
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA novel dual-release scaffold for fluorescent labels improves cyclic immunofluorescence†
Thorge
Reiber
ab,
Christian
Dose
a and
Dmytro A.
Yushchenko
 *a
*a
aDepartment of Chemical Biology, Miltenyi Biotec GmbH, Friedrich-Ebert Straße 68, Bergisch Gladbach 51429, Germany. E-mail: dmytroy@miltenyi.com
bDepartment of Chemistry, Humboldt-Universität zu Berlin, Brook-Taylor-Str. 2, Berlin 12489, Germany
First published on 28th May 2024
Abstract
Cyclic immunofluorescence is a powerful method to generate high-content imaging datasets for investigating cell biology and developing therapies. This method relies on fluorescent labels that determine the quality of immunofluorescence and the maximum number of staining cycles that can be performed. Here we present a novel fluorescent labelling strategy, based on antibodies conjugated to a scaffold containing two distinct sites for enzymatic cleavage of fluorophores. The scaffold is composed of a dextran decorated with short ssDNA that upon hybridization with complementary dye-modified oligos result in fluorescent molecules. The developed fluorescent labels exhibit specific staining and remarkable brightness in flow cytometry and fluorescence microscopy. We showed that the combination of DNase-mediated degradation of DNA and dextranse-mediated degradation of the dextran as two complementary enzymatic release mechanisms in one molecule, improves signal erasure from labelled epitopes. We envision that such dual-release labels with high brightness and efficient and specific erasure will advance multiplexed cyclic immunofluorescence approaches and thereby will contribute to gaining new insights in cell biology.
Introduction
Immunofluorescence is a powerful tool widely used for protein visualization and localization, where targets are typically stained with fluorophore-decorated protein binders.1 To guarantee reliable detection by fluorescence microscopy, considerable efforts have been made to develop bright labels, in particular based on fluorophore multimerization.2,3 However, in addition to the bright staining of the protein of interest, there is an increasing demand for multiplexing in immunofluorescence applications to generate high-content datasets needed to address the complex nature of cellular spatial relationships and heterogeneity in tissues. It is not surprising that in recent years a significant effort has been made to develop new fluorescent dyes4,5 and to optimize their photophysical properties to advance multiplexed imaging.6,7As one of the main methods in spatial proteomics, multiplexed cyclic immunofluorescence enables cellular and subcellular localization of large numbers of proteins within populations of cells. This approach has recently provided unprecedented insights into biological processes, not only in terms of cell biology, but also from a clinical perspective, e.g., describing localization changes of proteins in pathogenic cells.8–10
An essential step in multiplexed cyclic immunofluorescence is sufficient signal removal after each staining round. In recent years multiple techniques have been developed to erase fluorescent signals. Gavins et al. developed peptide nucleic acids (PNA), that bind fluorescently tagged nucleotides and can displace the fluorescent signal from the target within minutes. In this approach, increased brightness was obtained by hybridization with up to 5 fluorophores without compromising the erasure efficiency.11 Similarly, DNA-conjugated antibodies were used to label protein targets in fixed cells by formation of sequence-driven dynamic DNA complexes that allowed sequential analysis.12–14 In these reports, removal of DNA was achieved by competitive binding as well as by the introduction of instability to the complexes. Short fluorescently tagged DNA can also be removed within minutes by buffer exchange as demonstrated in cell and tissue experiments by Wang et al.15 Erasure of fluorescent signal using competitive binding is not restricted to PNA or DNA complexes, it can be also applied to ligand-receptor complexes as it was demonstrated in the example of receptor-specific glycans by Lin and coworkers.16
The fluorophore or protein binder can also be rendered non-functional by bleaching or by protein denaturation, respectively. The former is usually performed under oxidative conditions in alkaline medium, causing destruction of the fluorophore.17 Photobleaching can be universally applied on tissue and cells, and under alkaline conditions, several cycles of immunofluorescence are possible on fixed tissue slices and on cells within 1 to 4 hours. Radtke et al. demonstrated with “iterative bleaching extends multiplexity” (IBEX) that LiAlH4-mediated bleaching enables rapid erasure of several fluorophores, which is compatible with other techniques such as CITE-seq but affects epitopes on cells and tissue.18 In addition, stripping of antibodies from their targets for cyclic immunostaining using different denaturing agents (e.g., NaBH4, chaotropic salts, and a combination of 2-ME and SDS) was shown by several laboratories to be effective for multiplexed staining on tissue sections.19,20
Other groups have focused on cleavage of the linker between fluorophore and protein binder for signal erasure. Carlson and coworkers introduced scission-accelerated fluorophore exchange, a method based on rapid bioorthogonal click chemistry to remove immunofluorescent signals, and enable multiple rounds of staining of the same sample.21 Mondal et al. developed chemically cleavable azide-based linkers readily reduced by TCEP via Staudinger reaction and demonstrated their application for multiplexed single-cell protein analysis.22 Enzyme-mediated cleavage of linkers between dye and binder may offer a less harsh alternative to chemical cleavage. Gibbs and coworkers labelled cells with antibody-oligo conjugates and investigated several signal removal methods.23,24 They reported that approx. 80% of the signal was removed within minutes using enzymatic cleavage with restriction endonucleases.23,24 The authors also proposed several approaches to increase degree of labelling (DOL) of antibodies, and thus fluorescence signal of conjugates. Namely, they tested hybridization of several fluorescent imaging strands to a single docking strand on the antibody, and also tested imaging strands decorated with more than one fluorophore.25 Such fluorophore multimerization can lead to enhanced signal-to-background ratio, however this comes at the expense of incomplete removal of the signal after erasure. With a similar release mechanism, SeqStain was introduced by Rajagopalan and coworkers as an efficient method for multiplexed spatialomic profiling.26 In this work the authors demonstrated sequential analysis of cells and tissue by utilizing antibody-oligonucleotide conjugates that can be cleaved by specific endonucleases. This allowed fast staining times with efficient fluorophore cleavage (>90%) of more than 25 markers.
