
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Lipid–polymer hybrid-vesicles interrupt nucleation of amyloid fibrillation†‡
Newton
Sen
a,
Stephanie
Krüger
b and
Wolfgang H.
Binder
 *a
*a
aMacromolecular Chemistry, Institute of Chemistry, Faculty of Natural Science II (Chemistry, Physics and Mathematics), Martin Luther University Halle-Wittenberg, von-Danckelmann-Platz 4, Halle D-06120, Germany. E-mail: wolfgang.binder@chemie.uni-halle.de
bBiocenter, Martin-Luther University Halle-Wittenberg, Weinbergweg 22, Halle (Saale) D-06120, Germany
First published on 24th October 2024
Abstract
Solubility and aggregation of proteins are crucial factors for their functional and further biological roles. Aggregation of proteins in vivo, such as the amyloid beta (Aβ1–40) peptide into fibrils, is significantly modulated by membrane lipids, abundantly present in cells. We developed a model membrane system, composed of lipid hybrid-vesicles bearing embedded hydrophilic polymers to in vitro study the aggregation of the Aβ1–40 peptide. Focus is to understand and inhibit the primordial, nucleation stages of their fibrillation by added hybrid-vesicles, composed of a natural lipid and amphiphilic polymers. These designed hybrid-vesicles are based on 1-palmitoyl-2-oleoyl-glycero-3-phosphocholine (POPC), displaying embedded hydrophilic (EO)mPnA_EG polymers (m = 2 or 3; Pn = 10 to 52 with Mn = 2800–9950 gmol−1) in amounts ranging from 5–20 mol%, anchored to the POPC vesicles via hydrophobic hexadecyl-, glyceryl- and cholesteryl-moieties, affixed to the polymers as end-groups. All investigated hybrid-vesicles significantly delay fibrillation of the Aβ1–40 peptide as determined by thioflavin T (ThT) assays. We observed that the hybrid-vesicles interacted with early aggregating species of Aβ1–40 peptide, irrespective of their composition or size. A substantial perturbation of both primary (k+kn) and secondary (k+k2) nucleation rates of Aβ1–40 by the POPC–polymer vesicles compared to POPC vesicles was observed, particularly for the cholesteryl-anchored polymers, interfering with the fragmentation and elongation steps of Aβ1–40. Furthermore, morphological differences of the aggregates were revealed by transmission electron microscopy (TEM) images supported the inhibitory kinetic signatures.
Introduction
In humans, about 20,![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 distinct proteins are associated with the protein homeostasis,1 while the pathogenicity of amyloids is linked with over 50 various proteins and peptides.2 The pathogenicity of proteins manifests through the formation of highly ordered amyloid fibrillar structures, a densely packed cross β-strand stabilized by ‘steric zippers’, arising from the variable primary sequences of proteins which ultimately succumb to the formation of amyloid aggregate.3 These fibrillary forms of protein aggregation are involved in neurodegenerative diseases (Alzheimer's, Parkinson's)4 to insulin aggregation5 and eye lens protein aggregation.6
000 distinct proteins are associated with the protein homeostasis,1 while the pathogenicity of amyloids is linked with over 50 various proteins and peptides.2 The pathogenicity of proteins manifests through the formation of highly ordered amyloid fibrillar structures, a densely packed cross β-strand stabilized by ‘steric zippers’, arising from the variable primary sequences of proteins which ultimately succumb to the formation of amyloid aggregate.3 These fibrillary forms of protein aggregation are involved in neurodegenerative diseases (Alzheimer's, Parkinson's)4 to insulin aggregation5 and eye lens protein aggregation.6
The non-covalent polymerization of soluble proteins into solid amyloids can be initiated by a reversible liquid–liquid phase transition, which then irreversibly can lead to the formation of solid amyloid condensates. When the monomer concentration reaches supersaturation in amyloidogenesis,7,8 their polymerization involves a cascade of microscopic steps like homo- and heterogeneous primary nucleation, secondary nucleation (small aggregates dependent), fragmentation, and elongation to insoluble fibrillar solids. Secondary nucleation, catalytic in nature,9 and the subsequent microscopic steps in a single process required for amyloidosis, follow classical and non-classical nucleation theory to connect microscopic steps kinetically and thermodynamically.7,10,11 To deeper understand the complexity of those aggregation processes, model systems (in vitro) are important to understand, modulate, and if possible, control this aggregation process, finally aiming for its inhibition. Thus the amyloidogenesis of amyloid beta (Aβ1–40/42) peptides is intricately regulated by various factors, both often intracellular (like pH, ions) and extracellular (like air–water interfaces, lipid membranes, other proteins like α-synuclein,12 preformed fibrils, small aggregates, seed concentrations and temperature) with the precise role of each factor being largely unclear,13 in view of the complex role in Alzheimer's disease pathogenicity.14 Neuronal biomembranes, where amyloid aggregation takes place, are mainly composed of phospholipids (both zwitterionic and anionic), cholesterol, ganglioside (GM1),15,16 and sphingomyelin,17 all potentially involved in the first steps of this complex protein assembly process. In vitro studies of physiochemical properties of lipid bilayers like membrane's phase, thickness,18 curvature,19 oxidative stress,20–22 among many others, produce a significant impact on amyloid aggregation.23 The use of lipid model systems, such as lipid vesicles composed of natural lipids like zwitterionic lipids (POPC, DMPC, and POPE), anionic lipids (POPS, POPG), neutral lipids (DOPC), cholesterol24–26 or a mixture of lipids offers a powerful platform to decipher the membrane role on Aβ1–40/42 fibrillation in vitro.27,28 Early stages of fibrillation, such as nucleation (primary, secondary, and surface-mediated nucleation) are profoundly decided by model lipids14,29,30 that can induce both, acceleration or inhibition of the fibrillation.14,28,31–36 In this context, efforts are directed towards targeting the deleterious early forms (oligomers, fibrillar segments, small liquid condensates) of the amyloid transformation to reduce amyloid beta peptide (Aβ1–40/42) pathogenicity by inhibition of early nucleation stages.37,38
Similar to the counterintuitive effects of lipid bilayer physico–chemical properties on amyloid fibrillation, PEGylated lipids are interesting candidates representing pharmaceutically accepted drugs, with the ability to balance lipid hydrophobicity by PEG hydrophilicity.39 Therefore, they are potential candidates to inhibit amyloid-aggregation as determined in our previous work, displaying small, but significant effects40 with enhancements of the lag-times of Aβ1–40/42 peptide fibrillation by a factor of ∼12. We here investigate a set of potential inhibitors for Aβ1–40/42 peptide fibrillation by a novel approach based on hybrid-vesicles, composed of model peptide (POPC) hybridized with hydrophilic polymers (hydrophobic and membrane-anchored, see Fig. 1) bearing PEO-sidechains. In the current investigations we delved into the impact of fine-tuning the hydrophilic–hydrophobic profile of the polymers on hybridization and amyloid formation akin to the pegylated lipids. Biophysical approaches were employed to gain insights into how the hybrid system modulates the fibrillation and conformational changes, particularly upon binding with the hybrid-vesicles. Via in vitro approaches, we elucidated the inhibition potential of those vesicles, together with analyzing of early stages of fibrillation and the associated nucleation events.
