
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePd(II)-Catalyzed enantioselective C–H olefination toward the synthesis of P-stereogenic phosphinamides†
Zi-Jia
Chen‡
 a,
Ling-Jie
Fan‡
b,
Pei-Pei
Xie
a,
Pu-Fan
Qian
a,
Xinquan
Hu
b,
Tao
Zhou
*ac and
Bing-Feng
Shi
*acd
a,
Ling-Jie
Fan‡
b,
Pei-Pei
Xie
a,
Pu-Fan
Qian
a,
Xinquan
Hu
b,
Tao
Zhou
*ac and
Bing-Feng
Shi
*acd
aCenter of Chemistry for Frontier Technologies, Department of Chemistry, Zhejiang University, Hangzhou 310027, China. E-mail: taozhou.zju.edu.cn; bfshi@zju.edu.cn
bCollege of Chemical Engineering, Zhejiang University of Technology, Hangzhou 310032, China
cCollege of Material Chemistry and Chemical Engineering, Key Laboratory of Organosilicon Chemistry and Material Technology, Ministry of Education, Hangzhou Normal University, Hangzhou, 311121, Zhejiang, China
dCollege of Biological, Chemical Sciences and Engineering, Jiaxing University, Jiaxing, Zhejiang 314001, China
First published on 12th January 2024
Abstract
P-Stereogenic phosphorus compounds are important structural elements in chiral ligands or organocatalysts. Herein, we report a Pd(II)-catalyzed enantioselective C–H olefination toward the synthesis of P-stereogenic phosphinamides using cheap commercially available L-pGlu-OH as a chiral ligand. A broad range of P-stereogenic phosphinamides were gained in good yields with high enantioselectivities (33 examples, up to 77% yield, 99% ee) via desymmetrization and kinetic resolution.
Chiral phosphorus compounds play an important role in both laboratory and industrial synthesis due to their extensive applications in asymmetric synthesis as chiral ligands or organocatalysts.1 However, most of them have chirality located in their backbones, such as a chiral axis, planar-chirality, or a stereogenic carbon center (Scheme 1a). Although better chiral induction may be gained during the transformation because the stereogenic phosphorus atom is coordinated in closer proximity to the catalytic center,2 the application of P-stereogenic phosphorus compounds is less described due to the lack of general and efficient methods for their enantioselective preparation.3 Classical synthetic methods usually rely on resolution processes or the use of chiral auxiliaries.4 In recent years, more and more approaches including desymmetrization reactions5 and catalytic asymmetric methods have been developed.6
 | ||
| Scheme 1 Asymmetric synthesis of P-stereogenic phosphinamides via enantioselective C–H activation. | ||
In the past decade, transition metal-catalyzed asymmetric C–H functionalization has been developed rapidly and proved to be a powerful strategy to access chiral building blocks from simple starting materials.7 In particular, Pd(II)/mono-N-protected amino acid (MPAA)-catalyzed enantioselective C–H activation developed by Yu and coworkers has received extensive attention for the diverse reactivities of palladacycles and the use of cheap and commercially available ligands.8 In 2015, Han and coworkers successfully applied this catalytic system for the synthesis of P-stereogenic phosphinamides using Boc-Tyr(tBu)-OH as a chiral ligand (Scheme 1b).9 Phosphinamide bearing a 2,3,5,6-tetrafluoro-4-cyanophenylamino (ArF) directing group was important for this transformation. And it also should be noted that the arylation products could be used as chiral organocatalysts in the desymmetric enantioselective reduction of cyclic 1,3-diketones.10 Since then, efficient synthesis of P-stereogenic phosphorus compounds via asymmetric C–H functionalization has been developed enabled by different catalytic systems.11 However, until recently, another Pd(II)/MPAA-catalyzed enantioselective C–H activation towards the synthesis of chiral phosphorus compounds was reported by our group using cheap and commercially available L-pyroglutamic acid (L-pGlu-OH) as a chiral ligand.12 A newly developed N-ethyl-N-(3-methylpyridin-2-yl)amino directing group was crucial for the reactivity. Herein, we report the synthesis of P-stereogenic phosphinamides via Pd(II)-catalyzed enantioselective C–H olefination (Scheme 1c). A broad range of P-stereogenic phosphinamides were gained in good yields with high enantioselectivities (33 examples, up to 77% yield and 99% ee) via desymmetrization and kinetic resolution. The P-stereogenic phosphinamide products could be easily transformed to P-chiral phosphine oxides.
