
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A platform of ADAPTive scaffolds: development of CDR-H3 β-hairpin mimics into covalent inhibitors of the PD1/PDL1 immune checkpoint†
Sarah H.
Naylon
a,
Alexis D.
Richaud
a,
Guangkuan
Zhao
a,
Linda
Bui
a,
Craig P.
Dufresne
b,
Chunjing J.
Wu
c,
Medhi
Wangpaichitr
c,
Niramol
Savaraj
c and
Stéphane P.
Roche
 *a
*a
aDepartment of Chemistry and Biochemistry, Florida Atlantic University, Boca Raton, Florida 33431, USA. E-mail: sroche2@fau.edu
bThermo Fisher Scientific, West Palm Beach, Florida 33407, USA
cUniversity of Miami, Miller School of Medicine, Miami, Florida 33136, USA
First published on 4th November 2024
Abstract
Aberrant and dysregulated protein–protein interactions (PPIs) drive a significant number of human diseases, which is why they represent a major class of targets in drug discovery. Although a number of high-affinity antibody-based drugs have emerged in this therapeutic space, the discovery of smaller PPI inhibitors is lagging far behind, underscoring the need for novel scaffold modalities. To bridge this gap, we introduce a biomimetic platform technology – adaptive design of antibody paratopes into therapeutics (ADAPT) – that enables the paratope-forming binding loops of antibodies to be crafted into large β-hairpin scaffolds (ADAPTins). In this study, we describe a novel strategy for engineering native CDR-H3 “hot loops” with varying sequences, lengths, and rigidity into ADAPTins, ultimately transforming these compounds into irreversible covalent inhibitors. A proof-of-concept was established by creating a series of ADAPTin blockers of the PD1:PDL1 immune checkpoint PPI (blocking activity EC50 < 0.3 μM) which were subsequently modified into potent covalent PD1 inhibitors. The compelling rate of stable and folded ADAPTins above physiological temperature (21 out of 29) obtained across six different scaffolds suggests that the platform technology could provide a novel opportunity for high-quality peptide display and biological screening.
A. Introduction
Protein–protein interactions (PPIs) regulate a plethora of fundamental cellular processes and their misregulation has now been associated with a variety of diseases.1–4 Yet, most PPIs interfaces exhibit rather shallow, water-exposed, and sizeable surface areas (800–2000 Å2) which are challenging to disrupt with small molecules from conventional drug libraries (MW < 0.5 kDa, binding surface < 100 Å2).5,6 Moreover, theses interfaces can be either rugged or more dynamic, further contributing to their “undruggable” reputation. Over the past two decades, a new landscape of antibody drugs (Abs) and biological therapeutics of smaller size such as nanobobies,7 DARPins,8 and more recently BiTEs9 have truly revolutionized our clinical approach to targeted therapies.10 Despite their efficacy in modulating or blocking PPIs, the massive size of Abs (∼150 kDa) is often associated with delicate pharmacokinetic properties such as low bioavailability, but also poor tissue penetration, and slow clearance rates resulting in undesirable high systemic accumulations.11,12 Because of these drawbacks, a relatively unexplored therapeutic space between large biologics and low-molecular weight small-molecule drugs has attracted a lot of attention.13–15 In this uncharted space, cyclic peptides,16 bicycles,17 β-bracelets,18 and other helical peptides19–21 have laid the groundwork for the development of smaller size scaffolds as PPI inhibitors.22 Despite these advances, a pressing need persists for more robust and versatile scaffolding technologies capable of engineering peptide therapeutics with antibody-like structures, affinity, and potency.23 With this goal in mind, we created the ADAPT technology (short for adaptive design of antibody paratopes into therapeutics) that enables “hot loops” of antibodies with varying sequences, lengths, and rigidity to be crafted into short stand-alone β-hairpin scaffolds (ADAPTins).To substantiate the technological proof of concept, we selected the programmed cell death-1 protein (PD1) and its ligand-1 (PDL1) as our focal PPI target. Here, we outline a general strategy to engineer synthetic loop mimics into ADAPTins that mimic the native fold of antibody CDR-H3 loops. We showed that out of the six anti-PD1 antibodies evaluated, four distinct CDR-H3 scaffolds could be obtained without altering the original H3 loop sequence. Several standalone CDR-H3 mimics displayed a remarkably efficient inhibition of the PD1/PDL1 immune checkpoint interactions at sub-micromolar concentrations. Selected ADAPTins were subsequently crafted with electrophilic warheads to achieve a covalent and irreversible inhibition of PD1 and advance candidates for in vitro- and in cellulo-studies. Unlike conventional strategies of protein epitope mimicry (Fig. 1, Top panel), our technology offers a novel avenue for grafting large non-canonical CDR-H3 antibody loops into smaller ADAPTin scaffolds which are not accessible by other means (Fig. 1, bottom panel).
 | ||
| Fig. 1 ADAPT platform technology. A biomimetic approach for designing β-hairpin peptide inhibitors of PPIs (ADAPTins) based on the plasticity or rigidity of the protein of interest. The relative flexibility of PD1 (bRMSD of 1.68 Å) compared to its ligand PDL1 (bRMSD of 0.76 Å) was calculated from backbone alignments over +400 atoms using unbounded apo-PD1 (PDB: 3RRQ) and apo-PDL1 (PDB: 5C3T) as respective reference. Top panel depicts typical strategies for designing protein epitope mimics into peptide macrocycles or larger protein-derived scaffolds. Bottom panel depicts a novel general approach to mimic CDR-H3s found in antibodies paratopes into ADAPTin scaffolds of varying rigidity. These stand-alone scaffolds (β-strap + β-bulge motifs) can display a broad variety of CDR-H3 loops which can be modified to incorporate electrophilic warheads to covalently bind a protein target. | ||
B. Results and discussion
General design principles guiding CDR-H3 mimics
Despite the significant breakthrough by Baker and Craik in transforming computational models of protein motifs into large scaffolds (MW ∼ 12–25 kDa),24 the synthesis of sizeable and 3-dimensionally folded peptides outside of their protein context remains non-trivial.25 Although powerful, protein epitope mimics (PEMs) are inherently limited to canonical motifs found in proteins (Fig. 1, top panel).26,27 Similarly, the miniaturization of high-affinity antibody paratopes into smaller scaffolds, aka complementary determining regions (CDRs), has been essentially focused on the canonical loops found at the apexes of light-chain (L1-3) and heavy-chain (H1-2) CDRs.28,29 Yet, recent structural analyses of protein–protein complexes in the Protein Data Bank revealed that a large number of PPI “hot contacts” are in fact generated by nonregular secondary structures (∼50%) mainly from loops embedded in either β-hairpin structures or non-canonical forms.30,31 To bridge this gap, our approach innovatively repurposes the CDR-H3 hairpin and β-bulge motif into a unified scaffold that could withhold longer loops (>10-residue long) while closely mimicking the native fold found in high-affinity antibodies (Fig. 1, bottom panel).32In comparison to all other CDRs, CDR-H3 loops are known to possess the largest variability of sequence33,34 topology, and length (4 up to >21 residues)35 which drastically increases the span of conformational space accessible34,36 to maximize protein binding affinity and specificity.37–40 Strikingly, the vast majority of CDR-H3s possess a β-bulge motif edging their loops;41,42 Yet the role of this structural motif in CDR-H3 folding, stability, and rigidification remains mostly unknown.43 One could imagine that the promiscuity of bulges is a result of evolutionary optimization to favor the display of long and conformationally adaptable H3 loops to mutations and 3D-rigidification (Fig. 1, bottom panel conformational hinge).44–46 In addition, most H3 loops have unique ‘noncanonical’ topologies47,48 that may enhance antibody specificity to a protein target and therefore constitute an exciting starting point for the design of PPI inhibitors.49 For all these reasons, we and others became interested in mimicking CDR-H3 scaffolds to recreate miniaturized peptide loop displays either for protein loop grafting or standalone loop scaffolding.32,50–53
To validate a proof of concept of biomimetic CDR-H3 scaffolding platform, we selected the immune checkpoint PD1:PDL1 interaction. Indeed, anti-PD(L)1 antibody drugs have completely transformed our current approach to cancer therapy.54 The PD1:PDL1 interaction is nearly an ideal model to test our ADAPT technology because: (a) PD1 is inherently more flexible than PDL1 (backbone bRMSD of 1.68 Å vs. 0.76 Å) which is in line with an entropically-driven induced-fit binding mechanism of PD1 to PDL1 vs. PDL2,55 and (b) its low complex affinity between PD1 and PDL1 (KD of 8.2 μM) suggesting that a PEM strategy would be complicated. Indeed, the PD1·PDL1 interface lacks well-defined binding pockets and is highly dynamic, which explains why small-molecule intervention remains challenging. Since the initial report by Bristol-Myers Squibb scientists of anti-PDL1 macrocyclic peptides,56 only a handful of small molecules57 and larger peptide scaffolds have been discovered (Fig. 1, top panel).58,59 Likewise, only very few PD1 antagonists have been reported.60,61 Excitingly, a significant number of high-resolution crystal structures of PD1·AbDrug complexes are available, providing us with detailed structural information to rationally design anti-PD1 CDR-H3 mimics (ADAPTins) (Fig. 1, bottom panel).
