossRef CAS PubMed.
- T.-C. Lee, E. Kalenius, A. I. Lazar, K. I. Assaf, N. Kuhnert, C. H. Grün, J. Jänis, O. A. Scherman and W. M. Nau, Nat. Chem., 2013, 5, 376–382 CrossRef CAS PubMed.
- X. Qiu, Y. Wang, S. Leopold, S. Lebedkin, U. Schepers, M. M. Kappes, F. Biedermann and S. Bräse, Small, 2023, 2307318 CrossRef PubMed.
- S. Angelos, Y.-W. Yang, K. Patel, J. F. Stoddart and J. I. Zink, Angew. Chem., Int. Ed., 2008, 47, 2222–2226 CrossRef CAS PubMed.
- D. Tuncel, Ö. Özsar, H. B. Tiftik and B. Salih, Chem. Commun., 2007, 1369–1371, 10.1039/B616764K.
- T. G. Brevé, M. Filius, C. Araman, M. P. van der Helm, P.-L. Hagedoorn, C. Joo, S. I. van Kasteren and R. Eelkema, Angew. Chem., Int. Ed., 2020, 59, 9340–9344 CrossRef PubMed.
- Y. Kang, X. Tang, H. Yu, Z. Cai, Z. Huang, D. Wang, J.-F. Xu and X. Zhang, Chem. Sci., 2017, 8, 8357–8361 RSC.
- X. Tang, Z. Huang, H. Chen, Y. Kang, J.-F. Xu and X. Zhang, Angew. Chem., Int. Ed., 2018, 57, 8545–8549 CrossRef CAS PubMed.
- D. Li, Z. Feng, Y. Han, C. Chen, Q.-W. Zhang and Y. Tian, Adv. Sci., 2022, 9, 2104790 CrossRef CAS PubMed.
- N. Zhao, L. Liu, F. Biedermann and O. A. Scherman, Chem. – Asian J., 2010, 5, 530–537 CrossRef CAS PubMed.
- R. C. Laird, M. A. Sinnwell, N. P. Nguyen, D. C. Swenson, S. V. S. Mariappan and L. R. MacGillivray, Org. Lett., 2015, 17, 3233–3235 CrossRef CAS PubMed.
- D. Sigwalt, M. Šekutor, L. Cao, P. Y. Zavalij, J. Hostaš, H. Ajani, P. Hobza, K. Mlinarić-Majerski, R. Glaser and L. Isaacs, J. Am. Chem. Soc., 2017, 139, 3249–3258 CrossRef CAS PubMed.
- H. Wu, Y. Wang, L. O. Jones, W. Liu, B. Song, Y. Cui, K. Cai, L. Zhang, D. Shen, X.-Y. Chen, Y. Jiao, C. L. Stern, X. Li, G. C. Schatz and J. F. Stoddart, J. Am. Chem. Soc., 2020, 142, 16849–16860 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2309696. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc05783f |
| This journal is © The Royal Society of Chemistry 2024 |