To advance cyclic immunofluorescence with bright, specific and fully-erasable dye-conjugates, new technologies have to combine high-fluorescence labelling with efficient fluorophore release, avoid false-positive signals, removal of labels under mild conditions, and reduce the duration of imaging cycles. To address these requirements, we recently developed a dextranase-mediated release technology, for cyclic immunofluorescence of up to hundreds of markers in tissue sections.27 Here we sought to combine this release technology with DNase-mediated cleavage, to further increase the rate and efficiency of the release step and maintain high brightness by fluorophore multimerization on the dextran scaffold. In this report, we present the design and synthesis of dual-releasable conjugates and discuss their performance in comparison with respective mono-modal release reagents.
Results and discussion
Probe design and bioconjugation strategy
Previously we showed that some dextran molecules of specific sizes can act as suitable scaffolds for synthesis of releasable conjugates.27 In this study, we designed our conjugates based on 250 kDa dextran decorated with amino groups (AmDex). This allowed the attachment of several fluorophores and protein binders and enabled a release mechanism based on the enzyme dextranase. As the second release mechanism, we installed dsDNA as a linker between the fluorescent dye and the dextran scaffold, which can be digested by DNases (Fig. 1).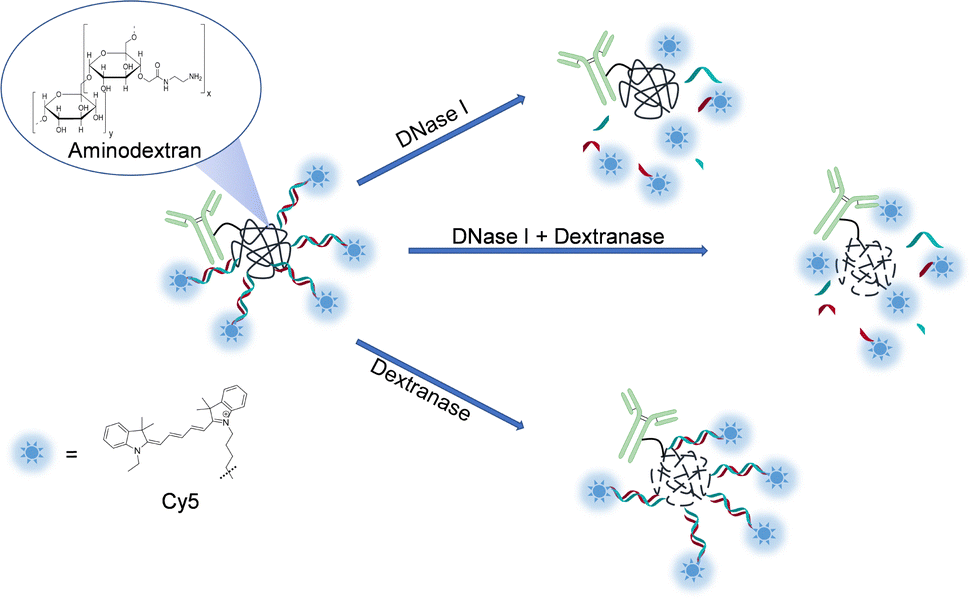 | ||
| Fig. 1 Conjugate design with proposed mechanisms of release. Conjugates are comprised of antibodies and oligonucleotides bound to dextran. Complementary oligos are modified with Cy5 fluorophore for direct primary staining. First mechanism (top): cleavage of dsDNA by DNase I. Second mechanism (bottom): cleavage of carbohydrate backbone by dextranase. Dual-release (middle) shows the combination of both enzymes acting simultaneously. | ||
In the first step, the dextran-oligonucleotide intermediates were prepared by activating AmDex with succinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylate (SMCC), following reaction with thiolated ssDNA oligonucleotides (see ESI†). AmDex with 50 amino groups was chosen due to its capacity to harbour several strands on a confined space. Indeed, we were able to conjugate an average of 18–19 primer strands per dextran, as determined by analytical size-exclusion chromatography (SEC, see Fig. S1 and Table S3, ESI†).