Recently, our laboratory brought forward a novel synthetic technology for the synthesis of acyclic β-hairpins with long loops. Prior to these studies, the access to hairpin peptides was essentially limited to short loops possessing an innate β-turn (4-AA long: ![[D with combining low line]](https://www.rsc.org/images/entities/char_0044_0332.gif) DATK
DATK![[T with combining low line]](https://www.rsc.org/images/entities/char_0054_0332.gif) and
and ![[N with combining low line]](https://www.rsc.org/images/entities/char_004e_0332.gif) PATG
PATG![[K with combining low line]](https://www.rsc.org/images/entities/char_004b_0332.gif) ).62–64 Inspired by the work of Andersen on long-loop closure,65–67 we created a series of minimalist β-straps (strap = strand + cap) RWVW⋯W(V/H)WE that enable regular hairpin folds with up to 10-AA loops. To compare hairpins’ stability, both regular (R) and bulged (B) scaffolds were crafted around a flexible 10-residue model loop (G4K2G4) and analyzed by CD (circular dichroism) spectroscopy (Fig. 2(A), see ESI,† Table S5). The tertiary structure of these model scaffolds 1avs.2a in solution were recorded in the far-UV CD spectra. The characteristic and very intense exciton couplet maxima at 214 and 228 ± 2 nm in the CD spectra of β-hairpins (π–π* transition) originates from an edge-to-face staking of tryptophans W2/W19 nearby the C-/N-β-strap termini. The CD-exciton intensity was therefore used as a global probe to determine the %-folding of ADAPTins and to obtain melting curves corresponding to the hairpin fraying mechanism upon a gradual increase of temperature (0 to 95 °C).68 The melting temperature (TM) upshift of about 10 °C calculated from CD-melts clearly indicates that the bulged scaffold 2a is significantly more resistant to thermal denaturation than the regular hairpin 1a. As shown in the CD-spectrum of the bulged scaffold 2a (Fig. 2(A)), a unique positive band at 202 ± 2 nm was observed in each bulged ADAPTin2 which was never observed in the spectra of regular scaffolds like 1a. This additional band was therefore exploited to monitor the unfolding of bulged ADAPTins2 and the melting data were found to be in general agreement with the global hairpin unfolding results. In addition, NMR (nuclear magnetic resonance) chemical shifts were measured to determine variations in backbone tertiary structures (Fig. 2(B)). Secondary chemical shift deviations (CSDs) were calculated against random coil values69 for both ADAPTin scaffolds 1vs.2 to verify that the hairpins were folded and if any variation of conformational rigidity exist within the loops. As expected, successive backbone CSDs of Hα protons within the β-straps of 1a–b and 2a–b are relatively large (0.5–0.9 ppm) confirming a β-sheet register. In addition, the β-bulge residues D and Y (marked with asterisk in Fig. 2(B)) appeared abnormally deshielded in 2a–b (up to 0.7 ppm) suggesting that bulge-like ADAPTins present an extension of β-sheet structure. Notably, a number of secondary chemical shifts observed within the loops of the scaffold 2a and ADAPTin2b differ by more than 0.1 ppm than their regular hairpins counterparts 1a and 1b respectively suggesting that the bulged scaffold generate a more structured and strained loop.70
).62–64 Inspired by the work of Andersen on long-loop closure,65–67 we created a series of minimalist β-straps (strap = strand + cap) RWVW⋯W(V/H)WE that enable regular hairpin folds with up to 10-AA loops. To compare hairpins’ stability, both regular (R) and bulged (B) scaffolds were crafted around a flexible 10-residue model loop (G4K2G4) and analyzed by CD (circular dichroism) spectroscopy (Fig. 2(A), see ESI,† Table S5). The tertiary structure of these model scaffolds 1avs.2a in solution were recorded in the far-UV CD spectra. The characteristic and very intense exciton couplet maxima at 214 and 228 ± 2 nm in the CD spectra of β-hairpins (π–π* transition) originates from an edge-to-face staking of tryptophans W2/W19 nearby the C-/N-β-strap termini. The CD-exciton intensity was therefore used as a global probe to determine the %-folding of ADAPTins and to obtain melting curves corresponding to the hairpin fraying mechanism upon a gradual increase of temperature (0 to 95 °C).68 The melting temperature (TM) upshift of about 10 °C calculated from CD-melts clearly indicates that the bulged scaffold 2a is significantly more resistant to thermal denaturation than the regular hairpin 1a. As shown in the CD-spectrum of the bulged scaffold 2a (Fig. 2(A)), a unique positive band at 202 ± 2 nm was observed in each bulged ADAPTin2 which was never observed in the spectra of regular scaffolds like 1a. This additional band was therefore exploited to monitor the unfolding of bulged ADAPTins2 and the melting data were found to be in general agreement with the global hairpin unfolding results. In addition, NMR (nuclear magnetic resonance) chemical shifts were measured to determine variations in backbone tertiary structures (Fig. 2(B)). Secondary chemical shift deviations (CSDs) were calculated against random coil values69 for both ADAPTin scaffolds 1vs.2 to verify that the hairpins were folded and if any variation of conformational rigidity exist within the loops. As expected, successive backbone CSDs of Hα protons within the β-straps of 1a–b and 2a–b are relatively large (0.5–0.9 ppm) confirming a β-sheet register. In addition, the β-bulge residues D and Y (marked with asterisk in Fig. 2(B)) appeared abnormally deshielded in 2a–b (up to 0.7 ppm) suggesting that bulge-like ADAPTins present an extension of β-sheet structure. Notably, a number of secondary chemical shifts observed within the loops of the scaffold 2a and ADAPTin2b differ by more than 0.1 ppm than their regular hairpins counterparts 1a and 1b respectively suggesting that the bulged scaffold generate a more structured and strained loop.70
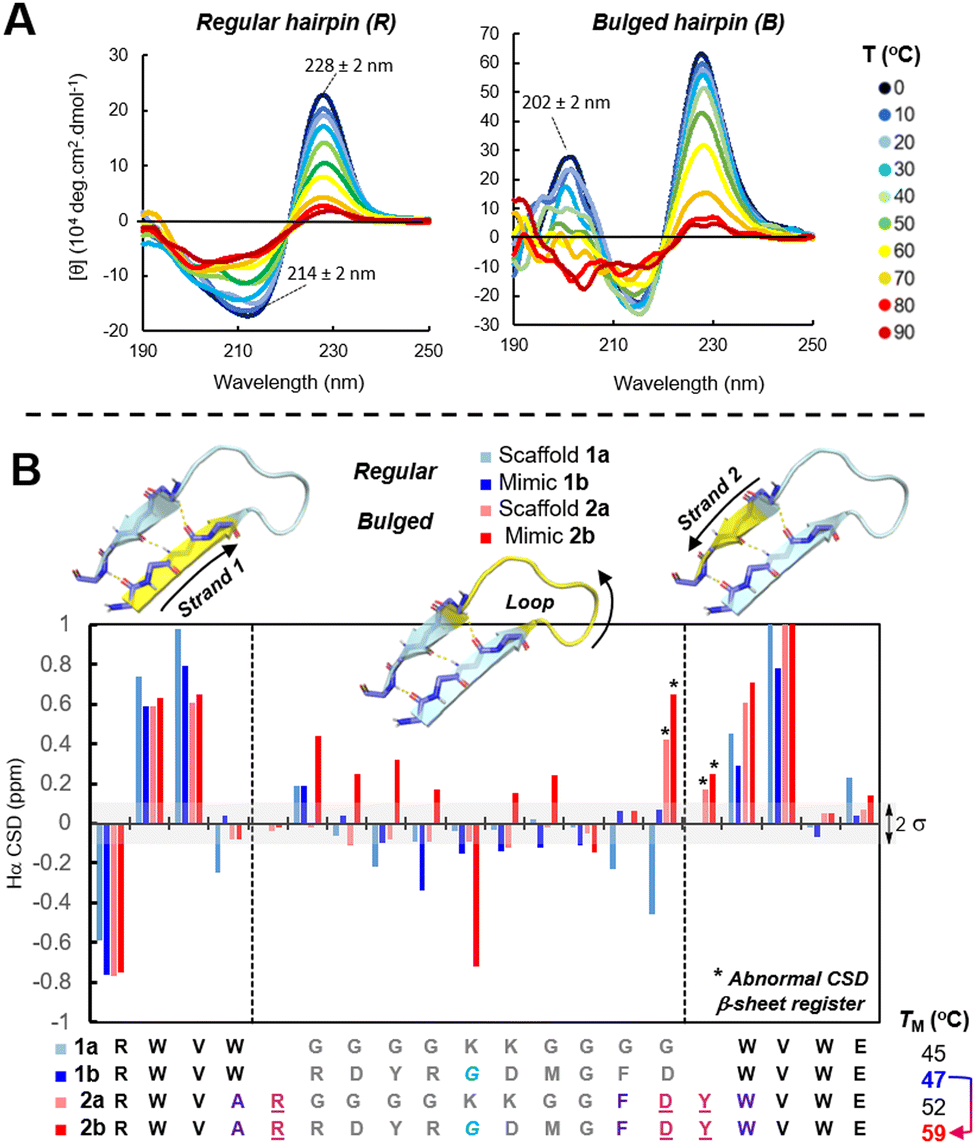 | ||
| Fig. 2 Structuration of stand-alone CDR-H3 scaffolds and their loops. Regular 1vs. bulge-like 2 β-hairpins. (A) Variable-temperature CD analysis of hairpin folds. (B) Hα NMR chemical shift deviations (CSDs) analysis with 2σ of standard deviation from random coil values (in shaded grey). | ||
Proof of concept: engineering a library of CDR-H3 mimics targeting the PD1:PDL1 interface
To rationally design our library of anti-PD1 ADAPTins, high-resolution crystal structures of PD(L)1·AbDrug complexes were analyzed. Using a series of bioinformatic tools in the Rosetta Suite, RosettaDock-4.071 and Peptiderive,72 the energy profile for each binding paratope was exploited to score the “hot loops” contacts (ESI,† Fig. S8–S13). These H3 loops were ranked based off their calculated binding free energy, the buried surface area of interaction (>20% total BSA), and the total number of contacts to PD1. The results summarized in Table 1 suggest that most CDR-H3s were found promising scaffolds in comparison to all the other CDRs (ESI,† Table S1). Ranking of H3 loops from overall scores of binding free energy, binding-surface areas, and RMSD was obtained as follows: pembrolizumab > tislelizumab ∼ GY-14 > mAb059c > MW11-h317. Overall, the pembrolizumab H3 loop was found to be unique presenting the largest surface of interaction of 460 Å2 (36% of the overall Ab·PD1 buried-surface area) encompassing 21 contact interactions across distal regions of the PD1 epitope (localized on C′D and FG loops of PD1).| a Individual CDR buried surface area (BSA) computed by dr_SASA with %-binding surface calculated as a ratio to the total BSA (TBSA) from Ab·PD1 cocrystal structures. b Total number of hydrophobic and polar binding contacts created at the CDR-H3 interface. c Binding Gibbs free energy (reu) calculated by Peptiderive to score binding interfaces. d Folded fraction (χF) at 25 °C and melting temperatures calculated from CD-melts based on the type of ADAPTin fold (B: bulged, R: regular). |