We initiated our investigation by optimizing the Pd(II)-catalyzed C–H olefination of phosphoramide 1a with butyl acrylate 2a. To our delight, the product 3a was obtained in 54% yield with 93% ee in the presence of Pd(OAc)2 (10 mol%), L-pGlu-OH (20 mol%), Ag2CO3 (0.05 mmol), and benzoquinone (0.1 mmol) in tBuOH for 12 h under air (Table 1). Next, we screened the solvents such as PhCl, dioxane and t-AmylOH, and t-AmylOH gave the desired product in a higher yield and enantioselectivity. Other silver salts were also investigated, and no better result was observed (entries 5–7). Increasing the temperature to 70 °C or decreasing the temperature to 50 °C resulted in a lower yield (entries 8 and 9). Control experiments showed that L-pGLu-OH and silver salt were crucial to this reaction (entries 10 and 11). In fact, the phosphinamide bearing a 2,3,5,6-tetrafluoro-4-cyanophenylamino directing group (ArF), which was used in Han's work, could not afford the olefination product (entry 12).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | [Ag] | T (°C) | Yield of 3ab (%) | ee of 3ac (%) |
| a Reaction conditions: 1a (0.1 mmol), 2a (0.2 mmol), Pd(OAc)2 (0.01 mmol), [Ag] (0.05 mmol), L-pGlu-OH (0.02 mmol), BQ (0.1 mmol), solvent (1.0 mL), air for 12 h. b Isolated yield. c The ee was determined by chiral HPLC. d Without L-pGlu-OH. e 2,3,5,6-Tetrafluoro-4-cyanophenylamino (ArF) directing group was used. | |||||
| 1 | t-BuOH | Ag2CO3 | 60 | 54 | 93 |
| 2 | PhCl | Ag2CO3 | 60 | 20 | 81 |
| 3 | Dioxane | Ag2CO3 | 60 | 63 | 89 |
| 4 | t-AmylOH | Ag2CO3 | 60 | 66 | 95 |
| 5 | t-AmylOH | Ag2SO4 | 60 | 32 | 92 |
| 6 | t-AmylOH | AgOAc | 60 | 41 | 92 |
| 7 | t-AmylOH | Ag3PO4 | 60 | 47 | 95 |
| 8 | t-AmylOH | Ag2CO3 | 50 | 43 | 94 |
| 9 | t-AmylOH | Ag2CO3 | 60 | 57 | 94 |
| 10 | t-AmylOH | No [Ag] | 60 | 10 | 53 |
| 11d | t-AmylOH | Ag2CO3 | 10 | 4 | 0 |
| 12e | t-AmylOH | Ag2CO3 | 70 | nr | — |

With the optimal reaction conditions in hand, we then investigated the scope of this enantioselective transformation (Table 2). Diaryl-phosphinamides with electron-donating groups (Me, tBu, Ph and OMe) or electron-withdrawing groups (OCF3, F, and Cl) on the para-position gave the corresponding olefination products in moderate to good yields with high enantioselectivities (3b–3h, 50–77% yield, 88–96% ee). meta-Me diarylphosphinamide 1i gave the corresponding product 3i in 62% yield with 96% ee. Diaryl-phosphinamides with steric properties were also tolerated well, and ortho-Me, 1-naphthyl phosphinamide and 2-naphthyl substituted phosphinamide afforded the respective olefination products in good yields with good to high enantioselectivities (3j–3l, 88–92% ee). The scope of the coupling partner, olefins, was also evaluated (Table 2b). A broad range of acrylates bearing Me, tBu, Ph, Bn and hydroxyethyl worked well, giving the desired olefination products in good yields with high ee values (3m–3q, 93–95% ee). In addition, acrylates with useful functional groups, trifluoromethyl and hydroxyl, were still compatible and gave 3r in 60% yield with 95% ee and 3s in 59% yield with 94% ee. Other electronically biased olefins, such as acrylaldehyde, α,β-unsaturated ketone, and acrylamide proceeded well to give the corresponding products in good results (3t–3v, 54–58% yield, 94–95% ee). Various styrenes also reacted smoothly using Ag2SO4 as an oxidant, affording the desired products in moderate yields with high enantioselectivities (3w–3aa, 48–57% yield, 91–94% ee). What's more, chiral phosphinamide bearing core structures of natural products, including L-menthol, tyrosine and estrone, were also obtained in good yields with high enantioselectivities (3ab–3ad, 54–69% yield, 95% ee). The absolute configuration of 3a was ascribed by X-ray diffraction analysis and extrapolated to the other products.13