The obtained dextran-oligonucleotide intermediates were modified in a way that allowed for antibody coupling via strain-promoted azide alkyne cycloaddition (SPAAC). Here we chose CD4, EGFR, and EpCAM as targets, which are highly expressed in immune and cancer cells. As a model system, we chose T cell lymphoma-derived SUP-T1 and pancreas adenocarcinoma-derived AsPC-1 cell cultures, which express high levels of these receptors of interest.28–30 To generate conjugates, the respective recombinant antibodies were modified with dibenzocyclooctyne (DBCO) and added to the DNA-decorated azido-dextran intermediates for further reaction in a SPAAC. The final conjugates were purified by SEC to separate the desired product from unconjugated antibody. The obtained conjugates contained on average three antibodies per dextran being in the desired range (Table S4, ESI†). In the last step, antibody-dextran-ssDNA conjugates were hybridized with commercially available Cy5-labelled DNA oligomers to generate final fluorescent conjugates. In a more universal approach, described later in this study, we additionally sought to keep flexibility of colour by using a biotinylated complementary oligonucleotide that could be incubated with fluorescent anti-biotin antibodies in a secondary staining step.
Brightness and release of oligo-based conjugates in flow cytometry
Flow cytometry was initially used to test and estimate functionality, brightness, and the second release mechanism of the prepared conjugates. The performance of the dextran-oligonucleotide scaffold was investigated with conjugates against the CD4 receptor of T helper cells. Here, we hybridized Cy5-decorated oligonucleotides complementary to DNA coupled to the aCD4-dextran conjugate. As controls we used conventional antibody-oligo-Cy5 and antibody-Cy5 conjugates. After incubation of SUP-T1 cells with the hybridized probes, the mean fluorescence intensity (MFI) of the cells has increased indicating the functional binding of antibodies to their targeted receptors (Fig. 2A). Notably, the cells stained with dextran-oligo-based construct were 3-fold brighter (3-fold higher MFI) in comparison to the cells stained with antibody-oligo conjugate. This is likely due to more available docking strands and thus dyes per conjugate for dextran-oligo-based conjugates. Dextran-oligo-based conjugates were also shown to be