|---|

|
Having selected a set of potential CDR-H3 binders (Table 1), we carried out the synthesis of a library of regular (R) and bulged (B) ADAPTin peptides 1b–f and 2b–r respectively, using a typical Fmoc-chemistry on solid support. The folding of these peptides was first verified by their CD signature (Fig. 2(A)) and CD-melts measurements. At the exception of peptides 2m and 2q which are characterized by a β-sheet structure (band at 214 ± 2 nm, and a lack of exciton at 228 ± 2 nm) suggesting a misalignment within the hairpin β-strap, most peptides folded as expected according to their designed (R)- or (B)-scaffolds (Table 1, detailed CD analysis provide in the ESI†). Strikingly, the introduction of a glycine residue within the loop mimics of pembrolizumab (F10G, 1b, 2b, 2d, 2f, 2h, and 2j, vs.1c, 2c, 2e, 2g, 2i, and 2k,), GY-14 (W12G, 2nvs.2o), and tislelizumab (W11G, 2qvs.2r) afforded in each case additional flexibility that enhanced the global folding. Within the entire library, 10 bulge-like ADAPTins out of 14 analogs 2a–r had a melting temperature above 37 °C (Table 1), showing that the β-strap design is adaptable to a large variety of CDR-H3 loops. While these results are consistent with the notion that the ADAPT technology can create hairpin with long loops (6 to 10-residue tested), our ability to fully extend and extrapolate these folding properties is currently limited by the nature of the loop sequences. It will therefore be necessary to further optimize the β-strap stability-potentially by mimicking the rigidification and maturation mechanisms of CDR-H3s-to improve and generalize the ADAPT technology to a broader range of loop sequences and lengths.
Given the significant structural differences between regular and bulged ADAPTins, we investigated if different loop topologies resulted in different PD1 binding affinity. Examination of interference of ADAPTins1 and 2 on the binding of PDL1 to PD1 was performed with a fluorescence-based ELISA (Fig. 3(A)). The blocking activity of these peptides was obtained and compared to a macrocyclic hairpin cyclo-2j (EC50 of 140 nM) and to the FDA-approved anti-PD1 antibody pembrolizumab (IC50 of 1 nM, positive control). Regarding regular ADAPTins, the MW11 mimic 1e offered the strongest activity (EC50 of 270 nM), while three different bulge-like scaffolds inspired by pembrolizumab (2c, 2g, 2i–k), but also by GY-14 (2n,p), and tislelizumab (2q–r) all presented promising blocking activities (EC50 < 300 nM). Given that all ADAPTins are approximately of same size, a ligand efficiency (LE) metric was calculated to better compare the H3 loops’ affinity to PD1, (Fig. 3(A)).73 By this measure, the LEs of ADAPTins studied herein (range 0.05–0.10, mean of 125 HA) were about two-fold lower than the comparably large peptide scaffold SFTI-1 (LE of 0.145 for 105 HA).18 This analysis suggested that ADAPTins1e, 2c, 2g, 2i–k, 2p, and 2r possess both potency and loop-display efficiency to block the PD1:PDL1 interaction. In addition, the plot of inhibitory activities of ADAPTins by congeneric pairs (Fig. 3(B)) revealed that the substitution of tryptophan or phenylalanine residues by glycines in the loops resulted in most cases in a substantial reduction of activity. These results mirror the membrane permeation measurements previously reported for those ADAPTins,74 highlighting the importance of hydrophobic residues to enhance both the passive diffusion and the pharmacological activity of long loops. Taken together, these results suggest that the ADAPT technology could become a new tool for mimicking antibody CDR-H3 structures; yet these miniaturized scaffolds have obviously lost a great deal of potency in comparison to their full-length parental antibodies (KD low nano- to picomolar range).75 Indeed, the kon/off kinetics of anti-PD1 mAbs are characteristic of very tight binders that attach almost irreversibly to PD1 (residence time in the order of v6 hours).76 To remediate to the lower potency of ADAPTins, we sought to further exploit these scaffolds to create irreversible covalent binders of PD1.
 | ||
| Fig. 3 Inhibitory activity and ligand efficient of non-covalent ADAPTins against the PD1:PDL1 interaction. (A) Mean EC50 values determined from dose-dependent binding curves obtained by ELISA in a PD1–PDL1 assay; n = 3, SD reported in the ESI.† (B) Comparative plot of PD1·PDL1 inhibitory activity by single side chain modulation within ADAPTins. Original loops with Phe/Trp and their Gly-derived analogs. Experimental EC50 values from inhibitory dose–response curves. Error bars indicate the mean ± SD, (n = 3). | ||
Optimization of electrophilic ADAPTin inhibitors
While downsizing a full antibody drug into a single CDR-H3 “hot loop”, one should anticipate a significant loss in binding affinity and specificity. We previously reported that the non-covalent ADAPTin2c inhibited the PD1:PDL1 interaction with an apparent Ki of 41 nM and a residence time on target of v30 minutes.77 Yet even at a 3 μM concentration, this competitive inhibitor was fully displaced by PDL1 within two hours of incubation (Fig. 4(D)). Therefore, we decided to strategically modify the initial ADAPTin hits into covalent binders to engage the PD1 target irreversibly (Fig. 4). The PD1 protein contains one free cysteine (Cys93) and displays three surface-exposed lysines (K78, K131, and K135). Based on the available crystallographic data of PD1 bound to antibody drugs (see ESI,† Fig. S14 and S15), lysine K131 positioned on the highly flexible PD1FG loop appeared to be the most attractive residues to target.78,79 Indeed, the FG loop was shown by us and others to have an important innate conformational plasticity and no hindering N-glycosylation sites that creates a large surface of direct contacts with anti-PD1 mAbs.77–81 This flexibility enables a shallow binding-groove to form upon contact with the anti-PD1 pembrolizumab CDR-H3 loop (Fig. 4(A)).82 In addition, the FG loop conformation was suggested to influence the downstream signaling of PD1.80,83 For these reasons, ADAPTins2c and 2p, respective mimics of pembrolizumab and GY-14 CDR-H3s, were selected to introduce electrophilic warheads for covalent binding. For 2c, the methionine M105 in van der Waals interaction with PD1K131 (within v4.0 Å) was deemed appropriately positioned for modification, while for 2p tyrosine Y106 was the closest interacting residue to the PD1K131 (see ESI,† Fig. S15). To modify ADAPTins 2c and 2p, we selected two anchoring amino acids of different side chain length 2,3-diaminopropionic acid (Dap, n = 1) and 2,4-diaminobutyric acid (Dab, n = 2) that can be readily installed by solid-phase peptide synthesis (SPPS). Due to their tunable electrophilicity,84,85 a series of acrylamide-type electrophilic warheads was generated on Dap/Dab residues at the selected positions of ADAPTin loops, including acrylamide (ACA), dimethylaminobutenamide (DMA), and methacrylic amide (MAA) (Fig. 4(A)). Acrylamides are physiologically stable, yet powerful and selective Michael acceptors that have demonstrated efficacy in a number of covalent drugs86,87 in particular targeting surface exposed lysines.88,89 | ||
Fig. 4 Design of lysine-targeted acrylamide-derived electrophilic inhibitors of PD1. (A) Pembrolizumab (orange) and GY-14 (green) bulged CDR-H3 hairpin loops in contact with PD1 (PDB codes: 5GGS and 6J14). Sequence of ADAPTins2c and 2p showing substitutions of M12 and Y13 respectively with Dap/Dab residues attached to acrylamide-derived electrophiles. M12 and Y13 were selected for the introduction of electrophilic warheads based on distances between the corresponding M105 and Y106 to the targeted PD1K131, see ESI,† Fig. S15. (B) CD spectra for the electrophilic ADAPTins2ca–cd/2pa,b and TM values obtained from a global fit of CD-melts representing the temperature at which each peptide retain 50% folding. (C) Inhibitory activity of the covalent inhibitors against the PD1:PDL1 interaction, n = 3 (mean ± SD). (D) Kinetic curves of competitive inhibition targeting the PD![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1:PDL1 interaction (1:12 ratio) showing the irreversible covalent nature of electrophilic inhibitors 2ca and 2cb. Error bars indicate the mean ± SD, n = 4. 1:PDL1 interaction (1:12 ratio) showing the irreversible covalent nature of electrophilic inhibitors 2ca and 2cb. Error bars indicate the mean ± SD, n = 4. | ||
First, the folding of these electrophilic peptides 2ca–d and 2pa–b was confirmed by CD spectroscopy (Fig. 4(B)). Excitingly, most analogs (at the exception of 2cd) were well folded (χF > 68%, TM > 35 °C) presenting both bands at 202 and 228 ± 2 nm characteristic of a bulged-like β-hairpin scaffold and their melting curves were in each case very similar to the corresponding non-covalent ADAPTin molecules. To obtain a more accurate estimate of folding, we developed a global fit protocol that allowed the CD-melts of parent ADAPTins2c and 2p to be fitted simultaneously to their covalent congeners (see ESI,† Fig. S18 and S19). The resulting denaturation curves and melting temperatures strongly suggest that the introduction of acrylamide-derived warheads on either Dap or Dab amino acids did not substantially interfere with the intended hairpin fold. Next, the inhibitory activity of these electrophilic analogs was measured on the PD1:PDL1 interaction by ELISA (Fig. 4(C)). The six covalent analogs inhibited the interaction in a dose dependent manner with IC50s in the low micromolar range. By comparison, the inhibitory activity of all these covalent inhibitors is about 10-fold weaker than the parent ADAPTins2c and 2p, which could presumably be imparted by either the steric hindrance of the warhead, a change in the loop topology, or a deceleration of binding kinetics. Next, we asked if these covalent inhibitors could still exhibit high binding affinity in our competitive assay under saturating conditions of PDL1 (Fig. 4(D)). Time-course experiments were repeated by preincubating the non-covalent inhibitor 2c in one case, or the covalent ADAPTins2ca and 2cb at the same concentration. Under these physiologically relevant conditions (PDL1 excess: 12-fold), the binding profile of both electrophilic peptides 2ca and 2cb is consistent with an irreversible inhibition affording a complete blockade of the PD1:PDL1 interaction. Over the course of two hours, both ADAPTins2ca and 2cb bonded covalently to PD1 leading to a complete blockade similar to the one observed with the full-length pembrolizumab antibody (at 70 pM). These results are in stark contrast to the non-covalent inhibitor 2c which was easily displaced by the excess ligand PDL1.
Covalent bonding to the PD1 protein
PD1 is heavily N- and O-glycosylated at positions N49/58/74/116 and T153/168, S157/159 respectively, therefore to assess the covalent binding of our inhibitors and generate a less diffused PD1 band on SDS-PAGE gel, the extracellular portion of the protein was first incubated with PNGase F. None of the glycosylation sites are near the binding contacts of pembrolizumab or GY-14 CDR-H3 loops, supporting the idea that such deglycosylated PD1 model protein remains relevant. Reaction of the electrophilic ADAPTin2cb with N-deglycosylated PD1 was investigated at 37 °C under different concentrations, and incubation times (Fig. 5(A)). It was found that the formation of covalent PD1-conjugates could be assessed in a dose-dependent manner at pH 6.5 using a minimum incubation of 12 h and 4 eq. of 2cb. Incubation with the non-covalent peptide 2c did not produce any higher negative molecular weight adduct on the SDS-PAGE 10% bis-tris gels (negative control), while the incubation of 2cb produced a slightly higher molecular conjugates (darker band) at peptide/protein ratios ranging from of 4:1 to 16:1 (Fig. S21 and S22, ESI†). Yet, due to the large number of O-glycans still present on PD1, smearing of the PD1 band (across ∼5 kDa) rendered the visualization and isolation of conjugated adducts difficult (Fig. S22, ESI†). Trypsin-digested bands of plausible conjugated adducts were subjected to MS/MS analysis, but none of the fragmentation patterns expected for the conjugation of ADAPTins to PD1 were observed. Therefore, we turned our attention to the direct incubation of PD1 to our electrophilic peptides (2 days at pH 8 and physiological temperature) and the detection by intact mass spectrometry (Fig. 5(B)). The deconvoluted mass spectrum of intact N-deglycosylated PD1 presented a number of masses ranging from 19995 to 21884 Da (Fig. 5(B), bottom red spectrum). The results of ADAPTin2pa incubation suggested that the peptide bonded covalently to certain glycol-forms of PD1 in a 1:1 complex affording new peaks with a Δm of 2600 Da. In contrast, peptide 2pb was less selective and found to form 1:1 and 1:2 complexes with PD1 (Fig. S24 and S25).
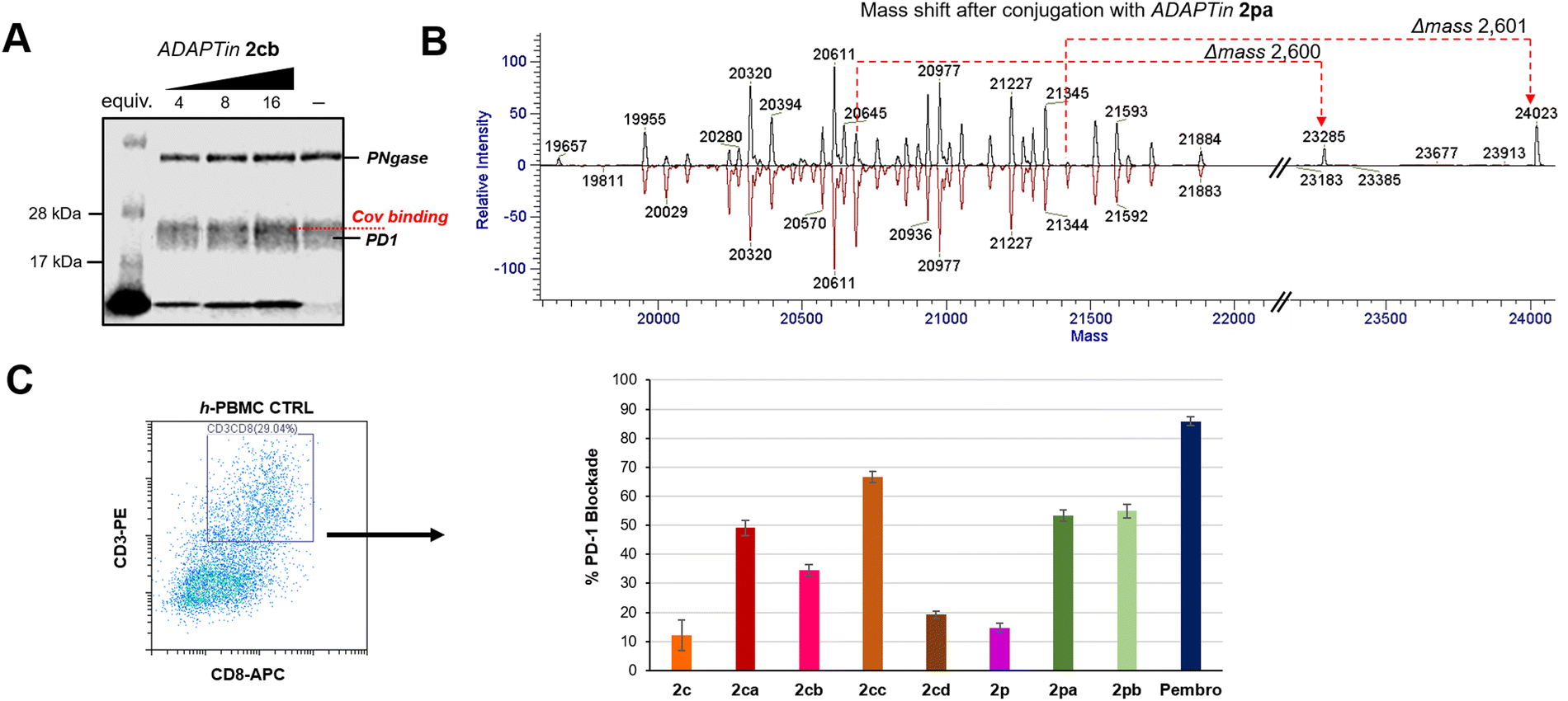 | ||
| Fig. 5 Electrophilic ADAPTins activity on the PD1 Protein. (A) Denaturing SDS-PAGE gel analysis showing a dose-dependence conjugation of 2cb to PD1 after incubation (12 h, 37 °C). No conjugation observed with 2c (negative control). (B) Mass spectrometry characterization of PD1 conjugation with 2pa (conjugation reaction with protein:peptide 55:220 μM, pH 8.0, 48 h). Deconvolution of the PD1 glycol-forms before (bottom red) and after (top black) conjugation with 2pa. (C) In cellulo PD1 blockade by covalent ADAPTins2ca–cd and 2pa–pbversus non-covalent analogs 2c and 2p and the full antibody control pembrolizumab. Cell surface level of PD1 blockade in PBMC cultures (30 h) determined by flow-cytometry analysis. Cells were gated on CD45+ and CD3+/CD8+ cells were selected from the CD45+ fraction; error bars indicate the mean PD1%-blockade ± SD (n = 3). | ||
Cell-based antitumor immunity rescue
To verify the binding efficacy of ADAPTins under more physiologically relevant conditions in cellulo, the binding of PD1 to non-covalent inhibitors 2c/2p and the corresponding electrophilic analogs 2ca–cd and 2pa–pb was examined on healthy human peripheral blood mononuclear cells (h-PBMCs) by flow cytometry (Fig. 5(C)).90 Levels of cell surface PD1 on h-PBMCs was measured after 30 hours of incubation with or without anti-PD1 peptides and compared to the levels measured with pembrolizumab (positive control, >95% blockade at 100 nM, see ESI,† Fig. S26). Compared to the non-covalent molecules 2c/2p (<20% binding), the efficacy of our electrophilic analogs 2ca, 2cc, and 2pa–pb on T-effector cells (CD3+/CD8+) were significantly higher, achieving 48 to 67 ±10% of PD1 blockade (Fig. 5(C)). These results confirmed that several electrophilic peptides not only irreversibly bind to free PD1 but also to CD8+ T cells expressing high levels of PD1 on their membrane. In addition, this cell-based assay suggests that ADAPTins achieved their covalent binding task while resisting (at least partially) to proteolytic degradation over the two-day experiments. Then, an ex vivo immune cell culture system using h-PBMCs and