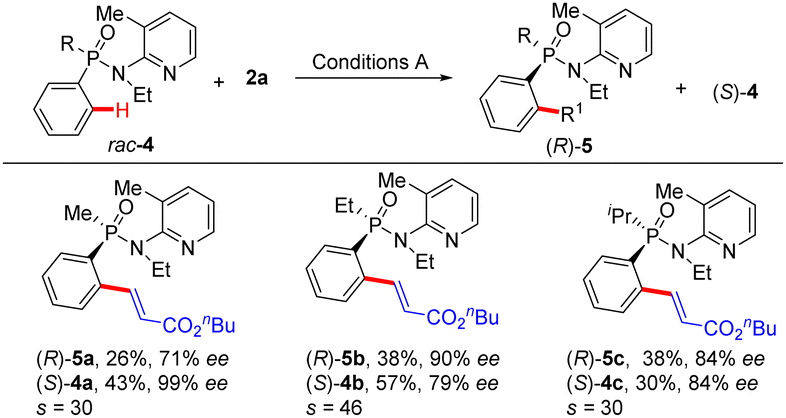
To our delight, kinetic resolution was also compatible under the same conditions (Table 3). Racemic phosphinamides rac-4 with Me, Et and iPr substituted afforded the olefination products (R)-5 and unreacted (S)-4 in good to excellent enantioselectivities.
| a Reaction conditions: rac-4 (0.2 mmol), 2a (0.4 mmol), Pd(OAc)2 (0.02 mmol), L-pGlu-OH (0.04 mmol), Ag2CO3 (0.1 mmol), BQ (0.2 mmol), t-AmylOH (1.0 mL) at 60 °C for 12 h under air in a sealed reaction tube, isolated yield. Selectivity factors: s = ln[(1 − C)(1 − ee4)]/ln[(1 − C)(1 + ee4)], C = (ee4)/(ee4 + ee5). |
|---|
|
We then conducted gram-scale preparation and derivatizations to demonstrate the synthetic utility of this protocol. The olefination of 1a on a 3.0 mmol scale with 2a afforded 3a in 66% yield (920.8 mg) with 90% ee (see the ESI† for details). The directing group of olefination product 3w could be removed in TfOH, giving product 6w in 72% yield with 92% ee (Scheme 2). The reaction of 6w with Grignard reagents afforded the desired chiral phosphine oxides 7a to 7d in good yields with minimum enantiomeric loss.
 | ||
| Scheme 2 Synthetic applications. | ||
Several experiments were conducted to gain mechanistic insights. The H/D exchange experiment was carried out firstly. The recovered 1a was incorporated with no deuterium when 1a reacted in the absence of 2a. The kinetic isotope effect (KIE) experiments were then conducted and a KIE value of kH/kD = 2.38 was obtained. These results indicate that the C–H cleavage step is irreversible and likely the rate-determining step (see the ESI† for details). We then revealed the chiral induction model through DFT calculations. The C–H bond activation transition states of the two phenyl groups (colored by blue and black) of phosphonamide are located (Fig. S2a, ESI†). In TS2-R, there is a significant steric repulsion between the methyl group on the directing group and the phenyl group of phosphamide, so the favorable conformer for generating the R enantiomer is TS1-R. In the transition states that generate the S enantiomer, due to the favorable π–π stacking in TS1-S, its energy is 1.6 kcal mol−1 lower than that of TS2-S. Therefore, the enantioselectivity is determined by TS1-R and TS1-S. The energy difference of 1.9 kcal mol−1 is consistent with the high ee value in the experiment. We also calculated the stereoselective outcome for the kinetic resolution result when one of the phenyl is replaced with methyl. The energy span that determines the enantioselectivity is reduced to 0.9 kcal mol−1 (TS1-R-(Me) vs. TS1-S-(Me)), which is consistent with the experimental results (71% ee).
In summary, we have developed the efficient synthesis of P-stereogenic phosphinamides via Pd(II)-catalyzed enantioselective olefination. A broad range of P-stereogenic phosphinamides were gained in moderate to good yields with high enantioselectivities via desymmetrization and kinetic resolution (33 examples, up to 77% yield, 99% ee). Gram-scale preparation was also compatible with good yield and high ee value. And the olefination P-stereogenic phosphinamide products could be transformed to potentially useful P-chiral phosphine oxides. The practical applications of these new P-stereogenic phosphinamides are currently being explored.