brighter than direct antibody-dye conjugates (Fig. 2A). In general, multimerization of fluorescent dyes directly on antibodies leads to increased dye self-quenching that results in lower conjugate brightness.2 Presumably due to dye self-quenching, cell staining with the direct aCD4-Cy5 conjugate having DOL of 6 was not brighter than the staining with aCD4-Oligo-Cy5 having DOL of 2 (Fig. 2A and Table S4, ESI†). However, dye multimerization on the oligo-dextran scaffold leads to conjugates that overperform antibody-oligo and direct antibody-dye conjugates making it a suitable approach to prepare bright conjugates.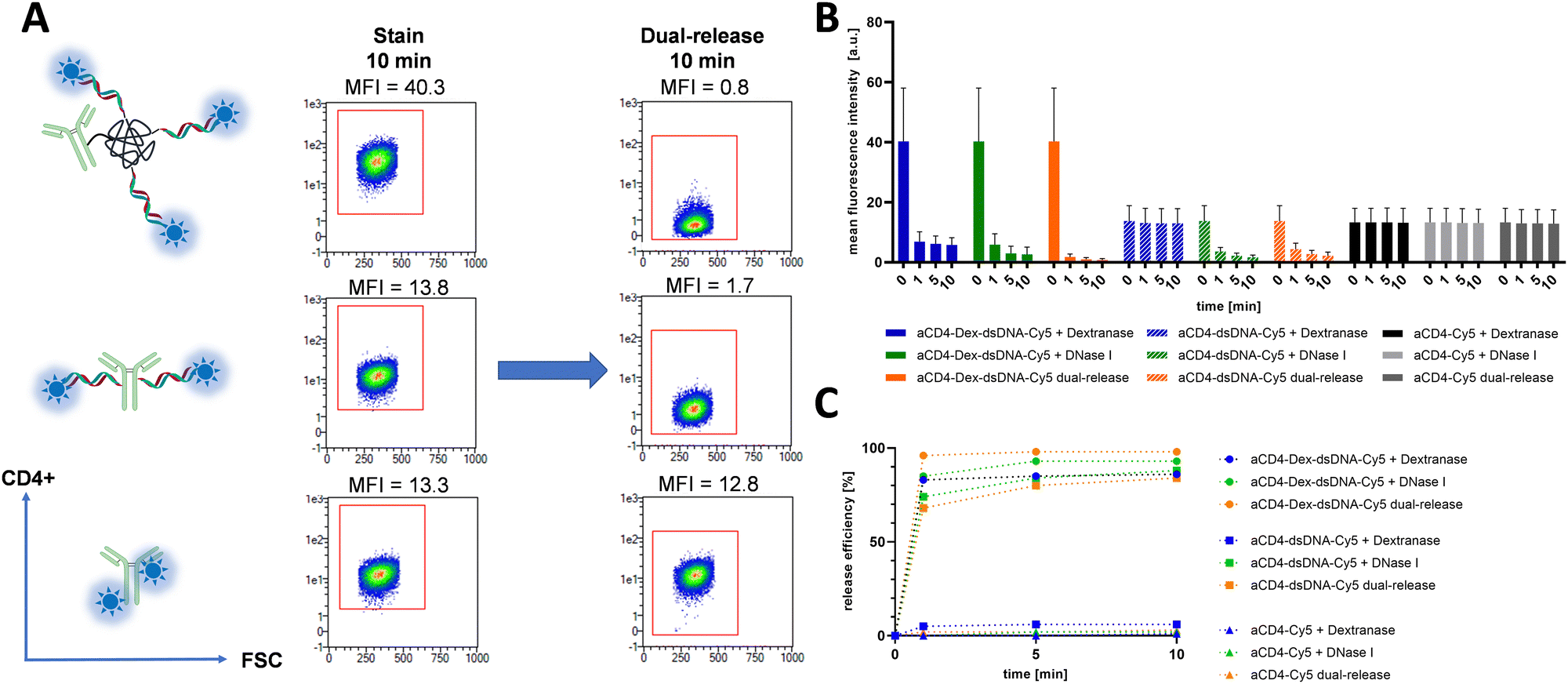 | ||
| Fig. 2 Stain and release flow cytometric analysis of SUP-T1 cells labelled with anti-CD4 conjugates (1 nM each). (A) Schematic representation of aCD4-Dex-dsDNA-Cy5 (top), aCD4-dsDNA-Cy5 (middle), and aCD4-Cy5 (bottom), the respective dot plots showing intensity after 10 min of staining and dual-release respectively. CD4+ signal is on the y-axis and forward scatter on the x-axis. aCD4-Dex-dsDNA-Cy5 shows approximately a 3-fold increase in staining intensity compared to the controls, while the dual-release in the presence of dextranase and DNase I leads to decrease of the fluorescence intensity for both oligo-based constructs. (B) Quantification of fluorescent signal at given time points and release pathway of flow cytometry experiment. Data is shown in MFI ± SD of the analyzed cell pool. (C) Quantification of release efficiency at given time points of release. Release efficiency is depicted in percent of signal reduction after release step. | ||
Next, as fast and efficient release of conjugates is an important factor in cyclic immunofluorescence, all probes were subjected to three possible release pathways: (1) dextranase release, (2) DNase I release, and (3) dual-release with both enzymes (Fig. 2). Notably, the second introduced cleavage step proved to increase the efficiency of erasure reaching 98 ± 1.5% after 10 min in contrast to mono-releases, as was shown on an example of aCD4-Dex-dsDNA-Cy5 (Fig. 2B and C). The dextran-free oligo-conjugates in the same conditions yielded only 88 ± 8.7% release efficiency, leaving higher residual background for next rounds of cyclic immunofluorescence. The direct antibody-fluorophore conjugate, as expected, did not show any release (Fig. 2B and C).