exhausted PBMCs from a melanoma patient (e-PBMCs) was used to determine the efficacy of selected ADAPTin peptides on T-effector cells (Fig. 6).91,92 T-cell exhaustion typically leads to a reduction in cytokine release, cytotoxic activity, as well as slower T-cell proliferation. Cytokine secretions were assayed after the reactivation of h-PBMCs and e-PBMCs with anti-CD2/28 antibodies and a six-hour incubation period with ADAPTins or pembrolizumab (positive control). As illustrated in Fig. 6(A), while non-covalent inhibitors 2c/2p had no detectable activity on cytokine secretions, an incubation with electrophilic ADAPTin analogs 2ca, 2cc, and 2pa–pb resulted in significant increases (2 to 5-fold) in TNF-α, IFN-γ, and perforin levels. In regards to the treatment of exhausted PBMCs (Fig. 6(B)), levels of inflammatory cytokines exhibited a substantial increase (TNF-α, IFN-γ, up by 3-fold to full recovery, IL-2 up by 1.5-fold) compared to untreated e-PBMCs and h-PBMCs (considered as two negative controls). Interestingly, the secretion of those cytokines are a hallmark of cytotoxic T lymphocytes (CTLs) response mediated by type-I natural killer (NK) cells.93 IL-2 concentrations reaching levels comparable to that observed upon treatment with pembrolizumab indicated that our anti-PD1 ADAPTins could promote the reactivation of an immune response and the rescue of effector T-cells proliferation. Along these lines, the significant increase in secreted cytoplasmic granule-associated proteins (perforin and granzyme B, levels >2-fold), particularly upon incubation with 2cb, suggested an activation of T-cell cytotoxicity, potentially related to NK cells activity.94 Cytokines concentrations measured at 72 h post-treatment were in most cases over the detection limit range further indicating the effectiveness of our compounds over time. Taken together, results from this ex vivo PBMC model suggest that ADAPTin2cb achieved a long-lasting PD1 inhibition and a potent rescue of T-cell proliferation and effector function. | ||
| Fig. 6 Ex vivo ADAPTin activity on the PD1 checkpoint pathway. Non-covalent ADAPTin2c/2p were used as negative control. (A) Levels of inflammatory cytokines measured by flow-cytometry (FACS) after 6 h incubation with electrophilic ADAPTins. Error bars indicate the mean ± SD, (n = 3). (B) Rescue of exhausted PBMCs from a melanoma patient. Levels of inflammatory cytokines and granule-associated proteins released (6 h) measured by FACS (healthy and untreated exhausted PBMCs: h-/e-PBMCs used as positive and negative controls respectively). Error bars indicate the avg ± SD, (n = 2). | ||
C. Conclusions
This study provides a proof-of-concept for transposing CDR-H3 structures found in antibodies into independent stand-alone ADAPTin peptide scaffolds. Our combined NMR and thermal CD-denaturation data demonstrated that diverse CDR-H3 loop sequences can be mounted into stable ADAPTins. Out of six anti-PD1 antibodies (pembrolizumab, MW11, M59c, camrelizumab, GY14, and tislelizumab), four native CDR-H3 loops were successfully transposed into folded scaffolds (1e, 1f, 2c, and 2n). Of the 29 peptides synthesized, over 70% demonstrated thermal stability above 37 °C. Although further NMR studies will be required to fully understand the structuration of β-bulges in ADAPTins,32 these motifs found at the apex of antibody CDR-H3s appear to play a pivotal role in structuring a large variety of loops. Notably, eight non-covalent ADAPTins effectively blocked the PD1/PDL1 protein–protein interaction at low nanomolar inhibitory concentrations (EC50 below 0.5 μM). Introducing acrylamide-based warheads to ADAPTins2c and 2p led to covalent binding despite a loss of affinity (∼10-fold). Even though electrophoresis gels and intact mass experiments further validated the covalent binding of 2cb and 2pa, peptide epitope mapping by MS/MS analyses were complicated by the high level of PD1 glycosylation. To establish the specificity of covalent bonding to the targeted PD1K131 residue and ultimately validate the antibody CDR-H3 biomimicry, further O-deglycosylation strategies will be evaluated95 as well as the introduction of more reactive lysine-targeting warheads.96,97 While further studies are needed to confirm specific PD1 residue targeting, covalent ADAPTins already showed superior PD1 binding in vitro and the rescue of exhausted PBMCs through cytokines secretion. The success of this scaffold highlights ADAPTin's potential for peptide display and biological screening, offering a novel biomimetic platform to target protein–protein interactions implicated in human diseases. With growing antibody structural databases, CDR-H3 mimics can be rationally designed and further optimized using high-throughput mRNA- or phage-display technologies53 to position these scaffolds in the therapeutic space of PPI inhibition.Author contributions
This project was conceived by S. P. R.; S. H. N., A. D. R., G. Z., and L. B. per-formed the research experiments and S. P. R. with S. H. N. analysed the data. Biological studies were coordinated by C. J. W, M. W., and N. S. and the mass spectrometry experiments were directed by C. P. D. The manuscript was written through contributions of S. P. R. and S. H. N.; all authors have given approval to the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.† The structural analysis of CDR-H3 binding surfaces was carried out using publicly available data of six different full-length antibodies from the worldwide protein data bank at [URL – https://www.rcsb.org/] with [PDB Entries: 5B8C, 5GGS, 6J14, 6JJP, 6K0Y, 7BXA, and 7CU5]. The NMR-guided 3D-structure and conformational ensemble of our model CDR-H3 ADAPTin2b is also available at the worldwide protein data bank [PDB Entry – 8W0Q].Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for this research was provided by the National Institutes of Health through an NIGMS Grant R21GM132754 to G. Z., L. B., A. D. R. and S. P. R., an NCI Grant R03CA286528 to S. H. N. and S. P. R. and by the Florida Department of Health through a Florida Cancer Innovation Grant MOAAP.Notes and references
- D. Ryan and J. Matthews, Protein–protein interactions in human disease, Curr. Opin. Struct. Biol., 2005, 15, 441–446 CrossRef CAS PubMed.
- J. D. Lapek, P. Greninger, R. Morris, A. Amzallag, I. Pruteanu-Malinici, C. H. Benes and W. Haas, Detection of dysregulated protein-association networks by high-throughput proteomics predicts cancer vulnerabilities, Nat. Biotechnol., 2017, 35, 983 CrossRef CAS PubMed.
- K. Lage, Protein–protein interactions and genetic diseases: The interactome, Biochim. Biophys. Acta, Mol. Basis Dis., 2014, 1842, 1971 CrossRef CAS PubMed.
- A. Laddach, J. C.-F. Ng, S. S. Chung and F. Fraternali, Genetic variants and protein–protein interactions: a multidimensional network-centric view, Curr. Opin. Struct. Biol., 2018, 50, 82 CrossRef CAS PubMed.
- D. E. Scott, A. R. Bayly, C. Abell and J. Skidmore, Small molecules, big targets: drug discovery faces the protein–protein interaction challenge, Nat. Rev. Drug Discovery, 2016, 15, 533 CrossRef CAS PubMed.
- X. Ran and J. E. Gestwicki, Inhibitors of protein–protein interactions (PPIs): an analysis of scaffold choices and buried surface area, Curr. Opin. Chem. Biol., 2018, 44, 75 CrossRef CAS PubMed.
- S. Steeland, R. E. Vandenbroucke and C. Libert, Nanobodies as therapeutics: big opportunities for small antibodies, Drug Discovery Today, 2016, 21, 1076 CrossRef CAS PubMed.
- Y. L. Boersma and A. Plückthun, DARPins and other repeat protein scaffolds: advances in engineering and applications, Curr. Opin. Biotechnol, 2011, 22, 849 CrossRef CAS PubMed.
- M.-E. Goebeler and R. C. Bargou, T cell-engaging therapies—BiTEs and beyond, Nat. Rev. Clin. Oncol., 2020, 17, 418 CrossRef PubMed.
- D. Schrama, R. A. Reisfeld and J. C. Becker, Antibody targeted drugs as cancer therapeutics, Nat. Rev. Drug Discovery, 2006, 5, 147 CrossRef CAS PubMed.
- A. I. Minchinton and I. F. Tannock, Drug penetration in solid tumours, Nat. Rev. Cancer, 2006, 6, 583 CrossRef CAS PubMed.
- K. T. Xenaki, S. Oliveira and P. M. P. van Bergen en Henegouwen, Antibody or Antibody Fragments: Implications for Molecular Imaging and Targeted Therapy of Solid Tumors, Front. Immunol., 2017, 8, 1287 CrossRef PubMed.
- M. Gebauer and A. Skerra, Engineered protein scaffolds as next-generation antibody therapeutics, Curr. Opin. Chem. Biol., 2009, 13, 245 CrossRef CAS PubMed.
- D. A. Richards, Exploring alternative antibody scaffolds: Antibody fragments and antibody mimics for targeted drug delivery, Drug Discovery Today: Technol., 2018, 30, 35 CrossRef PubMed.
- T. Wurch, A. Pierré and S. Depil, Novel protein scaffolds as emerging therapeutic proteins: from discovery to clinical proof-of-concept, Trends Biotechnol., 2012, 30, 575 CrossRef CAS PubMed.
- A. A. Vinogradov, Y. Yin and H. Suga, Macrocyclic Peptides as Drug Candidates: Recent Progress and Remaining Challenges, J. Am. Chem. Soc., 2019, 141, 4167 CrossRef CAS PubMed.
- C. A. Rhodes and D. Pei, Bicyclic Peptides as Next-Generation Therapeutics, Chem. – Eur. J., 2017, 23, 12690 CrossRef CAS PubMed.
- S. J. de Veer, A. M. White and D. J. Craik, Sunflower Trypsin Inhibitor-1 (SFTI-1): Sowing Seeds in the Fields of Chemistry and Biology, Angew. Chem., Int. Ed., 2021, 60, 8050 CrossRef CAS PubMed.
- L. D. Walensky, A. L. Kung, I. Escher, T. J. Malia, S. Barbuto, R. D. Wright, G. Wagner, G. L. Verdine and S. J. Korsmeyer, Activation of Apoptosis in Vivo by a Hydrocarbon-Stapled BH3 Helix, Science, 2004, 305, 1466 CrossRef CAS PubMed.
- A. L. Jochim and P. S. Arora, Systematic analysis of helical protein interfaces reveals targets for synthetic inhibitors, ACS Chem. Biol., 2010, 5, 919 CrossRef CAS PubMed.
- V. Azzarito, K. Long, N. S. Murphy and A. J. Wilson, Inhibition of α-helix–mediated protein–protein interactions using designed molecules, Nat. Chem., 2013, 5, 161 CrossRef CAS PubMed.
- M. Muttenthaler, G. F. King, D. J. Adams and P. F. Alewood, Trends in peptide drug discovery, Nat. Rev. Drug Discovery, 2021, 20, 309 CrossRef CAS PubMed.
- K. Van Holsbeeck, J. C. Martins and S. Ballet, Downsizing antibodies: Towards complementarity-determining region (CDR)-based peptide mimetics, Bioorg. Chem., 2022, 119, 105563 CrossRef CAS PubMed.
- G. Bhardwaj, V. K. Mulligan, C. D. Bahl, J. M. Gilmore, P. J. Harvey, O. Cheneval, G. W. Buchko, S. V. S. R. K. Pulavarti, Q. Kaas, A. Eletsky, P.-S. Huang, W. A. Johnsen, P. Greisen, Jr., G. J. Rocklin, Y. Song, T. W. Linsky, A. Watkins, S. A. Rettie, X. Xu, L. P. Carter, R. Bonneau, J. M. Olson, E. Coutsias, C. E. Correnti, T. Szyperski, D. J. Craik and D. Baker, Accurate de novo design of hyperstable constrained peptides, Nature, 2016, 538, 329 CrossRef CAS PubMed.
- W. S. Horne and T. N. Grossmann, Proteomimetics as protein-inspired scaffolds with defined tertiary folding patterns, Nat. Chem., 2020, 12, 331 CrossRef CAS PubMed.
- T. A. Hill, N. E. Shepherd, F. Diness and D. P. Fairlie, Constraining Cyclic Peptides To Mimic Protein Structure Motifs, Angew. Chem., Int. Ed., 2014, 53, 13020 CrossRef CAS PubMed.
- A. Luther, K. Moehle, E. Chevalier, G. Dale and D. Obrecht, Protein epitope mimetic macrocycles as biopharmaceuticals, Curr. Opin. Chem. Biol., 2017, 38, 45 CrossRef CAS PubMed.
- M. Favre, K. Moehle, L. Jiang, B. Pfeiffer and J. A. Robinson, Structural Mimicry of Canonical Conformations in Antibody Hypervariable Loops Using Cyclic Peptides Containing a Heterochiral Diproline Template, J. Am. Chem. Soc., 1999, 121, 2679 CrossRef CAS.
- D. Obrecht, E. Chevalier, K. Moehle and J. A. Robinson, β-Hairpin protein epitope mimetic technology in drug discovery, Drug Discovery Today: Technol., 2012, 9, e63 CrossRef CAS PubMed.
- M. Guharoy and P. Chakrabarti, Secondary structure based analysis and classification of biological interfaces: identification of binding motifs in protein–protein interactions, Bioinformatics, 2007, 23, 1909 CrossRef CAS PubMed.
- J. Gavenonis, B. A. Sheneman, T. R. Siegert, M. R. Eshelman and J. A. Kritzer, Comprehensive analysis of loops at protein–protein interfaces for macrocycle design, Nat. Chem. Biol., 2014, 10, 716 CrossRef CAS PubMed.
- G. Zhao, A. D. Richaud, R. T. Williamson, M. Feig and S. P. Roche, De Novo Synthesis and Structural Elucidation of CDR-H3 Loop Mimics, ACS Chem. Biol., 2024, 19, 1583 CrossRef CAS PubMed.
- J. Glanville, W. Zhai, J. Berka, D. Telman, G. Huerta, G. R. Mehta, I. Ni, L. Mei, P. D. Sundar, G. M. R. Day, D. Cox, A. Rajpal and J. Pons, Precise determination of the diversity of a combinatorial antibody library gives insight into the human immunoglobulin repertoire, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20216 CrossRef CAS PubMed.
- H.-J. Chang, J.-W. Jian, H.-J. Hsu, Y.-C. Lee, H.-S. Chen, J.-J. You, S.-C. Hou, C.-Y. Shao, Y.-J. Chen, K.-P. Chiu, H.-P. Peng, K. H. Lee and A.-S. Yang, Loop-Sequence Features and Stability Determinants in Antibody Variable Domains by High-Throughput Experiments, Structure, 2014, 22, 9 CrossRef CAS PubMed.
- H. Wang, K. Yan, R. Wang, Y. Yang, Y. Shen, C. Yu and L. Chen, Antibody heavy chain CDR3 length-dependent usage of human IGHJ4 and IGHJ6 germline genes, Antibody Ther., 2021, 4, 101 CrossRef CAS PubMed.
- W. K. Wong, J. Leem and C. M. Deane, Comparative Analysis of the CDR Loops of Antigen Receptors, Front. Immunol., 2019, 10, 2454 CrossRef CAS PubMed.
- I. Sela-Culang, S. Alon and Y. Ofran, A Systematic Comparison of Free and Bound Antibodies Reveals Binding-Related Conformational Changes, J. Immunol., 2012, 189, 4890 CrossRef CAS PubMed.
- S. E. Wong, B. D. Sellers and M. P. Jacobson, Effects of somatic mutations on CDR loop flexibility during affinity maturation, Proteins, 2011, 79, 821 CrossRef CAS PubMed.
- F. Schiele, J. van Ryn, T. Litzenburger, M. Ritter, D. Seeliger and H. Nar, Structure-guided residence time optimization of a dabigatran reversal agent, mAbs, 2015, 7, 871 CrossRef CAS PubMed.
- M. L. Fernández-Quintero, J. R. Loeffler, J. Kraml, U. Kahler, A. S. Kamenik and K. R. Liedl, Characterizing the Diversity of the CDR-H3 Loop Conformational Ensembles in Relationship to Antibody Binding Properties, Front. Immunol., 2019, 9, 3065 CrossRef PubMed.
- H. Shirai, A. Kidera and H. Nakamura, Structural classification of CDR-H3 in antibodies, FEBS Lett., 1996, 399, 1–8 CrossRef CAS PubMed.
- V. Morea, A. Tramontano, M. Rustici, C. Chothia and A. M. Lesk, Conformations of the third hypervariable region in the VH domain of immunoglobulins, J. Mol. Biol., 1998, 275, 269 CrossRef CAS PubMed.
- B. J. DeKosky, O. I. Lungu, D. Park, E. L. Johnson, W. Charab, C. Chrysostomou, D. Kuroda, A. D. Ellington, G. C. Ippolito, J. J. Gray and G. Georgiou, Large-scale sequence and structural comparisons of human naive and antigen-experienced antibody repertoires, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E2636 CrossRef CAS PubMed.
- A. G. Schmidt, H. Xu, A. R. Khan, T. O’Donnell, S. Khurana, L. R. King, J. Manischewitz, H. Golding, P. Suphaphiphat, A. Carfi, E. C. Settembre, P. R. Dormitzer, T. B. Kepler, R. Zhang, M. A. Moody, B. F. Haynes, H.-X. Liao, D. E. Shaw and S. C. Harrison, Preconfiguration of the antigen-binding site during affinity maturation of a broadly neutralizing influenza virus antibody, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 264 CrossRef CAS PubMed.
- N. G. Bozhanova, A. K. Sangha, A. M. Sevy, P. Gilchuk, K. Huang, R. S. Nargi, J. X. Reidy, A. Trivette, R. H. Carnahan, A. Bukreyev, J. E. Crowe and J. Meiler, Discovery of Marburg virus neutralizing antibodies from virus-naïve human antibody repertoires using large-scale structural predictions, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 31142 CrossRef CAS PubMed.
- M. L. Fernández-Quintero, J. R. Loeffler, L. M. Bacher, F. Waibl, C. A. Seidler and K. R. Liedl, Local and Global Rigidification Upon Antibody Affinity Maturation, Front. Mol. Biosci., 2020, 7, 182 CrossRef PubMed.
- B. D. Weitzner, R. L. Dunbrack and J. J. Gray, The Origin of CDR H3 Structural Diversity, Structure, 2015, 23, 302 CrossRef CAS PubMed.
- Z. Bahrami Dizicheh, I. L. Chen and P. Koenig, VHH CDR-H3 conformation is determined by VH germline usage, Commun. Biol., 2023, 6, 864 CrossRef CAS PubMed.
- J. Parkinson, R. Hard and W. Wang, The RESP AI model accelerates the identification of tight-binding antibodies, Nat. Commun., 2023, 14, 454 CrossRef CAS PubMed.
- M. Levi, M. Sällberg, U. Rudén, D. Herlyn, H. Maruyama, H. Wigzell, J. Marks and B. Wahren, A complementarity-determining region synthetic peptide acts as a miniantibody and neutralizes human immunodeficiency virus type 1 in vitro, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 4374 CrossRef CAS PubMed.
- B.-W. Park, H.-T. Zhang, C. Wu, A. Berezov, X. Zhang, R. Dua, Q. Wang, G. Kao, D. M. O'Rourke, M. I. Greene and R. Murali, Rationally designed anti-HER2/neu peptide mimetic disables P185HER2/neu tyrosine kinases in vitro and in vivo, Nat. Biotechnol., 2000, 18, 194 CrossRef CAS PubMed.
- A. M. Sevy, I. M. Gilchuk, B. P. Brown, N. G. Bozhanova, R. Nargi, M. Jensen, J. Meiler and J. E. Crowe, Computationally Designed Cyclic Peptides Derived from an Antibody Loop Increase Breadth of Binding for Influenza Variants, Structure, 2020, 28, 1114 CrossRef CAS PubMed.
- L. Misson Mindrebo, H. Liu, G. Ozorowski, Q. Tran, J. Woehl, I. Khalek, J. M. Smith, S. Barman, F. Zhao, C. Keating, O. Limbo, M. Verma, J. Liu, R. L. Stanfield, X. Zhu, H. L. Turner, D. Sok, P.-S. Huang, D. R. Burton, A. B. Ward, I. A. Wilson and J. G. Jardine, Fully synthetic platform to rapidly generate tetravalent bispecific nanobody–based immunoglobulins, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2216612120 CrossRef PubMed.
- C. Robert, A decade of immune-checkpoint inhibitors in cancer therapy, Nat. Commun., 2020, 11, 3801 CrossRef CAS PubMed.
- N. Lyu, K. Wang, F. Zhang, H. Qin, Y. Zhao, R. Wu, Y. Si and L. Wang, Recognition of PDL1/L2 by different induced-fit mechanisms of PD1: a comparative study of molecular dynamics simulations, Phys. Chem. Chem. Phys., 2020, 22, 1276 RSC.
- M. M. Miller, C. Mapelli, M. P. Allen, M. S. Bowshe, K. M. Boy, E. P. Gillis, D. R. Langley, E. Mull, M. A. Poirier, N. Sanghvi, L.-Q. Sun, D. J. Tenney, K.-S. Yeung, J. Zhu, P. C. Reid and P. M. Scola, Macrocyclic inhibitors of the pd-1/pd-l1 and cd80(b7-1)/pd-l1 protein/protein interactions, (Bristol-Myers Squibb Company), US Pat. 20140294898 A1, 2014 Search PubMed.
- A. Ganesan, M. Ahmed, I. Okoye, E. Arutyunova, D. Babu, W. L. Turnbull, J. Kundu, J. Shields, K. C. Agopsowicz, L. Xu, Y. Tabana, N. Srivastava, G. Zhang, T. C. Moon, A. Belovodskiy, M. Hena, A. S. Kandadai, S. N. Hosseini, M. Hitt, J. Walker, M. Smylie, F. G. West, A. G. Siraki, M. J. Lemieux, S. Elahi, J. A. Nieman, D. L. Tyrrell, M. Houghton and K. Barakat, Comprehensive in vitro characterization of PD-L1 small molecule inhibitors, Sci. Rep., 2019, 9, 12392 CrossRef PubMed.
- H. Yin, X. Zhou, Y.-H. Huang, G. J. King, B. M. Collins, Y. Gao, D. J. Craik and C. K. Wang, Rational Design of Potent Peptide Inhibitors of the PD-1 Interaction for Cancer Immunotherapy, J. Am. Chem. Soc., 2021, 143, 18536–18547 CrossRef CAS PubMed.
- C. M. Bryan, G. J. Rocklin, M. J. Bick, A. Ford, S. Majri-Morrison, A. V. Kroll, C. J. Miller, L. Carter, I. Goreshnik, A. Kang, F. DiMaio, K. V. Tarbell and D. Baker, Computational design of a synthetic PD-1 agonist, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2102164118 CrossRef CAS PubMed.
- S. Guardiola, M. Varese, X. Roig, M. Sánchez-Navarro, J. García and E. Giralt, Target-templated de novo design of macrocyclic D-/L-peptides: discovery of drug-like inhibitors of PD-1, Chem. Sci., 2021, 12, 5164 RSC.
- Q. Miao, W. Zhang, K. Zhang, H. Li, J. Zhu and S. Jiang, Rational design of a potent macrocyclic peptide inhibitor targeting the PD-1/PD-L1 protein–protein interaction, RSC Adv., 2021, 11, 23270 RSC.
- R. M. Hughes and M. L. Waters, Model systems for β-hairpins and β-sheets, Curr. Opin. Struct. Biol., 2006, 16, 514 CrossRef CAS PubMed.
- J. M. Anderson, B. Jurban, K. N. L. Huggins, A. A. Shcherbakov, I. Shu, B. Kier and N. H. Andersen, Nascent Hairpins in Proteins: Identifying Turn Loci and Quantitating Turn Contributions to Hairpin Stability, Biochemistry, 2016, 55, 5537 CrossRef CAS PubMed.
- C. M. Santiveri and M. A. Jiménez, Tryptophan residues: Scarce in proteins but strong stabilizers of β-hairpin peptides, Biopolymers, 2010, 94, 779 CrossRef CAS PubMed.
- B. L. Kier, I. Shu, L. A. Eidenschink and N. H. Andersen, Stabilizing capping motif for β-hairpins and sheets, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 10466 CrossRef CAS PubMed.
- J. M. Anderson and B. L. Kier, A. A. Shcherbakov and N. H. Andersen, An improved capping unit for stabilizing the ends of associated β-strands, FEBS Lett., 2014, 588, 4749 CrossRef CAS PubMed.
- J. M. Anderson, A. A. Shcherbakov, B. L. Kier, J. Kellock, I. Shu, A. L. Byrne, L. A. Eidenschink and N. H. Andersen, Optimization of a β-sheet-cap for long loop closure, Biopolymers, 2017, 107, e22995 CrossRef PubMed.
- J. M. Anderson, B. L. Kier, B. Jurban, A. Byrne, I. Shu, L. A. Eidenschink, A. A. Shcherbakov, M. Hudson, R. M. Fesinmeyer and N. H. Andersen, Aryl–aryl interactions in designed peptide folds: Spectroscopic characteristics and optimal placement for structure stabilization, Biopolymers, 2016, 105, 337 CrossRef CAS PubMed.
- M. Kjaergaard and F. M. Poulsen, Sequence correction of random coil chemical shifts: correlation between neighbor correction factors and changes in the Ramachandran distribution, J. Biomol. NMR, 2011, 50, 157 CrossRef CAS PubMed.
- J. Li, K. C. Bennett, Y. Liu, M. V. Martin and T. Head-Gordon, Accurate prediction of chemical shifts for aqueous protein structure on “Real World” data, Chem. Sci., 2020, 11, 3180–3191 RSC.
- B. D. Weitzner, J. R. Jeliazkov, S. Lyskov, N. Marze, D. Kuroda, R. Frick, J. Adolf-Bryfogle, N. Biswas, R. L. Dunbrack and J. J. Gray, Modeling and docking of antibody structures with Rosetta, Nat. Protoc., 2017, 12, 401 CrossRef CAS PubMed.
- S. Lyskov, F.-C. Chou, S. Ó. Conchúir, B. S. Der, K. Drew, D. Kuroda, J. Xu, B. D. Weitzner, P. D. Renfrew, P. Sripakdeevong, B. Borgo, J. J. Havranek, B. Kuhlman, T. Kortemme, R. Bonneau, J. J. Gray and R. Das, Serverification of Molecular Modeling Applications: The Rosetta Online Server That Includes Everyone (ROSIE), PLoS One, 2013, 8, e63906 CrossRef PubMed.
- A. L. Hopkins, G. M. Keserü, P. D. Leeson, D. C. Rees and C. H. Reynolds, The role of ligand efficiency metrics in drug discovery, Nat. Rev. Drug Discovery, 2014, 13, 105–121 CrossRef CAS PubMed.
- J. Moxam, S. Naylon, A. D. Richaud, G. Zhao, A. Padilla and S. P. Roche, Passive Membrane Permeability of Sizable Acyclic β-Hairpin Peptides, ACS Med. Chem. Lett., 2023, 14, 278–284 CrossRef CAS PubMed.
- M. E. Brown, D. Bedinger, A. Lilov, P. Rathanaswami, M. Vásquez, S. Durand, I. Wallace-Moyer, L. Zhong, J. H. Nett, I. Burnina, I. Caffry, H. Lynaugh, M. Sinclair, T. Sun, J. Bukowski, Y. Xu and Y. N. Abdiche, Assessing the binding properties of the anti-PD-1 antibody landscape using label-free biosensors, PLoS One, 2020, 15, e0229206 CrossRef CAS PubMed.
- R. A. Copeland, The drug–target residence time model: a 10-year retrospective, Nat. Rev. Drug Discovery, 2016, 15, 87 CrossRef CAS PubMed.
- A. D. Richaud, M. Zaghouani, G. Zhao, M. Wangpaichitr, N. Savaraj and S. P. Roche, Exploiting the Innate Plasticity of the Programmed Cell Death-1 (PD1) Receptor to Design Pembrolizumab H3 Loop Mimics, ChemBioChem, 2022, 23, e202200449 CrossRef CAS PubMed.
- L. Mittal, M. Srivastava, A. Kumari, R. K. Tonk, A. Awasthi and S. Asthana, Interplay among Structural Stability, Plasticity, and Energetics Determined by Conformational Attuning of Flexible Loops in PD-1, J. Chem. Inf. Model., 2021, 61, 358 CrossRef CAS PubMed.
- T. Lepir, M. Zaghouani, S. P. Roche, Y. Y. Li, M. Suarez, M. J. Irias and N. Savaraj, Nivolumab to pembrolizumab switch induced a durable melanoma response: A case report, Medicine, 2019, 98, e13804 CrossRef CAS PubMed.
- D. Chen, S. Tan, H. Zhang, H. Wang, W. He, R. Shi, Z. Tong, J. Zhu, H. Cheng, S. Gao, Y. Chai, J. Qi, M. Xiao, J. Yan and G. F. Gao, The FG Loop of PD-1 Serves as a “Hotspot” for Therapeutic Monoclonal Antibodies in Tumor Immune Checkpoint Therapy, iScience, 2019, 14, 113 CrossRef CAS PubMed.
- H. Liu, L. Guo, J. Zhang, Y. Zhou, J. Zhou, J. Yao, H. Wu, S. Yao, B. Chen, Y. Chai, J. Qi, G. F. Gao, S. Tan, H. Feng and J. Yan, Glycosylation-independent binding of monoclonal antibody toripalimab to FG loop of PD-1 for tumor immune checkpoint therapy, mAbs, 2019, 11, 681 CAS.
- S. Horita, Y. Nomura, Y. Sato, T. Shimamura, S. Iwata and N. Nomura, High-resolution crystal structure of the therapeutic antibody pembrolizumab bound to the human PD-1, Sci. Rep., 2016, 6, 35297 CrossRef CAS PubMed.
- L. F. Ponce, K. Leon and P. A. Valiente, Unraveling a Conserved Conformation of the FG Loop upon the Binding of Natural Ligands to the Human and Murine PD1, J. Phys. Chem. B, 2022, 126, 1441 CrossRef CAS PubMed.
- U. P. Dahal, A. M. Gilbert, R. S. Obach, M. E. Flanagan, J. M. Chen, C. Garcia-Irizarry, J. T. Starr, B. Schuff, D. P. Uccello and J. A. Young, Intrinsic reactivity profile of electrophilic moieties to guide covalent drug design: N-α-acetyl-L-lysine as an amine nucleophile, MedChemComm, 2016, 7, 864 RSC.
- A. Birkholz, D. J. Kopecky, L. P. Volak, M. D. Bartberger, Y. Chen, C. M. Tegley, T. Arvedson, J. D. McCarter, C. Fotsch and V. J. Cee, Systematic Study of the Glutathione Reactivity of N-Phenylacrylamides: 2. Effects of Acrylamide Substitution, J. Med. Chem., 2020, 63, 11602 CrossRef CAS PubMed.
- R. Lonsdale and R. A. Ward, Structure-based design of targeted covalent inhibitors, Chem. Soc. Rev., 2018, 47, 3816–3830 RSC.
- J. Singh, The Ascension of Targeted Covalent Inhibitors, J. Med. Chem., 2022, 65, 5886 CrossRef CAS PubMed.
- J. Pettinger, K. Jones and M. D. Cheeseman, Lysine-Targeting Covalent Inhibitors, Angew. Chem., Int. Ed., 2017, 56, 15200 CrossRef CAS PubMed.
- J. Pettinger, M. Carter, K. Jones and M. D. Cheeseman, Kinetic Optimization of Lysine-Targeting Covalent Inhibitors of HSP72, J. Med. Chem., 2019, 62, 11383 CrossRef CAS PubMed.
- S. M. Bacot, T. A. Harper, R. L. Matthews, C. J. Fennell, A. Akue, M. A. KuKuruga, S. Lee, T. Wang and G. M. Feldman, Exploring the Potential Use of a PBMC-Based Functional Assay to Identify Predictive Biomarkers for Anti-PD-1 Immunotherapy, Int. J. Mol. Sci., 2020, 21, 9023 CrossRef CAS PubMed.
- P. G. Sasikumar, N. S. Sudarshan, S. Adurthi, R. K. Ramachandra, D. S. Samiulla, A. Lakshminarasimhan, A. Ramanathan, T. Chandrasekhar, A. A. Dhudashiya, S. R. Talapati, N. Gowda, S. Palakolanu, J. Mani, B. Srinivasrao, D. Joseph, N. Kumar, R. Nair, H. S. Atreya, N. Gowda and M. Ramachandra, PD-1 derived CA-170 is an oral immune checkpoint inhibitor that exhibits preclinical anti-tumor efficacy, Commun. Biol., 2021, 4, 699 CrossRef CAS PubMed.
- A. Roberts, L. Bentley, T. Tang, F. Stewart, C. Pallini, J. Juvvanapudi, G. R. Wallace, A. J. Cooper, A. Scott, D. Thickett, S. T. Lugg, H. Bancroft, B. Hemming, C. Ferris, G. Langman, A. Robinson, J. Chapman, B. Naidu, T. Pinkney, G. S. Taylor, K. Brock, Z. Stamataki, C. A. Brady, S. J. Curnow, J. Gordon, O. Qureshi and N. M. Barnes, Ex vivo modelling of PD-1/PD-L1 immune checkpoint blockade under acute, chronic, and exhaustion-like conditions of T-cell stimulation, Sci. Rep., 2021, 11, 4030 CrossRef CAS PubMed.
- H. Ito and M. Seishima, Regulation of the Induction and Function of Cytotoxic T Lymphocytes by Natural Killer T Cell, J. Biomed. Biotechnol., 2010, 2010, 641757 Search PubMed.
- D. Krijgsman, M. Hokland and P. J. K. Kuppen, The Role of Natural Killer T Cells in Cancer—A Phenotypical and Functional Approach, Front. Immunol., 2018, 9, 367 CrossRef PubMed.
- P. Tit-oon, A. Wonglangka, K. Boonkanta, M. Ruchirawat, M. Fuangthong, R. Sasisekharan and A. Khongmanee, Intact mass analysis reveals the novel O-linked glycosylation on the stalk region of PD-1 protein, Sci. Rep., 2023, 13, 9631 CrossRef CAS PubMed.
- N. V. Mehta and M. S. Degani, The Expanding Repertoire of Covalent Warheads for Drug Discovery, Drug Discovery Today, 2023, 28, 103799 CrossRef CAS PubMed.
- L. Hillebrand, X. J. Liang, R. A. M. Serafim and M. Gehringer, Emerging and Re-Emerging Warheads for Targeted Covalent Inhibitors: An Update, J. Med. Chem., 2024, 67, 7668 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray analysis of CDR-H3s, electrophilic ADAPTins designs, experimental synthetic procedures and analytical data for all hairpin peptides, details of structural and binding studies (including variable temperature CD and NMR data, ELISA, SDS-PAGE and mass spectrometry data) are available online (PDF). A separate file contains raw CD-data and denaturation curves (XLSX) for all peptides. See DOI: https://doi.org/10.1039/d4cb00174e |
| This journal is © The Royal Society of Chemistry 2024 |