The authors wish to acknowledge the financial support from National Natural Science Foundation of China (U22A20388, 92256302, 21925109 for B.-F. S., 22271250 for T. Z.), Fundamental Research Funds for the Central Universities (226-2023-00115, 226-2022-00175, 226-2022-00224), and Zhejiang Provincial NSFC (LD22B030003), Open Research Fund of Key Laboratory of Organosilicon Chemistry and Material Technology of Ministry of Education, Hangzhou Normal University and Center of Chemistry for Frontier Technologies of Zhejiang University.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) W. Tang and X. Zhang, Chem. Rev., 2003, 103, 3029 CrossRef CAS PubMed; (b) S. J. Connon, Angew. Chem., Int. Ed., 2006, 45, 3909 CrossRef CAS PubMed; (c) H. Fernández-Pérez, P. Etayo, A. Panossian and A. Vidal-Ferran, Chem. Rev., 2011, 111, 2119 CrossRef PubMed; (d) P. W. N. M. van Leeuwen, P. C. J. Kamer, C. Claver, O. Pàmies and M. Diéguez, Chem. Rev., 2011, 111, 2077 CrossRef CAS PubMed; (e) D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047 CrossRef CAS PubMed.
- (a) A. Grabulosa, P-Stereogenic Ligands in Enantioselective Catalysis, RSC, Cambridge, 2011 Search PubMed; (b) W. Tang and X. Zhang, Chem. Rev., 2003, 103, 3029 CrossRef CAS PubMed.
- For select reviews, see: (a) J. S. Harvey and V. Gouverneur, Chem. Commun., 2010, 46, 7477 RSC; (b) I. Wauters, W. Debrouwer and C. V. Stevens, Beilstein J. Org. Chem., 2014, 10, 1064 CrossRef PubMed.
- (a) K. M. Pietrusiewicz and M. Zablocka, Chem. Rev., 1994, 94, 1375 CrossRef CAS; (b) A. Grabulosa, J. Granell and G. Muller, Coord. Chem. Rev., 2007, 251, 25 CrossRef CAS; (c) O. I. Kolodiazhnyi, Tetrahedron: Asymmetry, 2012, 23, 1 CrossRef CAS . For select examples, see: ; (d) Z. S. Han, N. Goyal, M. A. Herbage, J. D. Sieber, B. Qu, Y. Xu, Z. Li, J. T. Reeves, J.-N. Desrosiers, S. Ma, N. Grinberg, H. Lee, H. P. R. Mangunuru, Y. Zhang, D. Krishnamurthy, B. Z. Lu, J. J. Song, G. Wang and C. H. Senanayake, J. Am. Chem. Soc., 2013, 135, 2474 CrossRef CAS PubMed; (e) S. Rast, B. Mohar and M. Stephan, Org. Lett., 2014, 16, 2688 CrossRef CAS PubMed; (f) E. Bergin, C. T. O’Connor, S. B. Robinson, E. M. McGarrigle, C. P. O’Mahony and D. G. Gilheany, J. Am. Chem. Soc., 2007, 129, 9566 CrossRef CAS PubMed; (g) Z. S. Han, H. Wu, Y. Xu, Y. Zhang, B. Qu, Z. Li, D. R. Caldwell, K. R. Fandrick, L. Zhang, F. Roschangar, J. J. Song and C. H. Senanayake, Org. Lett., 2017, 19, 1796 CrossRef CAS PubMed.
- For selected examples, see: (a) Z. Huang, X. Huang, B. Li, C. Mou, S. Yang, B.-A. Song and Y. R. Chi, J. Am. Chem. Soc., 2016, 138, 7524 CrossRef CAS PubMed; (b) B. Pérez-Saavedra, N. Vázquez-Galiñanes, C. Saá and M. Fañanás-Mastral, ACS Catal., 2017, 7, 6104 CrossRef; (c) Y. Zhang, F. Zhang, L. Chen, J. Xu, X. Liu and X. Feng, ACS Catal., 2019, 9, 4834 CrossRef CAS; (d) B. M. Trost, S. M. Spohr, A. B. Rolka and C. A. Kalnmals, J. Am. Chem. Soc., 2019, 141, 14098 CrossRef CAS PubMed; (e) Y. Huang, Y. Li, P.-H. Leung and T. Hayashi, J. Am. Chem. Soc., 2014, 136, 4865 CrossRef CAS PubMed; (f) Y. Toda, M. Pink and J. N. Johnston, J. Am. Chem. Soc., 2014, 136, 14734 CrossRef CAS PubMed.