One-pot secondary staining approach for brighter and releasable conjugates
Next, we aimed to test the universality of the presented approach by enabling a simple plug-and-play principle of the developed conjugates. Here, we sought to recruit several protein-dye labeled antibodies targeted against biotin, that in conjunction with biotinylated complementary oligos form the sandwich-complexes in a secondary staining. Secondary staining usually includes labelling with a primary antibody, a washing step, another labelling step with a fluorescent secondary antibody, and a second washing step. Here, we sought to perform the primary and secondary labelling in a one-step (aka one pot) process, to deliver both faster staining protocols and brighter signals, due to more available binding sites of anti-biotin antibody. Compared to the controls, after 10 minutes of incubation, a 3-fold increase in MFI was achieved for both constructs (Fig. 3). This indicated a quick saturation of the dextran surface by secondary staining reagents, as a hypothetical 19-fold increase should be possible due to availability of biotins. However, antibody-APC/PE conjugates usually range from 300 kDa to 500 kDa depending on their construction, and it is thus not surprising that saturation of the dextran surface was quickly reached. This hypothesis is supported by the titration of complementary strands and anti-Biotin reagents, which showed no significant effect in staining performance, whereas saturation with the Cy5-labeled oligonucleotides resulted in higher staining intensities (see Fig. S10 and S11, ESI†). Nonetheless, we demonstrated that non-fluorescent conjugates can be successfully loaded with secondary antibodies to mimic a primary staining protocol. Additionally, cleavage carried out with both dextranase or DNase I as well as the combination of both for multiple labeled conjugates and different specificities (see ESI,† Fig. S12 and S13) were achieved despite bulkiness of the complexes. | ||
| Fig. 3 Flow cytometric analysis of SUP-T1 cells labeled with 5 nM aCD4-Dex-oligo hybridized with either aBiotin-PE or aBiotin-APC highlighted in red or blue, respectively. In both cases approximately a 3-fold increase in staining intensity compared to the controls was observed. Signal is given in mean fluorescence intensity (MFI) ± SD and release efficiency is depicted in percent of signal reduction after release step. | ||
Performance of dual-release conjugates in microscopy
Cell staining with conjugates based on dextran-oligonucleotide scaffold and their erasure were also tested in confocal laser-scanning microscopy. Firstly, we stained adherent AsPC-1 cells with aEGFR-Dex-dsDNA-Cy5 releasable conjugate. As non-releasable controls, we used respective anti-EGFR antibody aEGFR-Cy5 and aEGFR-APC. aEpCAM-PE was used as non-releasable reference in dual-release experiments to highlight exclusive release of EGFR signal. All anti-EGFR conjugates showed similar staining pattern and, as expected, aEGFR-Dex-dsDNA-Cy5 demonstrated higher fluorescent intensity than aEGFR-Cy5 and aEGFR-APC (Fig. 4A and Fig. S14, ESI†). | ||
| Fig. 4 Confocal laser-scanning microscopy images of fixed AsPC-1 cells stained with aEGFR-Dex-dsDNA-dye conjugates and signal quantification of initial fluorescence and during the course of release. (A) First row: release of aEGFR-Dex-dsDNA-Cy5 mediated by dextranase only; second row: release of aEGFR-Dex-dsDNA-Cy5 mediated by DNase only; third row: dual-release of aEGFR-Dex-dsDNA-Cy5 with both enzymes; fourth row: release of aEGFR-Dex-dsDNA-Cy5 conjugates while counterstained with non-cleavable aEpCAM-PE; fifth row: control with both non-cleavable anti-EGFR and anti-EpCAM conjugates. The columns indicate membrane staining after certain time points of release with t = 0 min being the initial staining before release. Cy5 signal is depicted in red, PE in green, and Vio515 in violet. Scale bar is 50 μm and is a representation for all images. LUTs for release with aEGFR-Dex-dsDNA-Cy5 are shown on the first panels and were kept constant for each release. (B) Signal quantification of (A), which showed faster and more efficient fluorophore cleavage when using both release reagents. Signal decrease of controls was also observed, however, it was accounted to photobleaching. (C) Quantification of dual-release mode of aBiotin-Vio515 labeled probes with aEpCAM-PE counterstaining. The releasable label was cleaved from the antibody while PE signal remains on the membrane. The data represent the mean average ± SE (aEGFR-Dex-dsDNA-Cy5 + Dextranase: n = 43; aEGFR-Dex-dsDNA-Cy5 + DNase I: n = 56; aEGFR-Dex-dsDNA-Cy5 dual release: n = 67; aEGFR-Dex-dsDNA-aBiotin-Vio515 + aEpCAM-PE dual release: n = 41; aEGFR-Cy5 + aEpCAM-PE dual release: n = 87). All signals are background-subtracted. | ||
Following the staining, the cells were treated by release reagents and each mode of release was quantified and compared to each other. Similarly to flow data, we observed superior efficiency of a dual-release compared to a single-release erasure by fluorescence microscopy. After 5 minutes of dual-release conditions, approximately 86 ± 2% erasure of fluorescence was reached, and up to 98 ± 1% after 15 min (Fig. 4A and B). In contrast, after 15 min dextranase-mediated erasure was below 90% for mono-release (Fig. 4A and B). Furthermore, this was reproducible with another antibody, namely Cetuximab, which showed fast cleavage when utilizing a dual release approach (Fig. S15, ESI†). As expected, staining of conventional non-releasable antibody-dye conjugates was stable after sample treatment with enzymes, demonstrating the excellent selectivity of the implemented cleavable moieties. Some non-significant reduction in signal intensity of conventional conjugates may be explained by dye photobleaching. When the conjugates with secondary aBiotin-dye antibodies were used, they showed reduced cleavage efficiency compared to the directly hybridized Cy5 counterpart (Fig. 4C and Fig. S16, ESI†). This may be due to an increase of steric hindrance caused by secondary antibodies rendering the respective enzymes inefficient due to lower access to the cleavage sites. In contrast, excellent cleavage was observed for the conjugates with Cy5 directly coupled to the complementary oligonucleotide. Importantly, we achieved remarkable brightness with Cy5 dyes compared to APC (Fig. S14, ESI†) demonstrating potential of this scaffold for uncompromised staining intensity. We next investigated the performance of these selective release mechanisms in cyclic immunofluorescence applications.
Fast cyclic immunofluorescence with dual-release conjugates
Finally, in order to determine the performance and specificity of a dual mode of release in multiplex cyclic immunofluorescence context, we iteratively imaged three markers expressed on the membrane of AsPC-1 cells: EGFR, EpCAM, and CD66c. For the staining of EGFR and EpCAM we prepared and used dual-release conjugates. CD66c was stained in the last cycle with non-releasable aCD66c-PE conjugate (Fig. 5A). The anti-EpCAM and anti-EGFR conjugates were prepared with either red-emitting Cy5 or green-emitting Vio515, which allowed (1) definite discrimination of targets by channels as well as (2) continuous detection of the residual signal following release, washing and re-staining. In agreement to our flow cytometry findings, we successfully stained targets on the membrane of AsPC-1 cells within 10 minutes and efficiently cleaved the fluorophores within 10 minutes, that resulted in no observable signal in the corresponding channel after wash and re-staining (Fig. 5A and B, see also ESI,† Fig. S17). It is worth to mention, that when imaged with a conventional microscope, like in this work, the region of interest can slightly shift due to taking out of the sample for washing and subsequent staining. Hence, imaging quality may further improve when a microscope with an automated liquid handling is used. | ||
| Fig. 5 Cyclic immunofluorescence of three targets on fixed AsPC-1 cells demonstrated in confocal microscopy with oligo-based conjugates. (A) Selective target staining located on the membrane observed in the appropriate channels. The first round of image acquisition was started with aEGFR-Dex-dsDNA-aBiotin-Vio515, followed by aEpCAM-Dex-dsDNA-Cy5, and ended with aCD66c-PE which is not releasable. The workflow of this experiment is indicated in the upper part of the figure. Duration of every staining and every release step was 10 min. Images were acquired with all three lasers to show successful release with the same ROI indicated within the white squares. Plates were taken out of the microscope for wash and re-staining, thus, slight shifts in ROI are visible. LUTs for each channel are shown on the right and were kept constant for each channel. Scale bar is 50 μm and is a representation for all images. (B) Quantification of (A) showing full release after 10 min. The data represent the mean average ± SE (each release with n = 47). | ||
Of note, conjugates were successfully applied for staining without purification from excess of complementary oligonucleotides. However, after hybridization minor non-specific labelling was sometimes observed. For example, aEpCAM-Dex-dsDNA-Cy5 exhibited minor cytosolic staining in addition to specific membrane staining (Fig. 5A, middle column). It is possible that fixation permeabilizes cells to a degree sufficient for penetration of small fluorescent oligonucleotides but not for full-length antibodies. Importantly, simple centrifugal filtration of conjugates removed the excess of complementary fluorescent oligonucleotides, resulting in pure conjugates that gave only specific membrane staining (Fig. S18, ESI†).
Conclusions
In this report, we developed conjugates with two distinct cleavable sites and demonstrated the advantage of implementing an additional release mechanism for cyclic immunostaining. While certain techniques require harsh conditions or long erasure times, and limited by the numbers of fluorophores, our method utilizes mild reaction conditions that allow for high brightness due to a high degree of labelling which does not compromise release efficiency.To prepare the dual-release conjugates, we inserted an oligo-based cleavable moiety as a linker between fluorophore and dextran, which acts as an orthogonal cleavage site. The obtained scaffold was then covalently coupled with antibodies to get conjugates for detecting different membrane receptors. Measuring brightness and releasability in flow cytometry and confocal microscopy demonstrated superior staining intensities of our conjugates and additive effects of release mechanisms. We further demonstrated cyclic immunofluorescence imaging of a tumour cell line with only several minutes between image acquisition. Importantly, for the targets tested in this work, our dual mode of release did not interfere with antibody functionality, or the respective epitope's structural integrity, which is crucial for specific labelling. Furthermore, the increased brightness of the developed conjugate design may simplify the imaging of low-abundant membrane proteins in cyclic immunofluorescence. We believe that the presented approach to prepare dual-release labels with high brightness and efficient erasure will help to advance multiplexed cyclic immunofluorescence imaging and thereby will contribute to gaining new insights in cell biology.
Author contributions
T. R. performed the experiments. T. R. and D. A. Y. designed the experiments and analysed the data. All authors discussed the results and contributed to the preparation and editing of the manuscript.Conflicts of interest
The authors declare the following competing interests. T. R., C. D. and D. A. Y. declare the filing of a patent application (EP 4 177 607 A1: Bright and Releasable Labels for Cell Staining Based on the Conjugates with Several Sites of Fluorophore Release), which was assigned to Miltenyi Biotec B.V. & Co. KG.Acknowledgements
We are grateful to Karen Möbius and Anna Baranska for assistance in experiments, Niels Werchau and Nele Knelangen for providing of cells and Nathan Brady for help in image analysis and proofreading the manuscript. We would like to thank Oleksandr Zavoiura, Travis Jennings and Antonina Kerbs for the insightful discussions. D. A. Y. would like to acknowledge Ukrainian people fighting for their country and for peace on the whole continent.Notes and references
- S. Joshi and D. Yu, in Basic Science Methods for Clinical Researchers, ed., M. Jalali, F. Y. L. Saldanha and M. Jalali, Academic Press, Boston, 2017, pp. 135–150 Search PubMed.
- T. Reiber, O. Zavoiura, C. Dose and D. A. Yushchenko, Eur. J. Org. Chem., 2021, 2817–2830 CrossRef CAS.
- A. H. Ashoka, I. O. Aparin, A. Reisch and A. S. Klymchenko, Chem. Soc. Rev., 2023, 52, 4525–4548 RSC.
- J. B. Grimm and L. D. Lavis, Nat. Methods, 2022, 19, 149–158 CrossRef CAS PubMed.
- J. V. Jun, D. M. Chenoweth and E. J. Petersson, Org. Biomol. Chem., 2020, 18, 5747–5763 RSC.
- K. Hoffmann, T. Behnke, D. Drescher, J. Kneipp and U. Resch-Genger, ACS Nano, 2013, 7, 6674–6684 CrossRef CAS.
- U. Resch-Genger, M. Grabolle, S. Cavaliere-Jaricot, R. Nitschke and T. Nann, Nat. Methods, 2008, 5, 763–775 CrossRef CAS PubMed.
- K. L. McNamara, J. L. Caswell-Jin, R. Joshi, Z. Ma, E. Kotler, G. R. Bean, M. Kriner, Z. Zhou, M. Hoang, J. Beechem, J. Zoeller, M. F. Press, D. J. Slamon, S. A. Hurvitz and C. Curtis, Nat. Cancer, 2021, 2, 400–413 CrossRef CAS PubMed.
- C. M. Schürch, S. S. Bhate, G. L. Barlow, D. J. Phillips, L. Noti, I. Zlobec, P. Chu, S. Black, J. Demeter, D. R. McIlwain, S. Kinoshita, N. Samusik, Y. Goltsev and G. P. Nolan, Cell, 2020, 182, 1341–1359.e1319 CrossRef PubMed.
- M. A. J. Gorris, A. Halilovic, K. Rabold, A. van Duffelen, I. N. Wickramasinghe, D. Verweij, I. M. N. Wortel, J. C. Textor, I. J. M. de Vries and C. G. Figdor, J. Immunology, 2018, 200, 347–354 CrossRef CAS PubMed.
- G. C. Gavins, K. Gröger, M. D. Bartoschek, P. Wolf, A. G. Beck-Sickinger, S. Bultmann and O. Seitz, Nat. Chem., 2021, 13, 15–23 CrossRef CAS PubMed.
- R. M. Schweller, J. Zimak, D. Y. Duose, A. A. Qutub, W. N. Hittelman and M. R. Diehl, Angew. Chem., Int. Ed., 2012, 51, 9292–9296 CrossRef CAS PubMed.
- D. Y. Duose, R. M. Schweller, W. N. Hittelman and M. R. Diehl, Bioconjugate Chem., 2010, 21, 2327–2331 CrossRef CAS PubMed.
- Y. Tang, Z. Wang, X. Yang, J. Chen, L. Liu, W. Zhao, X. C. Le and F. Li, Chem. Sci., 2015, 6, 5729–5733 RSC.
- Y. Wang, J. B. Woehrstein, N. Donoghue, M. Dai, M. S. Avendaño, R. C. J. Schackmann, J. J. Zoeller, S. S. H. Wang, P. W. Tillberg, D. Park, S. W. Lapan, E. S. Boyden, J. S. Brugge, P. S. Kaeser, G. M. Church, S. S. Agasti, R. Jungmann and P. Yin, Nano Lett., 2017, 17, 6131–6139 CrossRef CAS PubMed.
- N. Li, W. Zhang, L. Lin, S. N. A. Shah, Y. Li and J.-M. Lin, Anal. Chem., 2019, 91, 2600–2604 CrossRef CAS PubMed.
- J.-R. Lin, M. Fallahi-Sichani and P. K. Sorger, Nat. Commun., 2015, 6, 8390 CrossRef CAS PubMed.
- A. J. Radtke, E. Kandov, B. Lowekamp, E. Speranza, C. J. Chu, A. Gola, N. Thakur, R. Shih, L. Yao, Z. R. Yaniv, R. T. Beuschel, J. Kabat, J. Croteau, J. Davis, J. M. Hernandez and R. N. Germain, Proc. Natl. Acad. Sci., 2020, 117, 33455–33465 CrossRef CAS PubMed.
- M. M. Bolognesi, M. Manzoni, C. R. Scalia, S. Zannella, F. M. Bosisio, M. Faretta and G. Cattoretti, J. Histochem. Cytochem., 2017, 65, 431–444 CrossRef CAS PubMed.
- G. Gut, M. D. Herrmann and L. Pelkmans, Science, 2018, 361, eaar7042 CrossRef PubMed.
- J. Ko, M. Wilkovitsch, J. Oh, R. H. Kohler, E. Bolli, M. J. Pittet, C. Vinegoni, D. B. Sykes, H. Mikula, R. Weissleder and J. C. T. Carlson, Nat. Biotechnol., 2022, 40, 1654–1662 CrossRef CAS PubMed.
- M. Mondal, R. Liao, L. Xiao, T. Eno and J. Guo, Angew. Chem., Int. Ed., 2017, 56, 2636–2639 CrossRef CAS PubMed.
- J. A. Jones, N. P. McMahon, T. Zheng, J. Eng, K. Chin, S. Kwon, M. A. Nederlof, J. W. Gray and S. L. Gibbs, Sci. Rep., 2021, 11, 23844 CrossRef CAS PubMed.
- N. P. McMahon, J. A. Jones, S. Kwon, K. Chin, M. A. Nederlof, J. W. Gray and S. L. Gibbs, J. Biomed. Opt., 2020, 25, 1–18 Search PubMed.
- N. P. McMahon, J. A. Jones, A. N. Anderson, M. S. Dietz, M. H. Wong and S. L. Gibbs, Cancers, 2023, 15, 827 CrossRef CAS PubMed.
- A. Rajagopalan, I. Venkatesh, R. Aslam, D. Kirchenbuechler, S. Khanna, D. Cimbaluk, J. H. Kordower and V. Gupta, Cell Rep. Methods, 2021, 1, 100006 CrossRef CAS PubMed.
- A. Kinkhabwala, C. Herbel, J. Pankratz, D. A. Yushchenko, S. Rüberg, P. Praveen, S. Reiß, F. C. Rodriguez, D. Schäfer, J. Kollet, V. Dittmer, M. Martinez-Osuna, L. Minnerup, C. Reinhard, A. Dzionek, T. D. Rockel, S. Borbe, M. Büscher, J. Krieg, M. Nederlof, M. Jungblut, D. Eckardt, O. Hardt, C. Dose, E. Schumann, R.-P. Peters, S. Miltenyi, J. Schmitz, W. Müller and A. Bosio, Sci. Rep., 2022, 12, 1911 CrossRef CAS PubMed.
- D. Kozbor, R. Burioni, A. Ar-Rushdi, C. Zmijewski and C. M. Croce, Proc. Natl. Acad. Sci., 1987, 84, 4969–4973 CrossRef CAS PubMed.
- L. Wang, L. Wang, H. Zhang, J. Lu, Z. Zhang, H. Wu and Z. Liang, Oncol. Rep., 2020, 43 Search PubMed.
- Y. Sun, G. Wu, S. Cheng, A. Chen, K. Neoh, S. Chen, Z. Tang, P. F. Lee, M. Dai and R. Han, EBioMedicine, 2019, 46 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cb00007b |
| This journal is © The Royal Society of Chemistry 2024 |