 | ||
| Fig. 1 Lipid–polymer hybrid-vesicles and their influence on Aβ1–40 fibrillation. We focus on primary and secondary nucleation, elongation, and fragmentation steps (A; primary, secondary nucleation and B; fragmentation microscopic processes). These hybrid-vesicles can interfere with all steps of this Aβ1–40 fibrillation process, bearing the polymers, (EO)mPnA_EG, in variable molar ratios. Molecular structures of the lipid and polymers are displayed at the bottom of the figure. | ||
Result and discussion
Conceptually we have used vesicles, composed of POPC as the main lipid, bearing an added hydrophilic polymer, safely embedded into the vesicle via a lipid anchor (Fig. 1). Tuning of the polymer's hydrophilicity is accomplished by changing the length of the ethylene-units at the monomers from either two (DGME) or three (TGME) ethylene oxide (EO) units, with the overall polymer's molar mass ranging from 2800 to 9950 Da. Lipid anchors (hexadecyl, glyceryl and cholesteryl groups) were used to stably incorporate those hydrophilic polymers into the POPC vesicles, subsequently probing their influence on Aβ1–40 fibrillation as outlined in Fig. 1. Polymers were prepared by RAFT (Reversible Addition Fragmentation Chain Transfer) polymerization, finely tuning their hydrophilic–hydrophobic profile by controlling parameters such as the degree of polymerization (n), the number of EO (ethylene oxide) units in the polymer backbone, and the incorporation of anchoring groups (such as membrane lipids and hydrophobic moieties).40 The synthesis and characterization of the so tuned polymers is described in the ESI‡ (see Scheme S1 and Fig. S1–S2) together with structural details of the selected polymers presented in Table 1. Biophysical approaches such as thioflavin T (ThT) fluorescence kinetic study, CD-spectroscopy, and TEM were employed to quantitatively and qualitatively explicate the impact of hybrid-vesicles on Aβ1–40 fibrillation. Moreover, the mechanistic insights of how these hybrid-vesicles influence Aβ1–40 aggregation were further investigated using the established Nature Protocol adopted in Amylofit.41| Entry | Core lipid conc. (mM) | Embedded polymer | Anchored group | M n (gmol−1) | Polymer incorporation (mol%) |
|---|---|---|---|---|---|
| a Molar mass (Mn) of the polymers confirmed via1H-NMR in CDCl3. | |||||
| 1 | POPC (1.5 mM) | (EO)2P19A_Hy | Hexadecyl (Hy) | 3800 | (5–20)% |
| 2 | (EO)2P39A_Hy | 7250 | |||
| 3 | (EO)3P12A_Hy | 3100 | |||
| 4 | (EO)3P26A_Hy | 6150 | |||
| 5 | (EO)2P22A_Gl | Glyceryl (Gl) | 4600 | (5–20)% | |
| 6 | (EO)2P44A_Gl | 8450 | |||
| 7 | (EO)3P11A_Gl | 3200 | |||
| 8 | (EO)3P42A_Gl | 9950 | |||
| 9 | (EO)2P23A_Co | Cholesteryl (Co) | 4600 | (5–20)% | |
| 10 | (EO)2P48A_Co | 9000 | |||
| 11 | (EO)3P10A_Co | 2800 | |||
| 12 | (EO)3P18A_Co | 4550 | |||
| 13 | (EO)3P52A_Co | 8000 | |||
Formation of hybrid-vesicles
Hybrid-vesicles were obtained by mixing POPC (as a core lipid component) with the respective polymers, carefully tuned by embedding anchoring groups, modulating their chain length (molecular weight), and add a specific number of EO units onto the polymer backbone. These so-tuned polymer characteristics allow embedding into the POPC-vesicles and further tune their outside-surface properties, resulting in hybrid-vesicles. This allowed a variable amount (5 to 20 mol%) of polymers in the hybrid-vesicles, modulating the hydrophilicity of the outer surface of these hybrid-vesicles.Incorporating polymers with variable properties and amounts into the hybrid-vesicles posed a challenge as this alters the physio–chemical properties of the resulting hybrid-vesicles. We employed a modified extrusion process for the hybridization of POPC vesicles to generate hybrid-vesicles within a desirable size range of 55–200 nm (see Table S1, ESI‡). Lipids and polymers were mixed in water-free chloroform/methanol (2/1) at room temperature, followed by film formation and solvent removal. Subsequently, hybrid-vesicles were prepared in a 50 mM Na2HPO4 buffer solution supplemented with 150 mM NaCl at pH 7. 4 and a modified extrusion process. The buffer solution of the lipid–polymer mixture was extruded through a 400 nm polycarbonate (PC) membrane, followed by 200 nm and 100 nm PC membranes to obtain the desired size range of the hybrid-vesicles (details in ESI‡).42 The maxima of the narrow size distribution curves of dynamic light scattering (DLS) displayed the desired size range, while cryo transmission electron microscopy (Cryo-TEM) images shown in Fig. 2 and Fig. S4 (ESI‡) further confirmed the formation of hybrid-vesicles. The hybrid-vesicles exhibited stability at 4 °C for several hours, providing a sufficiently good timeframe for utilizing them in subsequent Aβ1–40 fibrillation kinetic investigations.
 | ||
| Fig. 2 Cryo-TEM images of hybrid-vesicles with a scale bar of 250 nm. (A) (EO)3P26A_Hy_20 mol%, (B) (EO)3P11A_Gl_5%, (C) (EO)3P42A_Gl_5%, (D) (EO)3P42A_Gl_20%, (E) (EO)2P48A_Co_10%, (F) (EO)3P18A_Co_10% and (G) (EO)3P52A_Co_10% polymers embedded in POPC lipid. | ||
To ensure the complete embedding of the polymers into the hybrid-vesicles and quantify the amount of polymers incorporated inside the hybrid-vesicles, 1H- and 31P-NMR of the hybrid-vesicles bearing 5–15 mol% of (EO)2P48A_Co were performed and compared to the pure POPC vesicles (presented in Fig. S3, ESI‡). Proton NMR of the hybrid-vesicles confirmed the incorporation of the polymers into the hybrid-vesicles and enabled quantification of their amounts (see Fig. S3, ESI‡) by integration of resonances of the polymer vs. the lipid. The lipid head groups and their surrounding environment, especially their polarity are also sensitive to 31P-NMR spectroscopy; therefore, the chemical shift of 31P-NMR clearly indicates the incorporation of polymers in hybrid-vesicles.43–45 Consequently, the chemical shift for pure POPC vesicles shifted from approximately −0.6 ppm to 0.3 ppm upon incorporation of the polymers inside the POPC-vesicles in amounts of up to 15%. This change in the chemical shift serves as a clear indicator of successful polymer incorporation, with the amphiphilic environment introduced by the polymers likely contributing to this effect. Moreover, confocal fluorescence microscopy of Rh-DPPE (Lissamine rhodamine B-1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine) fluorescently dye-labeled giant unilamellar vesicles (GUVs) composed of pure POPC and hybrid GUVs containing 5 mol% of (EO)2P48A_Co revealed no unusual membrane inhomogeneities in the POPC membrane caused by the polymer's incorporation (Fig. S20, ESI‡).46 The physical integrity of the hybrid-GUVs played a crucial role in the functionality, together with the proven stability of the polymers when embedded inside the membrane (as checked by MALDI-ToF measurements, see Fig. S22 and S23, ESI‡). This was further corroborated by monitoring the zeta potential (Fig. S21C, ESI‡) and size measurements (Fig. S21A, ESI‡) of the hybrid-vesicles over time and temperature (Fig. S21B, ESI‡).
Modulating fibrillation with hybrid-vesicles
We studied the influence of the hybrid-vesicles on Aβ1–40 fibrillation, focussing on macroscopic changes (determining lag time; tlag and half time; t1/2 of fibrillation by ThT assays) and morphological characteristics of the resulting aggregates via TEM. For the ThT assays purified Aβ1–40 peptide was dissolved and sonicated in buffer, the same buffer used for hybrid-vesicles preparation, to achieve a nucleation-free monomeric Aβ1–40 for the subsequent ThT assay.47 The freshly prepared POPC and hybrid-vesicles were added to the monomeric fibrillating amyloid-Aβ1–40 proteins, using a ratio of [lipid/peptide] = [150/1], shortly before the ThT-assays (details of the fibrillation kinetics in ESI‡).42 Utilizing in vitro biophysical assays by ThT fluorescence allowed tracking of the fibrillation kinetics and provided a deeper insight into both, quantitative parameters (from tlag and t1/2) and further qualitative aspects (including the mechanism of interactions). In all cases, the hybrid-vesicles, containing the embedded polymers, interfered significantly with the fibrillation processes and resulted in elongated fibrillation times, proving that all investigated hybrid-vesicles exerted a pronounced influence on Aβ1–40 fibrillation, as evidenced by the graphical representation of Aβ1–40 fibrillation kinetics shown in Fig. 3 and Fig. S5 (ESI‡). It was noted that fibrillation of Aβ1–40 had accelerated slightly (t1/2 ≈ 2.5 hours and tlag ≈ 2 hours) in the presence of native POPC vesicles devoid of polymers compared with the native Aβ1–40 (t1/2 ≈ 4.5 hours and tlag ≈ 4 hours). The fibrillation kinetics of both native Aβ1–40 and polymer-free POPC vesicles were considered as a reference (see Fig. 3, 4 and Fig. S5, ESI‡). Subsequently, the tlag and t1/2 obtained from the fitting of kinetics are summarized in Fig. 4 and Table S1 (ESI‡). | ||
| Fig. 3 Analysis of the effects of hybrid-vesicles on the aggregation profile of Aβ1–40. Thioflavin T (ThT) monitored kinetic profiles for the aggregation of Aβ1–40 in phosphate buffer solutions of pH 7.4 in the presence of POPC and hybrid-vesicles at 37 °C are shown in (A) to (D). The black error bars represent the standard deviation of three normalized independent time-resolved ThT fluorescence. Fibrillation kinetics of native Aβ1–40 (black diamond shape) and in the presence of POPC- and hybrid-vesicles embedded with comparable low (A) and (B) and high (C) and (D) molecular weight polymers bearing two and three ethylene oxide (EO) units, respectively. The polymers and their respective molar fraction in the hybrid-vesicles are presented in each panel with distinctive colors. Hy, Gl, and Co represent the hexadecyl, glyceryl, and cholesteryl end groups of the embedded polymers. | ||
 | ||
| Fig. 4 Quantitative determination of the impact of hybrid-vesicles on Aβ1–40 aggregation. The lag time, tlag, and half time, t1/2 are shown in each panel to represent the tlag and t1/2 estimated from the fitting of three individual Aβ1–40 fibrillation kinetics in the presence of POPC and POPC–polymer vesicles. tlag and t1/2 are plotted against the concentration of the incorporated polymers inside the hybrid-vesicles and compared with the t1/2 of Aβ1–40 in the absence of any vesicles in the solution. Impact of (A) hexadecyl; Hy, (B) glyceryl group; Gl and (C) cholesteryl group; Co anchored polymers in hybrid-vesicles on Aβ1–40 fibrillation. | ||
We firstly focussed on a set of POPC; polymer hybrid-vesicles bearing 5 mol% polymers to study the impact of polymer properties like hydrophilicity (number of EO units, degree of polymerization; n) and the hydrophobicity (nature of the anchoring groups; hexadecyl (Hy), glyceryl (Gl) and cholesteryl (Co)) on amyloid fibrillation. Thus, the nature of the tethering groups exhibited a profound impact on Aβ1–40 fibrillation, in all cases inducing a significant elongation of the fibrillation. In the presence of the hexadecyl group bearing polymer, (EO)2P19A_Hy; 3800 Da inhibition of fibrillation was observed with a t1/2 of ≈ 40 hours and tlag ≈ 15 hours, whilst the (EO)2P22A_Gl; 4600 Da polymer with a glyceryl group exhibited lower retardation effects (tlag ≈ 18 hours and t1/2 ≈ 28 hours) of fibrillation. The strongest retardation was observed when the cholesteryl group anchored (EO)2P23A_Co; 4600 Da polymer, incorporated in the POPC-hybrid-vesicles with t1/2 ≈ 143 hours and tlag ≈ 67 hours (Fig. 3A, 4 and Table S1, ESI‡).
There also was a strong influence of the length of the side-chain-EO-groups (2, 3) on fibrillation, when comparing similar polymers of otherwise similar structure. Among the previously mentioned polymers, the (EO)2P22A_Gl polymer exhibited a shorter retardation time compared to the other two mentioned counterparts. However, when the number of EO units increased from 2 to 3 a significant fibrillation inhibition was attained in (EO)3P11A_Gl, which was quantified with a t1/2 of 28 hours to 47 hours and tlag of 18 hours to 31 hours (see Fig. 3A, B, 4 and Table S1, ESI‡). We further studied the influence of chain length of the polymer upon the fibrillation of Aβ1–40.40 By comparing (EO)3P42A_Gl with increasing molar mass from 3200 Da to 9950 Da but otherwise identical number of EO units and the anchoring group, a substantial elongation of the lag time ≈76 hours for 5 mol% (EO)3P42A_Gl polymer bearing hybrid-vesicles compared with the native Aβ1–40 lag time (≈4 hours) was observed (details in Fig. 3C, 4 and Table S1, ESI‡).
Other than the above-mentioned inherent properties of polymers, the amount of incorporated polymers into hybrid-vesicles was also crucially important for altering the physio–chemical properties of the vesicles and concomitantly the fibrillation behaviour. In every tested lipid–polymer composition, 5 to 20 mol% polymers within POPC lipid, the inhibition of fibrillation persisted. However, no clear trend emerged correlating an increase in polymer content with greater fibrillation retardation. Focusing on a single set of hybrid-vesicles containing (EO)3P10A_Co, a tlag ≈ 45 hours was observed when the hybrid-vesicles were bearing only 5 mol%, changing only slightly the tlag (≈46 hours) when using 20 mol%. A more pronounced shift in the aggregation t1/2, from ≈69 hours to ≈75 hours was found between these two concentrations. Further, quantitative analysis (tlag, and t1/2) of the fibrillation kinetics are displayed in Fig. 4, Fig. S6 and Table S1 (ESI‡).
Besides the quantitate estimation of tlag and t1/2 from the fibrillation kinetics, the total amount of fibrillar aggregates can also be calculated from the raw ThT intensities upon reaching their maximum (see Fig. S7 and S8, ESI‡). The collective amount of fibrils was reduced substantially with the quantity of mature fibrils further reduced in the presence of all hybrid-vesicles regardless of their embedded polymer amount and properties (see Fig. S9, ESI‡).
Fibrillation kinetics and mechanistic understanding of the hybrid-vesicles interaction
Time-resolved ThT fluorescence kinetics can unveil microscopic processes such as nucleation (primary and secondary), fragmentation, elongation, and mature fibril formation during the transformation of the functional soluble state of the proteins to insoluble pathogenic solid aggregates. Mathematical models incorporating these microscopic processes offer a numerical survey of the fibrillation kinetics, including both seeded (involving small fibrils or pre-fibrillar aggregates) and unseeded (dependent on monomers) models, all contributing to the mechanistic understanding of the chemical progression to amyloids over time quantitatively.48–52 We here have used the open-access online fitting platform Amylofit41 to decipher the mechanism underlying amyloid aggregation and how the added hybrid-vesicles could interfere with fibrillation as observed in the ThT fluorescence kinetics. Fitting of the experimental ThT kinetics allows extraction of the integrated rate laws of the specific microscopic steps. Comparison of the rate laws enables interpretation of the interference by the materials semi-quantitatively. Our experimental kinetic data were fitted with the microscopic step-variable kinetic models to identify the most suitable model. Testing several models, the global analysis of the ThT fluorescence traces obtained for most hybrid-vesicles fitted reasonably with the unseeded version of the secondary nucleation dominated model. However, for some hybrid-vesicles bearing mainly the cholesteryl-anchored polymers, the unseeded version of fragmentation and secondary nucleation dominated model globally fitted suitably (Fig. 5 and Fig. S6, ESI‡). A brief summary of the hybrid-vesicles influence on Aβ1–40 fibrillation is presented in Table 2. All corresponding integrated rate laws from a quantitative analysis using Amylofit are listed in Table S2 (ESI‡) and graphically represented in Fig. 6. The combined rate constants k+kn (primary nucleation rate) and k+k2 (secondary nucleation rate) of Aβ1–40 fibrillation in the presence of hybrid-vesicles allow a rough correlation of inhibition of fibrillation in microscopic processes. POPC-vesicles only marginally accelerated the fibrillation in comparison to native Aβ1–40, while all hybrid-vesicles delayed fibrillation significantly. Subsequently, a substantial perturbation of both primary (k+kn) and secondary (k+k2) nucleation rates of Aβ1–40 in the presence of POPC–polymer vesicles compared to POPC vesicles was observed (see Fig. 6 and Table S2, ESI‡). Additionally, some of the hybrid-vesicles, particularly those embedding cholesteryl-anchored polymers, interfered with the fragmentation and elongation steps compared to the other investigated hybrid-vesicles as reflected in the combined rate laws (k−k+) listed in Table S2 (ESI‡). The extended fibrillation, as indicated by their microscopic rate constants, was affected by the lipid's hydrophobicity or proposedly by steric hindrance arising from non-productive interactions with the hydrophilic portion of the polymer to the hybrid-vesicles surface.28,39 These interactions could potentially alter the microscopic processes of fibrillation, including the morphological transition of the aggregates. | ||
| Fig. 5 Correlation between nature of the hybrid-vesicles and Aβ1–40 aggregation. Mean normalized kinetic reaction profiles fitted globally in Amylofit keeping the nucleus size (nc, primary = n2, secondary = 2)30,51,53,54 and initial monomer concentration (10 μM) of Aβ1–40 constant while fitting. The red dotted lines represent the fitting of the curve. Most hybrid-vesicles (presented in A, B and C panels) interfered the primary (1°) and secondary (2°) nucleation pathways of Aβ1–40 aggregation, whereas the hybrid-vesicles bearing (EO)3P26A_Hy_20% (panel A) and (EO)2P48A_Co_10% (panel C) polymers interfered the elongation pathway along with the nucleation pathways. Both POPC and hybrid-vesicles including polymers embedded are presented at the bottom of each panel with distinctive colours of the circle. | ||
| Polymer | Polymer mole fraction (%) incorporated in POPC: Polymer hybrid system | Mechanism of Aβ1–40 aggregation interference via hybrid-vesicles |
|---|---|---|
| (EO)2P19A_Hy | (5–20) % | Primary and secondary nucleation of Aβ1–40 aggregation interfered via hybrid-vesicles |
| (EO)2P39A_Hy | (5–20) % | |
| (EO)3P12A_Hy | (5–20) % | |
| (EO)3P26A_Hy | 5%, 10% | |
| (EO)2P22A_Gl | (5–20) % | |
| (EO)2P44A_Gl | (5–20) % | |
| (EO)3P11A_Gl | (5–20) % | |
| (EO)3P42A_Gl | (5–20) % | |
| (EO)2P23A_Co | (5–20) % | |
| (EO)3P10A_Co | 5%, 10%, 20% | |
| (EO)3P18A_Co | (5–20) % | |
| (EO)3P52A_Co | 5%, 10%, 20% | |
| (EO)3P26A_Hy | 15%, 20% | Primary, fragmentation and secondary nucleation of Aβ1–40 aggregation interfered via hybrid-vesicles |
| (EO)3P10A_Co | 15% | |
| (EO)2P48A_Co | (5–20) % | |
| (EO)3P52A_Co | 15% |
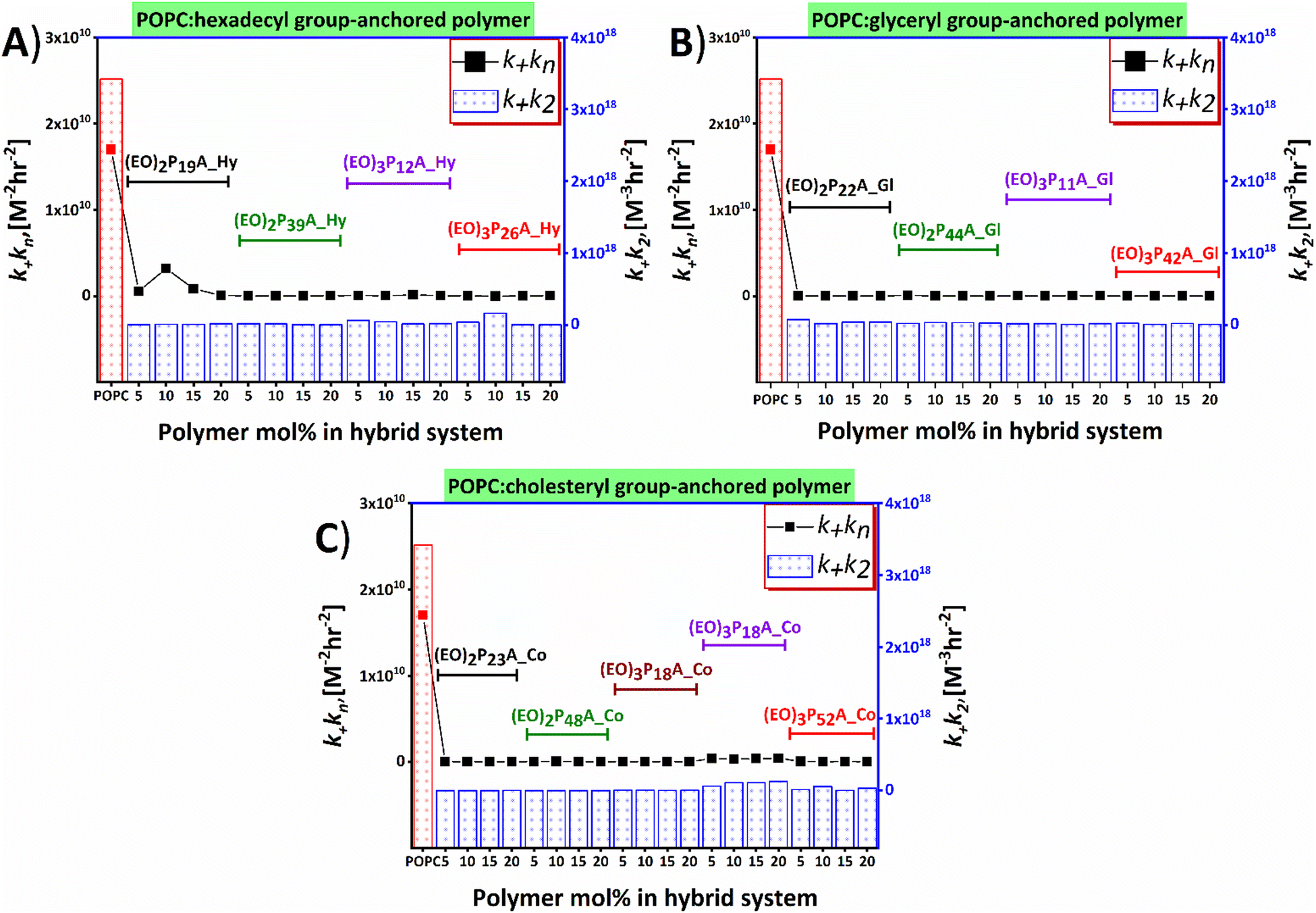 | ||
| Fig. 6 Influences of lipid–polymer hybrid-vesicles on primary and secondary nucleation process of Aβ1–40 aggregation as analysed by Amylofit. Amount of polymers embedded in hybrids plotted against the k+kn and k+k2 for variable end groups hexadecyl (Hy), glyceryl (Gl), and cholesteryl (Co) bearing polymers are presented in panels (A), (B), and (C), respectively. Polymers are marked with specific colors. The k+kn (for primary nucleation) and k+k2 (for secondary nucleation) of Aβ1–40 in the presence of hybrid-vesicles are compared with the POPC vesicles and obtained from the global fitting of ThT fluorescence kinetics. | ||
Morphological transitions of aggregates upon interaction with hybrid-vesicles
The reduced ThT fluorescence was a clear indication of structural changes in Aβ1–40 aggregates upon interaction with the hybrid-vesicles by changing into the aggregated β-sheets structures. To track eventual morphological changes in the Aβ1–40 aggregates induced by the POPC and hybrid-vesicles, circular dichroism (CD) spectroscopy and transmission electron microscopy (TEM) imaging experiments were performed on the fibrillar aggregates. The images presented in Fig. 7 and Fig. S15 (ESI‡) revealed the presence of secondary structures of Aβ1–40 aggregates after interactions with the vesicles. The incubation of monomeric Aβ1–40 with hybrid-vesicles is accompanied by a structural shift from enriched β-sheets to random coil conformations. This shift was distinct from the structural characteristics observed for native Aβ1–40 (as evidenced by CD-spectra presented in Fig. S12–S14, ESI‡). Furthermore, the CD-spectra of the aggregates validated the findings of fibril load, through structural elucidation via the BeStSel (Beta Structure Selection) algorithm, an online platform to determine secondary structures of proteins,55 unveiled the presence of α-helix and irregular structures along with compact β-sheets in the aggregates (see summary in Table S3, ESI‡). Subsequently, TEM images were used to crosscheck the structural transitions of the Aβ1–40 aggregates, revealing the presence of short, less compact aggregates to dense long entwined fibrils or no apparent fibrillar structures. Hybrid-vesicles with respective polymers and compositions are presented according to the microscopic steps of the Aβ1–40 aggregation interrupted upon interactions with the hybrid-vesicles. | ||
| Fig. 7 Morphological analysis hybrid-vesicles interactions with Aβ1–40 fibrils. Aβ1–40 fibrils formed in ThT assays in the presence of hybrid-vesicles containing (A) (EO)3P12A_Hy_10%, (B) (EO)3P26A_Hy_5%, (C) (EO)2P44A_Gl_20%, (D) (EO)2P44A_Gl_20%, (E) (EO)2P23A_Co_20% and (F) (EO)3P10A_Co_10% polymers. A negative stain of uranyl acetate was used to record TEM images. The scale bar of the images represents 100 nm. | ||
Conclusion
As neuronal membrane components and surfaces play a crucial role in nucleating the pathogenicity of the Aβ1–40,14,32,33,56,57 we here studied the impact of artificial hybrid-vesicles on amyloid fibrillation. Amphiphilic polymers were embedded into hybrid-POPC vesicles and were probed as an in vitro strategy to modulate Aβ1–40 fibrillation. Upon incubation of non-aggregated Aβ1–40 with hybrid-vesicles, a significant inhibition of fibrillation by an increase in the fibrillation times (both tlag and t1/2) was observed for all hybrid-vesicles bearing the embedded amphiphilic polymers with factors of up to 30-fold, compared to the native vesicles devoid of the incorporated polymer. Compared to our previous investigations with the same polymers being non-embedded inside the liposomes,40 we here observed a significant elongation of the fibrillation times of a factor 1.5–2, which hints at a contribution of the liposomal surfaces. A systematic mechanistic analysis by the program Amylofit allowed to track kinetic parameters (k+kn and k+k2) and thus primordial states of the Aβ1–40 aggregation. The polymer-modified liposomes interfere with amyloid nucleation processes, affecting both primary and secondary nucleation rate constants (k+kn and k+k2). This suggests an interaction on the molecular level between the hybrid-vesicles and the proteins also visible by the reduced fibril loads and the morphological transition of the highly compact long unbranched fibrils to very short uncompacted fibrils or amorphous aggregates. This remarkable inhibitory behaviour of the hybrid-vesicles may be attributed to an umbrella effect over the hybrid-vesicles created by the tuned hydrophilic–hydrophobic profile of the embedded polymers.58 Polymer-induced shielding effects over the hybrid-vesicles surface could ultimately resist the interactions (both electrostatic and hydrophobic interactions) between the monomers and hybrids, thereby mitigating amyloid fibrillation.It can also be considered that attenuated fibrillation is linked to the net charge of the POPC-hybrid-vesicles surface, altered by the amphiphilicity of the polymers that potentially facilitates repulsion to accumulate the negatively charged Aβ1–40 monomers.59–63 This overall stresses the importance of the outer decoration of vesicular, membrane-like surfaces in the fibrillation kinetics of the Aβ1–40 protein, further supported by CD and TEM. We think that our current work significantly extends the understanding of the role of tailored membrane physio–chemical properties in the context of their inhibitory role in Aβ1–40 pathogenesis via in vitro studies, stressing the influence of the early stages of Aβ1–40 aggregation and underscoring the impact of vesicular membrane in the process of amyloid disease progression.
Author contributions
N. S. did the synthesis of polymers, hybrid-vesicles preparation, and fibrillation kinetics assays. S. K. performed cryo TEM and TEM. The research was designed and the data was analyzed by N. S. and W. H. B. The manuscript was written by N. S. and W. H. B.Data availability
Materials, experimental procedures, NMR and mass spectra, raw data have been included in the ESI.‡Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the state of Saxony-Anhalt for support in the POliFaces initiative; as well as the SFB TRR 102/TP A012, project Nr 189853844. Open access funding enabled and organized by Projekt DEAL. The authors express their gratitude to Prof. Dr Jochen Balbach and Dr Anja Marinow for their invaluable assistance and to Dr Sven Rothemund for the synthesis of the native Aβ1–40 peptide. The authors would like to thank Prof. Dr Kirsten Bacia and Dr Caroline Haupt for providing the confocal microscopy images displayed in the ESI.‡References
- R. Aebersold, J. N. Agar, I. J. Amster, M. S. Baker, C. R. Bertozzi, E. S. Boja, C. E. Costello, B. F. Cravatt, C. Fenselau, B. A. Garcia, Y. Ge, J. Gunawardena, R. C. Hendrickson, P. J. Hergenrother, C. G. Huber, A. R. Ivanov, O. N. Jensen, M. C. Jewett, N. L. Kelleher, L. L. Kiessling, N. J. Krogan, M. R. Larsen, J. A. Loo, R. R. Ogorzalek Loo, E. Lundberg, M. J. MacCoss, P. Mallick, V. K. Mootha, M. Mrksich, T. W. Muir, S. M. Patrie, J. J. Pesavento, S. J. Pitteri, H. Rodriguez, A. Saghatelian, W. Sandoval, H. Schlüter, S. Sechi, S. A. Slavoff, L. M. Smith, M. P. Snyder, P. M. Thomas, M. Uhlén, J. E. Van Eyk, M. Vidal, D. R. Walt, F. M. White, E. R. Williams, T. Wohlschlager, V. H. Wysocki, N. A. Yates, N. L. Young and B. Zhang, Nat. Chem. Biol., 2018, 14, 206–214 CrossRef PubMed.
- M. G. Iadanza, M. P. Jackson, E. W. Hewitt, N. A. Ranson and S. E. Radford, Nat. Rev. Mol. Cell Biol., 2018, 19, 755–773 CrossRef CAS PubMed.
- P. Wittung-Stafshede, Biochem. Soc. Trans., 2023, 51, 1967–1974 CrossRef CAS.
- C. M. Dobson, Cold Spring Harbor Perspect. Biol., 2017, 9(6), a023648 CrossRef.
- A. Das, M. Shah and I. Saraogi, ACS Bio Med Chem Au, 2022, 2, 205–221 CrossRef CAS PubMed.
- K. L. Moreau and J. A. King, Trends Mol. Med., 2012, 18, 273–282 CrossRef CAS PubMed.
- A. K. Buell, Chem. Sci., 2022, 13, 10177–10192 RSC.
- S. Linse and T. Knowles, Chem. Sci., 2023, 14, 6491–6492 RSC.
- A. J. Dear, G. Meisl, T. C. T. Michaels, M. R. Zimmermann, S. Linse and T. P. J. Knowles, J. Chem. Phys., 2020, 152, 045101 CrossRef CAS PubMed.
- T. C. T. Michaels, D. Qian, A. Šarić, M. Vendruscolo, S. Linse and T. P. J. Knowles, Nat. Rev. Phys., 2023, 5, 379–397 CrossRef CAS.
- S. Cohen, M. Vendruscolo, C. Dobson and T. Knowles, The Kinetics and Mechanisms of Amyloid Formation, in Amyloid Fibrils and Prefibrillar aggregates, Wiley-VCH, Germany, 2013 Search PubMed.
- E. Chau and J. R. Kim, Arch. Biochem. Biophys., 2022, 717, 109120 CrossRef CAS.
- T. Sinnige, Chem. Sci., 2022, 13, 7080–7097 RSC.
- J. Habchi, S. Chia, C. Galvagnion, T. C. T. Michaels, M. M. J. Bellaiche, F. S. Ruggeri, M. Sanguanini, I. Idini, J. R. Kumita, E. Sparr, S. Linse, C. M. Dobson, T. P. J. Knowles and M. Vendruscolo, Nat. Chem., 2018, 10, 673–683 CrossRef PubMed.
- Y. Zhang, L. A. Borch, N. H. Fischer and M. Meldal, J. Am. Chem. Soc., 2024, 146, 2654–2662 CrossRef PubMed.
- D. Y. Zhang, J. Wang, R. M. Fleeman, M. K. Kuhn, M. T. Swulius, E. A. Proctor and N. V. Dokholyan, ACS Chem. Neurosci., 2022, 13, 1979–1991 CrossRef.
- J. H. Viles, Angew. Chem., Int. Ed., 2023, 62, e202215785 CrossRef PubMed.
- S. Meker, H. Chin, T. N. Sut and N. J. Cho, Langmuir, 2018, 34, 9548–9560 CrossRef PubMed.
- M. S. Terakawa, Y. Lin, M. Kinoshita, S. Kanemura, D. Itoh, T. Sugiki, M. Okumura, A. Ramamoorthy and Y. H. Lee, Biochim. Biophys. Acta, Biomembr., 2018, 1860, 1741–1764 CrossRef PubMed.
- T. John, S. Piantavigna, T. J. A. Dealey, B. Abel, H. J. Risselada and L. L. Martin, Chem. Sci., 2023, 14, 3730–3741 RSC.
- C. Cheignon, F. Collin, L. Sabater and C. Hureau, Antioxidants, 2023, 12, 472 CrossRef CAS PubMed.
- C. Cheignon, M. Tomas, D. Bonnefont-Rousselot, P. Faller, C. Hureau and F. Collin, Redox Biol., 2018, 14, 450–464 CrossRef CAS.
- K. Matsuzaki, Biochim. Biophys. Acta, 2007, 1768, 1935–1942 CrossRef CAS PubMed.
- M. Hashemi, S. Banerjee and Y. L. Lyubchenko, Int. J. Mol. Sci., 2022, 23(5), 2803 CrossRef CAS.
- S. Banerjee, M. Hashemi, K. Zagorski and Y. L. Lyubchenko, ACS Chem. Neurosci., 2021, 12, 506–516 CrossRef CAS.
- C. L. Dias, S. Jalali, Y. Yang and L. Cruz, J. Phys. Chem. B, 2020, 124, 3036–3042 CrossRef CAS PubMed.
- S. Andrade, J. A. Loureiro and M. C. Pereira, Chem. Phys. Chem., 2021, 22, 1547–1565 CrossRef CAS PubMed.
- M. S. Terakawa, H. Yagi, M. Adachi, Y. H. Lee and Y. Goto, J. Biol. Chem., 2015, 290, 815–826 CrossRef CAS PubMed.
- T. Weiffert, G. Meisl, P. Flagmeier, S. De, C. J. R. Dunning, B. Frohm, H. Zetterberg, K. Blennow, E. Portelius, D. Klenerman, C. M. Dobson, T. P. J. Knowles and S. Linse, ACS Chem. Neurosci., 2019, 10, 2374–2384 CrossRef CAS PubMed.
- D. J. Lindberg, E. Wesen, J. Bjorkeroth, S. Rocha and E. K. Esbjorner, Biochim. Biophys. Acta, Biomembr., 2017, 1859, 1921–1929 CrossRef CAS.
- T. John, L. L. Martin and B. Abel, Macromol. Biosci., 2023, 23, e2200576 CrossRef.
- X. Yu and J. Zheng, J. Mol. Biol., 2012, 421, 561–571 CrossRef CAS PubMed.
- P. Flagmeier, S. De, T. C. T. Michaels, X. Yang, A. J. Dear, C. Emanuelsson, M. Vendruscolo, S. Linse, D. Klenerman, T. P. J. Knowles and C. M. Dobson, Nat. Struct. Mol. Biol., 2020, 27, 886–891 CrossRef PubMed.
- M. Sanguanini, K. N. Baumann, S. Preet, S. Chia, J. Habchi, T. P. J. Knowles and M. Vendruscolo, ACS Chem. Neurosci., 2020, 11, 1347–1352 CrossRef PubMed.
- B. Mannini, J. Habchi, S. Chia, F. S. Ruggeri, M. Perni, T. P. J. Knowles, C. M. Dobson and M. Vendruscolo, ACS Chem. Neurosci., 2018, 9, 2959–2971 CrossRef PubMed.
- F. Grigolato and P. Arosio, Biophys. Chem., 2021, 270, 106533 CrossRef PubMed.
- E. Karran and B. De Strooper, Nat. Rev. Drug Discovery, 2022, 21, 306–318 CrossRef.
- A. Khursheed and J. H. Viles, J. Mol. Biol., 2024, 436, 168464 CrossRef PubMed.
- J. Adler, H. A. Scheidt, K. Lemmnitzer, M. Krueger and D. Huster, Phys. Chem. Chem. Phys., 2017, 19, 1839–1846 RSC.
- N. Sen, G. Hause and W. H. Binder, Macromol. Rapid Commun., 2021, 42, e2100120 CrossRef PubMed.
- G. Meisl, J. B. Kirkegaard, P. Arosio, T. C. T. Michaels, M. Vendruscolo, C. M. Dobson, S. Linse and T. P. J. Knowles, Nat. Protoc., 2016, 11, 252–272 CrossRef PubMed.
- N. Sen, C. Haupt, G. Hause, K. Bacia and W. H. Binder, Macromol. Biosci., 2023, 23, e2200522 CrossRef.
- E. London and G. W. Feigenson, J. Lipid Res., 1979, 20, 408–412 CrossRef.
- I. M. Armitage, D. L. Shapiro, H. Furthmayr and V. T. Marchesi, Biochemistry, 1977, 16, 1317–1320 CrossRef PubMed.
- P. L. Yeagle, eMagRes, 2019, 1115, 99 Search PubMed.
- M. Schulz, S. Werner, K. Bacia and W. H. Binder, Angew. Chem., Int. Ed., 2013, 52, 1829–1833 CrossRef PubMed.
- Z. Evgrafova, B. Voigt, A. H. Roos, G. Hause, D. Hinderberger, J. Balbach and W. H. Binder, Phys. Chem. Chem. Phys., 2019, 21, 20999–21006 RSC.
- S. I. Cohen, S. Linse, L. M. Luheshi, E. Hellstrand, D. A. White, L. Rajah, D. E. Otzen, M. Vendruscolo, C. M. Dobson and T. P. Knowles, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 9758–9763 CrossRef CAS PubMed.
- T. Scheidt, U. Łapińska, J. R. Kumita, D. R. Whiten, D. Klenerman, M. R. Wilson, S. I. A. Cohen, S. Linse, M. Vendruscolo, C. M. Dobson, T. P. J. Knowles and P. Arosio, Sci. adv., 2019, 5, eaau3112 CrossRef PubMed.
- M. Tornquist, T. C. T. Michaels, K. Sanagavarapu, X. Yang, G. Meisl, S. I. A. Cohen, T. P. J. Knowles and S. Linse, Chem. Commun., 2018, 54, 8667–8684 RSC.
- S. I. Cohen, M. Vendruscolo, C. M. Dobson and T. P. Knowles, J. Mol. Biol., 2012, 421, 160–171 CrossRef PubMed.
- S. Linse, Biophys. Rev., 2017, 9, 329–338 CrossRef PubMed.
- N. V. Dovidchenko, A. V. Glyakina, O. M. Selivanova, E. I. Grigorashvili, M. Y. Suvorina, U. F. Dzhus, A. O. Mikhailina, N. G. Shiliaev, V. V. Marchenkov, A. K. Surin and O. V. Galzitskaya, J. Struct. Biol., 2016, 194, 404–414 CrossRef PubMed.
- G. Meisl, X. Yang, E. Hellstrand, B. Frohm, J. B. Kirkegaard, S. I. Cohen, C. M. Dobson, S. Linse and T. P. Knowles, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 9384–9389 CrossRef PubMed.
- A. Micsonai, F. Wien, L. Kernya, Y. H. Lee, Y. Goto, M. Refregiers and J. Kardos, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E3095–3103 CrossRef PubMed.
- C. Galvagnion, J. W. Brown, M. M. Ouberai, P. Flagmeier, M. Vendruscolo, A. K. Buell, E. Sparr and C. M. Dobson, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 7065–7070 CrossRef PubMed.
- M. Kuragano, S. Yamanaka and K. Tokuraku, Colloids Surf., B, 2022, 214, 112449 CrossRef CAS.
- N. Xiang, Y. Lyu, X. Zhu and G. Narsimhan, Phys. Chem. Chem. Phys., 2018, 20, 6817–6829 RSC.
- P. Ghosh and P. De, ACS Appl. Bio Mater., 2020, 3, 6598–6625 CrossRef.
- D. Ghosh, P. Dutta, C. Chakraborty, P. K. Singh, A. Anoop, N. N. Jha, R. S. Jacob, M. Mondal, S. Mankar, S. Das, S. Malik and S. K. Maji, Langmuir, 2014, 30, 3775–3786 CrossRef PubMed.
- X. Yang, G. Meisl, B. Frohm, E. Thulin, T. P. J. Knowles and S. Linse, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E5849–E5858 CrossRef.
- Y. Kim, J. H. Park, H. Lee and J. M. Nam, Sci. Rep., 2016, 6, 19548 CrossRef PubMed.
- K. Yokoyama and D. R. Welchons, Nanotechnology, 2007, 18, 105101 CrossRef.
Footnotes |
| † Prof. Dr Wolfgang H. Binder, Dr Stephanie Krüger, MSci. Newton Sen. |
| ‡ Electronic supplementary information (ESI) available: Synthesis and experimental procedures, all substance analyses (NMR, MS) and further analyses and assays, AmyloFit-analyses, EM pictures, preparation and characterization of hybrid-vesicles. See DOI: https://doi.org/10.1039/d4cb00217b |
| This journal is © The Royal Society of Chemistry 2024 |