- For selected examples, see: (a) V. S. Chan, I. C. Stewart, R. G. Bergman and F. D. Toste, J. Am. Chem. Soc., 2006, 128, 2786 CrossRef CAS PubMed; (b) C. Scriban and D. S. Glueck, J. Am. Chem. Soc., 2006, 128, 2788 CrossRef CAS PubMed; (c) C. Scriban, D. S. Glueck, J. A. Golen and A. L. Rheingold, Organometallics, 2007, 26, 1788 CrossRef CAS; (d) B. J. Anderson, M. A. Guino-o, D. S. Glueck, J. A. Golen, A. G. DiPasquale, L. M. Liable-Sands and A. L. Rheingold, Org. Lett., 2008, 10, 4425 CrossRef CAS PubMed; (e) V. S. Chan, M. Chiu, R. G. Bergman and F. D. Toste, J. Am. Chem. Soc., 2009, 131, 6021 CrossRef CAS PubMed; (f) X.-T. Liu, Y.-Q. Zhang, X.-Y. Han, S.-P. Sun and Q.-W. Zhang, J. Am. Chem. Soc., 2019, 141, 16584 CrossRef CAS PubMed; (g) J. R. Moncarz, N. F. Laritcheva and D. S. Glueck, J. Am. Chem. Soc., 2002, 124, 13356 CrossRef CAS PubMed; (h) C. Korff and G. Helmchen, Chem. Commun., 2004, 530 RSC; (i) S. Pican and A.-C. Gaumont, Chem. Commun., 2005, 2393 RSC; (j) T. J. Brunker, B. J. Anderson, N. F. Blank, D. S. Glueck and A. L. Rheingold, Org. Lett., 2007, 9, 1109 CrossRef CAS PubMed; S. Zhang, J.-Z. Xiao, Y.-B. Li, C.-Y. Shi and L. Yin, J. Am. Chem. Soc., 2021, 143, 9912 Search PubMed; (k) N. F. Blank, J. R. Moncarz, T. J. Brunker, C. Scriban, B. J. Anderson, O. Amir, D. S. Glueck, L. N. Zakharov, J. A. Golen, C. D. Incarvito and A. L. Rheingold, J. Am. Chem. Soc., 2007, 129, 6847 CrossRef CAS PubMed; (l) V. S. Chan, R. G. Bergman and F. D. Toste, J. Am. Chem. Soc., 2007, 129, 15122 CrossRef CAS PubMed; Q. Dai, W. Li, Z. Li and J. Zhang, J. Am. Chem. Soc., 2019, 141, 20556 Search PubMed; (m) R. Beaud, R. J. Phipps and M. J. Gaunt, J. Am. Chem. Soc., 2016, 138, 13183 CrossRef CAS PubMed; (n) Q. Dai, L. Li and J. Zhang, Angew. Chem., Int. Ed., 2021, 60, 27247 CrossRef CAS PubMed; (o) X.-T. Liu, X.-Y. Han, Y. Wu, Y.-Y. Sun, L. Gao, Z. Huang and Q.-W. Zhang, J. Am. Chem. Soc., 2021, 143(30), 11309 CrossRef CAS PubMed; (p) W.-H. Wang, Y. Wu, H.-T. Wang, P.-J. Qi, W.-N. Lan and Q.-W. Zhang, Nat. Synth., 2022, 1, 738 CrossRef.
- (a) J. Wencel-Delord and F. Colobert, Chem. – Eur. J., 2013, 19, 14010–14017 CrossRef CAS PubMed; (b) C. Zheng and S.-L. You, RSC Adv., 2014, 4, 6173 RSC; (c) C. G. Newton, S.-G. Wang, C. C. Oliveira and N. Cramer, Chem. Rev., 2017, 117, 8908 CrossRef CAS PubMed; (d) T. G. Saint-Denis, R.-Y. Zhu, G. Chen, Q.-F. Wu and J.-Q. Yu, Science, 2018, 359, eaao4798 CrossRef PubMed; (e) J. Loup, U. Dhawa, F. Pesciaioli, J. Wencel-Delord and L. Ackermann, Angew. Chem., Int. Ed., 2019, 58, 12803 CrossRef CAS PubMed; (f) G. Liao, T. Zhang, Z.-K. Lin and B.-F. Shi, Angew. Chem., Int. Ed., 2020, 59, 19773 CrossRef CAS PubMed; (g) T. Yoshino, S. Satake and S. Matsunaga, Chem. – Eur. J., 2020, 26, 7346 CrossRef CAS PubMed; (h) T. K. Achar, S. Maiti, S. Jana and D. Maiti, ACS Catal., 2020, 10, 13748 CrossRef CAS; (i) O. Vyhivskyi, A. Kudashev, T. Miyakoshi and O. Baudoin, Chem. – Eur. J., 2021, 27, 1231 CrossRef CAS PubMed; (j) Q. Zhang and B.-F. Shi, Acc. Chem. Res., 2021, 54, 2750 CrossRef CAS PubMed; (k) Y.-Q. Han and B.-F. Shi, Acta Chim. Sin., 2023, 81, 1522 CrossRef.
- (a) Q. Shao, K. Wu, Z. Zhuang, S. Qian and J.-Q. Yu, Acc. Chem. Res., 2020, 53, 833 CrossRef CAS PubMed; (b) K. M. Engle and J.-Q. Yu, J. Org. Chem., 2013, 78, 8927 CrossRef CAS PubMed; (c) R. Giri, B.-F. Shi, K. M. Engle, N. Maugel and J.-Q. Yu, Chem. Soc. Rev., 2009, 38, 3242 RSC; (d) B.-F. Shi, N. Y.-H. Maugel, Y.-H. Zhang and J.-Q. Yu, Angew. Chem., Int. Ed., 2008, 47, 4882 CrossRef CAS PubMed.
- Z.-J. Du, J. Guan, G.-J. Wu, P. Xu, L.-X. Gao and F.-S. Han, J. Am. Chem. Soc., 2015, 137, 632 CrossRef CAS PubMed.
- (a) X.-L. Qin, A. Li and F.-S. Han, J. Am. Chem. Soc., 2021, 143, 2994 CrossRef CAS PubMed; (b) G.-J. Wu, D.-X. Tan and F.-S. Han, Acc. Chem. Res., 2021, 54, 4354 CrossRef CAS PubMed.
- (a) J. Diesel and N. Cramer, ACS Catal., 2019, 9, 9164 CrossRef CAS; (b) P.-F. Qian, J.-Y. Li, T. Zhou and B.-F. Shi, Synthesis, 2022, 4784 CrossRef CAS . For selected examples see: ; (c) Z.-Q. Lin, W.-Z. Wang, S.-B. Yan and W.-L. Duan, Angew. Chem., Int. Ed., 2015, 54, 6265 CrossRef CAS PubMed; (d) L. Liu, A. A. Zhang, Y. Wang, F. Zhang, Z. Zuo, W. X. Zhao, C. L. Feng and W. Ma, Org. Lett., 2015, 17, 2046 CrossRef CAS PubMed; (e) G. Xu, M. Li, S. Wang and W. Tang, Org. Chem. Front., 2015, 2, 1342 RSC; (f) Y. Sun and N. Cramer, Angew. Chem., Int. Ed., 2017, 56, 364 CrossRef CAS PubMed; (g) Y. Sun and N. Cramer, Chem. Sci., 2018, 9, 2981 RSC; (h) P. Hu, L. Kong, F. Wang, X. Zhu and X. Li, Angew. Chem., Int. Ed., 2021, 60, 20424 CrossRef CAS PubMed; (i) C.-W. Zhang, X.-Q. Hu, Y.-H. Dai, P. Yin, C.-Y. Wang and W.-L. Duan, ACS Catal., 2022, 12, 193 CrossRef CAS; (j) S.-Y. Song, Y. Li, Z. Ke and S. Xu, ACS Catal., 2021, 11, 13445 CrossRef CAS; (k) G. R. Genov, J. L. Douthwaite, A. S. K. Lahdenperä, D. C. Gibson and R. J. Phipps, Science, 2020, 367, 1246 CrossRef CAS PubMed; (l) Q.-J. Yao, J.-H. Chen, H. Song, F.-R. Huang and B.-F. Shi, Angew. Chem., Int. Ed., 2022, e202202892 CAS; (m) J.-H. Chen, M.-Y. Teng, F.-R. Huang, H. Song, Z.-K. Wang, H.-L. Zhuang, Y.-J. Wu, X. Wu, Q.-J. Yao and B.-F. Shi, Angew. Chem., Int. Ed., 2022, e202210106 CAS.
- T. Zhou, L.-J. Fan, Z.-J. Chen, M.-X. Jiang, P.-F. Qian, X. Hu, K. Zhang and B.-F. Shi, Org. Lett., 2023, 25, 5724 CrossRef CAS PubMed.
- CCDC 2183918 (3a) contain the supplementary crystallographic data for this paper†.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2183918. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc05052a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |