
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInsights into docking in megasynthases from the investigation of the toblerol trans-AT polyketide synthase: many α-helical means to an end†
Serge
Scat
 ,
Kira J.
Weissman
* and
Benjamin
Chagot
*
,
Kira J.
Weissman
* and
Benjamin
Chagot
*
Université de Lorraine, CNRS, IMoPA, F-54000 Nancy, France. E-mail: kira.weissman@univ-lorraine.fr; benjamin.chagot@univ-lorraine.fr
First published on 16th May 2024
Abstract
The fidelity of biosynthesis by modular polyketide synthases (PKSs) depends on specific moderate affinity interactions between successive polypeptide subunits mediated by docking domains (DDs). These sequence elements are notably portable, allowing their transplantation into alternative biosynthetic and metabolic contexts. Herein, we use integrative structural biology to characterize a pair of DDs from the toblerol trans-AT PKS. Both are intrinsically disordered regions (IDRs) that fold into a 3 α-helix docking complex of unprecedented topology. The C-terminal docking domain (CDD) resembles the 4 α-helix type (4HB) CDDs, which shows that the same type of DD can be redeployed to form complexes of distinct geometry. By carefully re-examining known DD structures, we further extend this observation to type 2 docking domains, establishing previously unsuspected structural relations between DD types. Taken together, these data illustrate the plasticity of α-helical DDs, which allow the formation of a diverse topological spectrum of docked complexes. The newly identified DDs should also find utility in modular PKS genetic engineering.
Introduction
The megaenzyme polyketide synthases (PKSs) of bacteria are biosynthetic assembly lines whose products are heavily exploited in the clinical application, notably as anti-cancer and anti-microbial agents.1,2 These extraordinarily complex types of machinery incorporate repeated sets of functional domains called modules, which act consecutively to elongate and chemically modify acyl-CoA-derived building blocks.3,4 Chain extension requires a minimum of three domains (acyl transferase (AT), ketosynthase (KS) and acyl carrier protein (ACP)) while those involved in tailoring (e.g. ketoreductase (KR), dehydratase (DH) and enoylreductase (ER)) vary between modules (Fig. 1). Two distinct classes of modular PKSs have been identified, which appear to have evolved both independently and convergently.5–8 Among the multiple differences between the two systems, the cis-AT PKS multienzymes possess integrated acyl transferase domains responsible for monomer selection, while the trans-AT PKSs interact in trans with discrete ATs.9,10 The modules of trans-AT PKSs also boast much greater functional diversity and organizational heterogeneity (Fig. 1), including duplicated domains and non-canonical domain ordering.6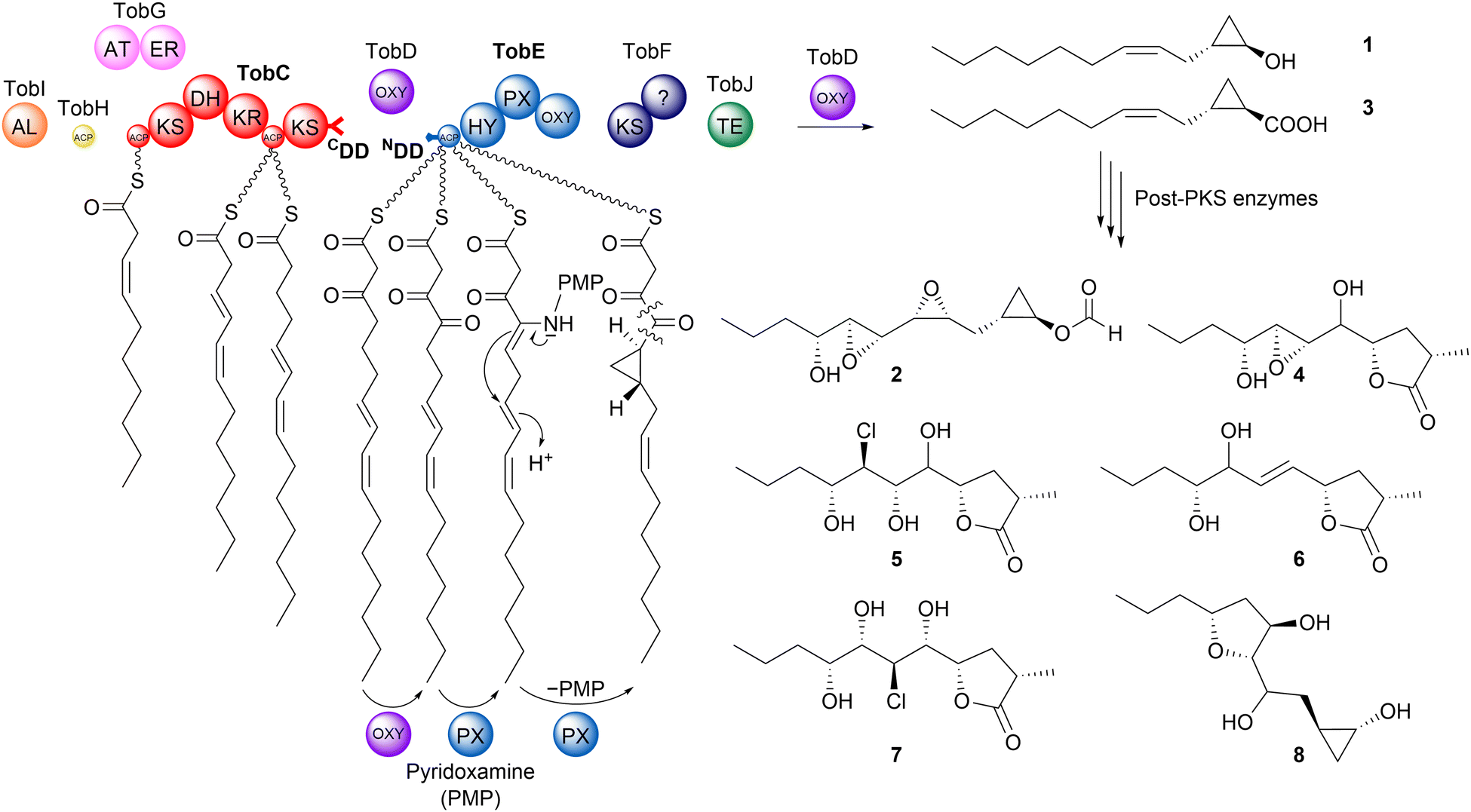 | ||
| Fig. 1 Hypothetical organization of the toblerol trans-AT PKS.11 The names of the proteins are indicated, and each domain is symbolized as a sphere. TobC CDD and TobE NDD investigated in this study/work are highlighted in bold. Key: AL, acyl-ligase; OXY, FAD-dependent oxygenase; KS, ketosynthase; AT, acyl (malonyl) transferase; ER, enoylreductase; KR, ketoreductase; ACP, acyl carrier protein; C, condensation; PX, pyridoxal phosphate-dependent enzyme; HY, hydrolase; TE, thioesterase. X indicates a domain of unknown function. Compounds 1–8 represent the characterized toblerols A–H. | ||
A feature common to both cis-AT and trans-AT PKSs is the distribution of modules among multiple polypeptide subunits (Fig. 1). Although this arrangement likely mitigates problems arising from the misfolding, proteolysis or mutational inactivation of even more massive proteins,4 it necessitates that recognition elements be present allowing for the faithful transfer of growing acyl chains across the inter-subunit interfaces. Work over many years in our laboratory12–15 and others16–22 has demonstrated that such non-covalent interactions are mediated by sequences present at the extreme C- and N-termini of the polypeptides called ‘docking domains (DDs)’ (CDD indicates a C-terminal docking domain, and NDD, an N-terminal docking domain). Notably, these portable protein–protein interaction elements have demonstrated utility for splitting PKS multienzymes into smaller units,23 creating novel interprotein interfaces,24–26 and facilitating interactions between non-PKS proteins.27,28 However, the effective use of DDs requires that two criteria be satisfied: (i) knowledge of the boundaries of the functional domains to allow for their transplantation; and (ii) information as to DD compatibility, so that biosynthetic fidelity can be maintained within a specific PKS/metabolic pathway context. The latter depends on determining the structural (sub)type to which a DD complex belongs, as these various categories are intrinsically orthogonal.
Multiple DD types have been identified from cis-AT PKSs and their hybrids with nonribosomal peptide synthetases (NRPSs). These include 1a (originally derived from the erythromycin (DEBS) PKS12), 1b (pikromycin17), 2 (curacin18), and 3 (or SLiM-βhD, derived from both tubulysin13 and enacyloxin29 PKS-NRPS systems and the rhabdopeptide NRPS19) (Fig. 2). More recently, we structurally characterized ‘type 1-related’ and ‘type 2-related’ DDs14 (Fig. 2) from the hybrid cis-AT/trans-AT hybrid system responsible for enacyloxin biosynthesis. These designations reflect the fact that although the individual DDs globally resemble classical type 1 and type 2 elements, the detailed overall topologies of the complexes differ. In terms of the molecular basis for docking, complex formation by type 1a, 1b, 1-related, and 2(-related) domains involves interactions between α-helices, while for type 3, the CDD includes a short linear motif (SLiM) that is inserted as a β-strand into a β-hairpin formed by the NDD,19,29 and in certain cases, an additional N-terminal α-helix19 (Fig. 2).
 | ||
| Fig. 2 Structures of docking domain complexes (DDs) from PKS and NRPS systems (the corresponding PDB codes are indicated in parentheses). The C-terminal DDs (CDDs) are colored in red, and the NDDs in blue, with α-helices represented as cylinders and β-sheets as arrows. NC indicates the N-terminus of a CDD, and CC, the C-terminus of a CDD, while NN indicates the N-terminus of an NDD, and CN, the C-terminus. (a) Type 1a DDs from the cis-AT DEBS PKS and type 1b from the cis-AT pikromycin PKS. (b) Type 2 DDs (left) represented by CurG/CurH (top) and CurK/CurL (bottom) from the cis-AT curacin PKS, and type 2-related (right) Bam_5925/Bam_5924 from the hybrid cis-AT/trans-AT PKS-NRPS enacyloxin pathway. (c) Type 3, as found in the rhabdopeptide NRPS. (d) 4 α-helix bundle-type DDs, as described from the VirA/VirFG interface in the virginiamycin trans-AT hybrid PKS-NRPS. (e) Three α-helix bundle represented by PaxB/PaxC (now designated 3HBa), and (f) PCP/4 α-helix bundle-type complex represented by PaxA/PaxB, from the PAX NRPS (the first three α-helices of the PaxA peptidyl carrier protein (PCP) are colored light red and are labeled). | ||
Trans-AT PKSs incorporate additional types of DDs, including the heterodimeric 4 α-helix bundle (4HB) class characterized from the virginiamycin15 and macrolactin16 systems. This type operates at both classical and so-called ‘split module’ junctions, in which the domains comprising a chain extension module are located on separate subunits. Notably, the NDDs of this class show convincing characteristics of intrinsically disordered regions (IDRs), demonstrating that such motifs operate in bacterial specialized metabolism.15 An extended IDR region called a dehydratase docking domain (DHD) was also found to mediate communication between KS and DH domains constituting a split module interface.22 Finally, two distinct types of α-helix-mediated docking have also been reported for the purely NRPS peptide-antimicrobial-Xenorhabdus (PAX) system, involving three (PaxB CDD/PaxC NDD, referred to as type 3HBa (vide infra)) or four α-helices (PaxA T1-CDD/PaxB NDD), respectively20,21 (Fig. 2). Despite the heterogeneity of characterized docking complexes, measured affinities all lie within the low–mid μM range, indicating that this is a sweet spot for such interactions.14,15,18,20
While putative DD-containing regions at the extreme C- and N-terminal ends of PKS subunits can readily be discerned, it remains challenging to predict the precise length and architecture of many DDs from the primary sequence alone.14 This situation reflects characteristics of the DDs themselves, including strong residue heterogeneity even within the same nominal structural types,14 as well as a high proportion of IDRs,14,15,22 but also the inability of AlphaFold230 to make confident predictions for short (<100 residues) sequences.31 AlphaFold2 readily predicts α-helices even in so-called ‘spurious proteins’ (i.e. sequences predicted from non-protein coding regions).31 This situation is particularly problematic for analyzing PKS multienzymes, as the majority of DDs are α-helical. In this context, the best way forward is to directly characterize additional DD pairs to improve our understanding of their structure–function relationships, and the attributes that define the various (sub-)types.
In this work, we aimed to extend our relatively limited understanding of docking in trans-AT PKSs by studying intersubunit interactions in the toblerol PKS (Fig. 1) of the model methylotroph Methylorubrum extorquens AM1.11 The toblerol system incorporates 8 polypeptides, and thus a relatively high number of potential docking interfaces compared to other trans-AT PKSs.32 This fragmented, largely non-modular architecture stymied the automated analysis of the pathway.11 As a first step, we focused on the central junction between TobC and TobE since direct evidence for its functionality would lend weight to the literature biosynthetic hypothesis.11 Herein, we describe a matched pair of DDs operating at this interface, report the NMR solution structure of the non-covalent docked complex, and identify key residues contributing to the interaction. Although docking is again mediated by α-helices, the overall topology of the DD complex is unprecedented. This result, which we contextualize via comparison to other solved DD structures, further highlights the versatility and plasticity of the α-helix as an intersubunit recognition motif in assembly line systems.
Results and discussion
In silico analysis of docking in the toblerol trans-AT PKS
The starting point for this work was to determine suitable boundaries for expressing candidate DDs from the TobC/TobE interface as recombinant proteins in E. coli. For this, we first identified the regions corresponding to the adjacent functional domains (TobC (WP_003598535), KS; TobE (WP_003598535), ACP (Fig. 1)) by comparison to solved structures of homologous domains in the PDB.15,33–35 Next, we analyzed the putative DD-containing sequences at the C-terminus of TobC (L2225–R2278) and the N-terminus of TobE (M1–D44), using several secondary structure and disorder propensity prediction programs.36,37 These analyses predicted a 10 residue α-helix at the end of the TobC C-terminal region (Q2267–A2276), and a 10 residue α-helix at the beginning of the TobE N-terminal region (ESI,† Fig. S1). Without further guidance (it must be noted that this work was initiated before the availability of AlphaFold230), we opted to clone and express the entire regions down- and upstream of the functional domains.Expression and purification of DD constructs
The TobE NDD and TobC CDD constructs were amplified from a Methylorubrum extorquens AM1 cosmid (gift from J. Piel, ETH Zurich, CH) and cloned into vector pBG102 (Center for Structural Biology, Vanderbilt University) for expression as His6-SUMO-tagged proteins in E. coli Rosetta 2 (DE3) (ESI,† Tables S1 and S2). In the absence of aromatic residues in the native DD sequences, a tyrosine residue was added to the N-terminus of the constructs (Table S1, ESI†) to facilitate protein detection and concentration measurement by UV-vis. As it has previously been reported that certain docking interactions include the adjacent carrier protein,21 we additionally cloned a version of TobE NDD incorporating the downstream ACP. Following Ni-NTA chromatography, the His6-SUMO tag was cleaved off using human rhinovirus 3C protease, resulting in a non-native GPGSY N-terminal sequence in the case of TobE NDD, GPGSPNSY for TobC CDD, and GPGS for the di-domain TobE NDD-ACP construct. In previous work, such additional short sequences did not negatively impact the native docking interactions.14,15Characterization of the interaction between TobC CDD and TobE NDD using size-exclusion chromatography
To demonstrate that the docking domains interact, we carried out analytical gel filtration in the presence of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of TobC CDD and TobE NDD (both at 400 μM) and compared the results to the profiles of the individual DDs. TobC CDD and TobE NDD eluted at volumes of 12.9 and 13.7 mL, respectively, whereas the mixture gave a single peak with a lower elution volume of 12.3 mL, providing initial evidence for complex formation (ESI,† Fig. S2).
:1 mixture of TobC CDD and TobE NDD (both at 400 μM) and compared the results to the profiles of the individual DDs. TobC CDD and TobE NDD eluted at volumes of 12.9 and 13.7 mL, respectively, whereas the mixture gave a single peak with a lower elution volume of 12.3 mL, providing initial evidence for complex formation (ESI,† Fig. S2).
Investigation of docking between TobC CDD and TobE NDD via isothermal titration calorimetry
To confirm DD complex formation, we characterized their interactions by ITC (Fig. 3). These experiments were carried out in both possible configurations by titrating TobE NDD in the cell (60 μM) with TobC CDD (600 μM), and TobC CDD in the cell (60 μM) with TobE NDD (600 μM). The determined KDs were in strong agreement: 4 ± 1 μM (n = 3) when TobE NDD was titrated with TobC CDD, and 3.2 ± 0.3 μM (n = 2) for the reverse situation. The values are also fully consistent with previous measurements of DD affinities (2–90 μM14,15,17,18). In terms of stoichiometry, we obtained values of N = 1.0 ± 0.2 and 0.8 ± 0.1 using TobE NDD and TobC CDD, respectively, as the titrant, indicative of 1:1 complex formation. The slight discrepancy from 1 in the latter case can be explained either by small errors in the concentration measurements due to the low molar extinction coefficients of the constructs (1490 M−1 cm−1), or the intrinsic difficulty of measuring interactions involving IDRs (vide infra).38 In any case, the decomposition of the ITC thermodynamic profile revealed that the docking interaction is mainly enthalpic, with an unfavorable (negative) entropic contribution. These latter data suggested that complex formation is accompanied by a loss of conformational freedom, which would be consistent with the induced folding of an IDR.
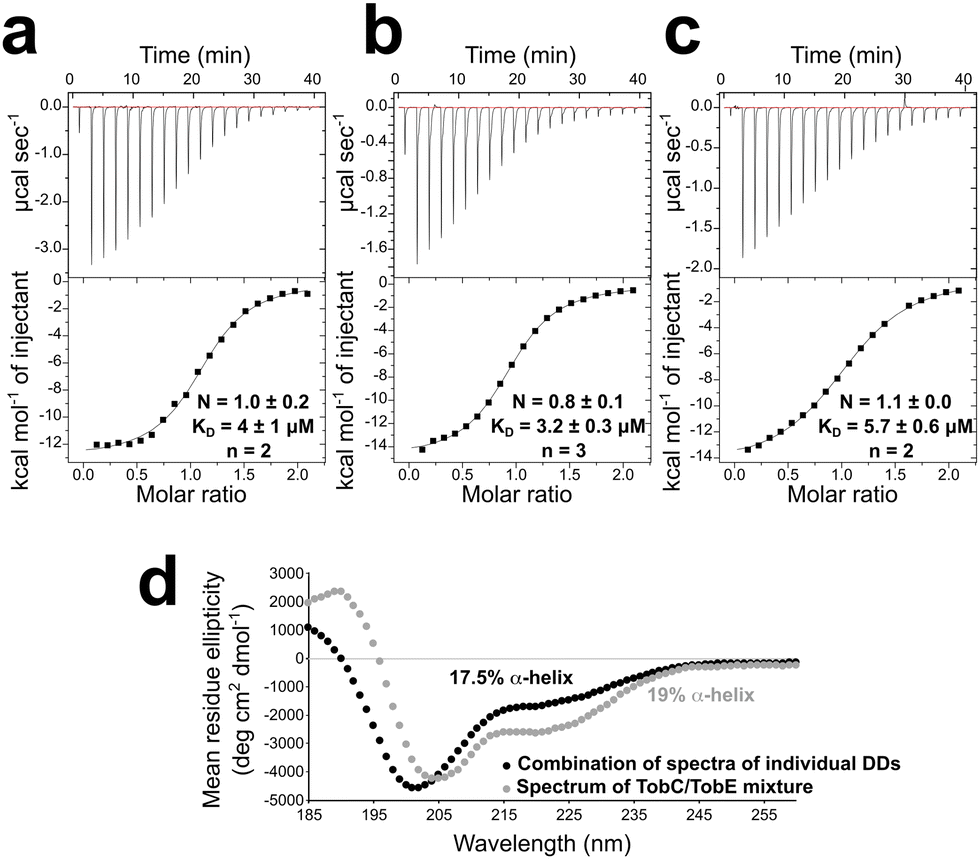 | ||
| Fig. 3 The characterization of TobC CDD and TobE NDD binding by isothermal titration calorimetry (ITC) and circular dichroism. (a) Titration of TobE NDD (in the cell) with TobC CDD (in the syringe). (b) Titration of TobC CDD (in the cell) with TobE NDD (in the syringe). (c) Titration of TobE NDD-ACP (in the cell) with TobC CDD (in the syringe). The stoichiometry (N), KD and error values represent the average of two or three measurements (the number of experiments (n) is indicated). (d) Differential CD analysis of the TobC CDD and TobE NDD pair. The combined spectrum (black) was obtained by simultaneously analyzing the two isolated DDs (both at 100 μM). Comparison with the spectrum of a mixture of the two docking domains (gray), again at an overall concentration of 100 μM, was used to measure increased structuration. | ||
To determine if residues of the flanking ACP contribute to the docking interface,21 we titrated Tob CDD with the TobE NDD-ACP didomain. This experiment yielded essentially the same interaction parameters (KD = 5.7 ± 0.6 μM (n = 2); N = 1.1 ± 0.0) (Fig. 3), demonstrating that the ACP is not involved in the interaction.
Analysis of docking between TobC CDD and TobE NDD using circular dichroism
Since the ITC experiments provided evidence for the IDR character of the DDs, we next aimed to investigate their secondary structure by CD, as previously described.14,15 Consistent with the in silico prediction, the CD spectrum of TobC CDD revealed minima around 205 and 220 nm characteristic of the presence of the α-helix (ESI,† Fig. S3). Deconvolution of the spectrum (180–260 nm) with Pro-Data Viewer software (Applied Photophysics) indicated an α-helical content of 25% (vs. 16% α-helix predicted using PSIPRED39). In contrast to the prediction, the CD spectrum of TobE NDD showed that it was substantially less structured in solution (ESI,† Fig. S3), with only 17% α-helical content (vs. 20% predicted by PSIPRED39).Next, we used CD to obtain more direct evidence for the induced folding of the DDs upon complex formation. For this, we acquired a combined spectrum of the two isolated DDs and compared it to the spectrum of a 1:1 TobC CDD/TobE NDD mixture (Fig. 3). The two spectra do not superimpose, consistent with the induced folding of one or both of the DDs in the presence of a partner. The overall secondary structure increased from 17.5% to 19% α-helix content when the DDs were combined. This increase is less substantial than that seen with previous IDR-type DDs,14,15 but the folding is likely masked by the extensive, fully-disordered regions present in the constructs. We interpreted this as evidence of the induced folding of one or both of the domains.
Characterization of docking between TobE NDD and TobC CDD by NMR
To investigate the docking interaction in detail, we analyzed complex formation by NMR. The 1H–15N HSQC spectra of the isolated DDs revealed narrow proton chemical shift dispersion, with values between 7.6 and 8.8 ppm, showing that the isolated DDs are indeed highly disordered (Fig. 4). Chemical shift assignment was performed using a standard series of double- and triple-resonance experiments (reviewed in ref. 40). Overall, the majority of HN, NH, Cα, Cβ, and CO chemical shifts were assigned for residues M1–D44 of TobE NDD and L2225–R2278 of TobC CDD, allowing for chemical shift-index-derived secondary structure identification using the TALOS-N software.41 The exceptions in TobC CDD included the prolines (P2226, P2232, P2244, P2249, and P2253) for which only Cα, Cβ, and CO were available (the Cβ chemical shifts are consistent with trans configuration), and P2243, K2261, A2262, and M2265 for which no assignments could be obtained. In accordance with the CD results, isolated TobC CDD and TobE NDD both contain α-helical regions (Fig. 4). In TobC CDD, these correspond to residues I2257–V2260 and Q2267–A2276 (α-helix propensity > 0.6, with the second α-helix having higher formation probability41), for an overall α-helix content of 24%. We were not able to assign chemical shifts to multiple residues within the intervening loop, indicative of its dynamic character. Only residues E24–S27 of TobE NDD exhibited chemical shifts consistent with the α-helix (α-helix propensity > 0.6), and an overall α-helix content (8%) that was lower than estimated by both CD and PSIPRED. Overall, these results are consistent with the idea that both DDs, while mostly disordered, contained pre-structured motifs (PreSMos) which reduce the entropic penalty of partner binding.42 | ||
| Fig. 4 NMR studies of isolated TobC CDD and TobE NDD. (a) Assigned 1H, 15N HSQC spectrum of TobC CDD alone, with the residues indicated. (b) Assigned 1H, 15N HSQC spectrum of TobE NDD alone, with the residues indicated. (c) and (d) TALOS-N chemical shift based α-helix propensity of the DDs. The identified secondary structures (defined by α-helix propensity ≥ 0.6) are shown above the data. (e) and (f) {1H}–15N heteronuclear NOEs measured at 14.1 T on TobC CDD and TobE NDD, respectively. All of the residues for which the heteronuclear NOEs fall in the grey zone are identified as dynamic. Residues for which NOE data could not be calculated are indicated by a black dot. Unumbered residues were introduced during cloning. A description of the value and error calculations is given in the Materials and methods section. | ||
We next investigated the dynamical properties of the two DDs in the absence of their partners (Fig. 4). The obtained data show that both DDs excised from their subunit context exhibit flexibility on a ps–ns timescale, with heteronuclear NOE values below 0.5 for all residues. Within TobC CDD, the dynamics are not limited to the residues in the loop region (K2261–M2265) as all residues exhibit low heteronuclear NOE values (<0.2), except for those showing higher α-helical propensity (i.e. I2257–A2276) with values between 0.2 and 0.5. The heteronuclear NOE values of all amino acids within TobE NDD are <0.2, including those within the predicted α-helical region. Overall, this analysis revealed both excised and unbound DDs to be highly dynamic in solution, consistent with the low content of secondary structure.
We next analyzed the interaction between TobC CDD and TobE NDD by titrating both DDs in 13C,15N-labeled form with different ratios of the unlabeled partner (Fig. 5). The heteronuclear NOE spectra of the complexed DDs were also recorded at the endpoint of each titration (Fig. 5). The first major observation was that upon titration, many of the amide signals of both DDs underwent large chemical shift changes. This suggests that complex formation induces substantial structural modification of both proteins (Fig. 5).
 | ||
| Fig. 5 NMR studies of bound TobC CDD and TobE NDD. (a) Assigned 1H, 15N HSQC spectrum of 1H, 15N-TobC CDD in complex with unlabeled TobE NDD. (b) Assigned 1H, 15N HSQC spectrum of 1H, 15N-TobE NDD in complex with unlabeled TobC CDD. Folded peaks are marked with an asterisk. (c) Residue chemical shift perturbation Z-scores calculated for TobC CDD at the endpoint of the titration with TobE NDD. The gray zone represents Z-scores below 1. (d) Residue chemical shift perturbation Z-scores calculated for TobE NDD at the endpoint of the titration with TobC CDD. (e) TALOS-N chemical shift-based α-helix propensity of TobC CDD. Identified secondary structures (defined by α-helix propensity ≥ 0.6) are shown above the data. (f) TALOS-N chemical shift-based α-helix propensity of TobE NDD. (g) {1H}–15N heteronuclear NOEs measured at 14.1 T on TobC CDD. The gray zone corresponds to values below 0.5. (h) {1H}–15N heteronuclear NOEs measured at 14.1 T on TobE NDD. In (c), (d), (g), and (h), residues for which data could not be calculated are indicated by a black dot. A description of the error calculation is given in the Materials and methods. | ||
To interpret these results, chemical shift assignments of the peptide backbone atoms were completed for residues L2225–R2278 of TobC CDD and M1–D44 of TobE NDD in their complexed forms (Fig. 5). This analysis revealed that residues T2252–R2278 of TobC CDD and I20–G33 of TobE NDD underwent the largest chemical shift perturbations (CSP Z-score > 1) (Fig. 5), and must therefore lie at the interface of the complex and/or be situated in regions experiencing induced structuration. Heteronuclear NOE coupled with NMR-based secondary structure analysis (Fig. 5) showed that the first α-helix in TobC CDD in complex with TobE NDD was extended relative to the isolated form (helix α1: P2253–A2262 (10 residues vs. 4 in isolated TobC CDD)), whereas the extent of helix α2 (Q2267–M2277) corresponds to the prediction. The dynamic behavior of the TobC CDD α-helices was also reduced, as the T2252–R2278 region exhibited heteronuclear NOE values above 0.5. In bound form, TobE NDD similarly contains an extended α-helix comprising residues A22–I32, with the overall region showing heteronuclear NOE values above 0.4 and residues S21–D30 exhibiting values above 0.5. Consistent with this increase in structuration, the % α-helix for TobC CDD and TobE NDD was calculated to be 39% and 23%, respectively. Thus, binding with a partner induces the transient PreSMos identified in the isolated DDs to fold into stable α-helical elements (so-called molecular recognition features (MoRFs)43). This result confirms that TobC CDD and TobE NDD are IDRs as observed previously,14,15,22 albeit never for both members of a matched DD pair.
The remaining amino acids within the DDs remained highly dynamic as evidenced by the low heteronuclear NOE values, and thus do not contribute directly to the docking interface, consistent with the chemical shift perturbation data (Fig. 5). Nonetheless, we cannot exclude that the interaction incorporates ‘fuzzy’ elements, i.e., the flanking disordered regions also kinetically promote docking.44 Approximately one-third of the residues within the unstructured portions are charged (ESI,† Fig. S1), and thus may restrict the conformational freedom in the individual IDRs via long-range intramolecular contacts and/or boost association rates via transient, non-specific electrostatic interactions.44
AlphaFold2-based modelling of the TobE NDD/TobC CDD complex
AlphaFold230 is highly accurate for predicting the 3D structures of isolated proteins. However, its reliability for shorter sequences (<100 residues), which include DDs, is lower. Indeed, it can predict α-helical structure for hypothetical proteins from non-coding genomic regions,31 and it has shown limited success to date at modeling the structures of protein complexes.45,46 Thus, it was of interest to evaluate its capacity to predict the structures of the isolated DDs, as well as that of the relatively simple complex between them. As anticipated, the program converged to a near-native model of the folded region within TobC CDD, incorporating two antiparallel α-helices P2253–A2262 and Q2267–A2276 (pLDDT > 80) (ESI,† Fig. S4). However, it was less accurate concerning the intrinsic flexibility of the isolated CDD, as the “predicted aligned error” (PAE) output suggested a rigid architecture in which the two α-helices are situated close together (ESI,† Fig. S4). In the case of the isolated TobE NDD, structure prediction resulted in three distinct sets of models, one composed of two non-interacting α-helices H3–D17 and A22–G35 (pLDDT >60), one comprising two α-helices S11–A16 and A22–G33 (pLDDT >60, except for S11, D12, and R13, for which pLDDT >57), and one including a single α-helix A22–G33 (pLDDT >60) (ESI,† Fig. S4). Overall, the third model is most consistent with the TobE NDD structure observed by NMR, even though the predicted α-helix is longer than that identified by NMR based on the NMR chemical shift data.The prediction of the TobC CDD/TobE NDD complex structure yielded a model in which the single α-helix of TobE NDD interacts with the two antiparallel α-helices of TobC CDD to form a three-α-helix bundle comprising P2253–A2262 and Q2267–A2276 of TobC CDD and A22-T34 of TobE NDD (pLDDT >60, except for T34 for which pLDDT >57) (ESI,† Fig. S4). However, as for the isolated DD, the single TobE NDD α-helix is longer in the model than that identified by NMR.
Elucidation of the NMR solution structure of the TobC CDD/TobE NDD complex
We next aimed to provide biophysical proof for the novel DD complex structure predicted by AlphaFold2. In contrast to our earlier studies,14,15 we were able to saturate the non-covalent complex of the domains and solve the resulting structure, thus obviating the need to covalently fuse them to stabilize their interaction, as previously described.12,14,15,17,18,20Structural elucidation of the TobC CDD/TobE NDD complex was carried out by multidimensional heteronuclear NMR spectroscopy using two samples in which either 15N–13C-labeled TobC CDD or TobE NDD was analyzed in the presence of a 2-fold excess of the unlabeled partner. Using standard double- and triple-resonance experiments, 1H, 13C, and 15N resonance assignments were completed to 98.7% for the backbone atoms, 98.2% for sidechains, and 100% for aromatic moieties (as calculated using the PSVS server47). Structure calculation was then performed using a two-stage procedure consisting of initial structure generation using CYANA48 followed by restrained molecular dynamics refinement within Amber,49 as reported previously.14,15,50 The 1349 nuclear Overhauser effect (NOE) distance restraints obtained from three-dimensional NOESY-HSQC experiments including 171 intermolecular NOEs, were distributed mainly between residues L2251–R2278 of Tob C CDD and I20–G33 of Tob E NDD, whose residues exhibited the highest heteronuclear NOE values. Here, 68 backbone angle restraints derived from the chemical shifts and stereospecific group assignments for 12 pairs of Leu and Val methyl groups determined from the 1H–13C constant-time HSQC (recorded on a 10% 13C-enriched sample),51 were also used to calculate the NMR solution structure of the DD complex.
The selected final ensemble of 20 conformers had no violation of NOE restraints greater than 0.2 Å and no angle violations greater than 5°. The large number of restraints (>12 per residue on average and >23 per residue when only considering residues with heteronuclear NOE value > 0.4 (i.e., L2251 and R2278 of Tob C CDD and I20–G33 of TobE NDD)) yielded a high-precision ensemble with root-mean-square deviation (RMSD) values of 0.6 ± 0.1 Å for the backbone and 1.1 ± 0.1 Å for all heavy atoms calculated on ordered residues (TobC CDD L2251–R2278 and TobE NDD E19–G33). The input and refinement statistics, as well as the structural statistics specified by the NMR-validation task force,52 are shown in ESI,† Table S3. The quality of the structure was assessed using the PROCHECK-NMR,53 PSVS,47 and MolProbity54 servers. The coordinates of the 20 structures defining the NMR structure ensemble and the full list of NMR restraints used for calculation and refinement were deposited in the Protein Data Bank under accession number 8RAJ.
Within the docking complex, TobC CDD comprises two consecutive short α-helices (P2253–K2261 and Q2267–M2277) oriented at an angle of 176 ± 4° with respect to each other, while the upstream residues L2225–D2250 are unstructured (Fig. 6). TobC CDD contains 6 prolines in the trans configuration,55 and thus there is likely no change in geometry relative to the unbound form. TobE NDD is composed of one unique α-helix (A22–A31), while the flanking residues M1–E19 and T34–D44 are disordered (Fig. 6). Notably, TobE NDD represents the first case, to our knowledge, in which a docking domain follows a relatively extended unstructured region at the subunit N-terminus (the extreme N-terminal sequence is encoded by GC-rich codons, which argues that the start site has been correctly assigned11). Docking of the TobE NDD α-helix on top of the two TobC CDD α-helices results in an overall parallel three-α-helix bundle. We thus propose to name this type of complex 3HBb, relative to that reported for the PAX NRPS (now designated 3HBa). Globally, the experimental structure of the DD complex is close to the model predicted by AlphaFold2, with an RMSD of 0.51 ± 0.05 Å calculated for the backbone of residues P2253–M2277 and A22–D30.
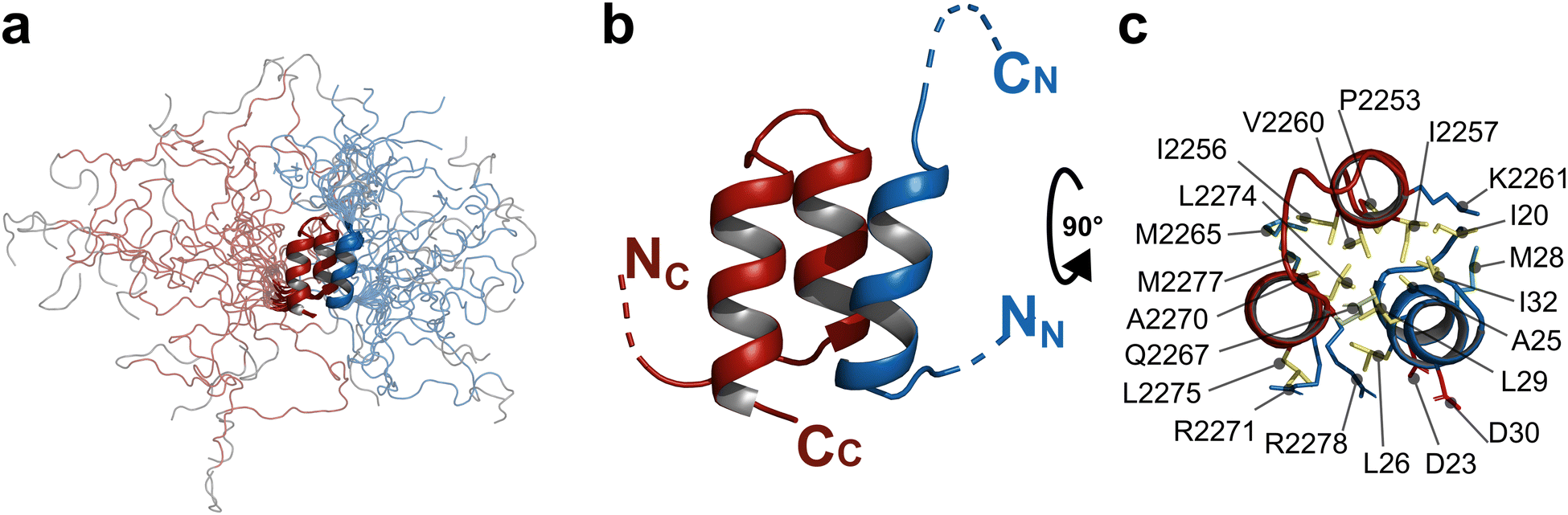 | ||
| Fig. 6 NMR structure of the TobC CDD/TobE NDD complex. TobC CDD and TobE NDD are colored red and blue, respectively. (a) NMR ensemble of the 20 best conformers that represent the solution structure of the TobC CDD/TobE NDD complex. Except for the regions comprising the α-helices, the residues are highly dynamic. TobE NDD is colored in blue, TobC CDD in red and the non-native sequence (tag) is shown in grey. (b) The representative conformer of the complex in the NMR ensemble. The unstructured regions have been removed and symbolized as dashed lines. (c) The interface between the two docking domains. Residues important for the interaction are colored according to their nature: hydrophobic in yellow, basic in blue, acidic in red, non-polar in light green, and methionine in olive green. | ||
The total buried surface area at the docking interface measures approximately 1140 Å2, with more than 75% composed of hydrophobic residues. These include P2253, I2256, I2257, V2260, M2265, A2270, L2274, L2275 and M2277 of TobC CDD and I20, A22, A25, L26, M28, L29 and I32 of TobE NDD (Fig. 6). The structure also identifies two pairs of charged residues at the interface, which may contribute to interaction specificity:12,14,15,17 TobC CDD R2271/TobE NDD D30, and TobC CDD R2278/TobE NDD D23.
Evaluation of charged residues potentially involved in TobC CDD/TobE NDD complex formation
We aimed to directly investigate the roles of the identified charged residues in mediating the docking interaction. However, this experiment was complicated by the IDR character of both DD partners as the introduced mutations could impact the ability of the DD domains to interact and to undergo induced folding. We therefore focused our attention on the more substantially structured TobC CDD, which we judged would be more resistant to mutation-induced perturbation. Nonetheless, mutation of R2278 to alanine (ESI,† Table S1) resulted in a detectable alteration of the TobC CDD secondary structure even in the absence of complex formation, with the structural perturbation even more pronounced in an R2271A/R2278A double mutant (ESI,† Fig. S5). However, as the R2271A single mutant exhibited no substantial structural difference when compared to the wild type (ESI,† Fig. S5), we investigated its binding to TobE NDD.Analysis of the interaction by ITC yielded variable results, with either no binding detected or very weak affinity (the KD was estimated at 769 μM due to the absence of saturation of the complex, and with forcing the stoichiometry to 1). While we cannot exclude an effect of the mutation on TobC CDD-induced structuration, the substantial decrease in affinity supports a role for this residue in driving complex formation with TobE NDD (kon). Consistent with this proposal, no evident increase in secondary structure was observed by CD when TobC CDD R2271A and wild-type TobE NDD were combined (ESI,† Fig. S5).
TobC CDD resembles 4HB-type CDDs
To our knowledge, no 3 α-helix bundle docking interaction analogous to the PAX 3HBa type20 has been described from a PKS system. However, the detailed topology differs between the Tob and PAX structures, as in the TobC CDD/TobE NDD complex, the three α-helices are parallel, while in the PaxB CDD/PaxC NDD case, the α-helical NDD binds to the center of the V-shaped α-helices of the CDD (Fig. 2). The TobC CDD fold more closely resembles the individual docking domains giving rise to the 4 α-helix bundle (4HB)-type DDs described from other trans-AT PKSs VirA CDD (PDB ID: 2N5D15), VirFG NDD (PDB ID: 2N5D15) and MlnE NDD (PDB ID: 5D2E16) (ESI,† Fig. S6). In the 4HB family, the two parallel α-helices contributed by both the CDD and NDD interact perpendicularly to form the 4 α-helix bundle (Fig. 2). The angles between the α-helices (163 ± 4°, 172° ± 3° and 166° respectively, for VirA CDD, VirFG NDD, and MlnE NDD) are close to that within folded TobC CDD (i.e., 176 ± 2°) (ESI,† Table S4). A comparison of the DD backbones (TobC CDD (G2255–L2274) vs. VirA CDD (A6941–L6960), VirFG NDD (E5–L24) and MlnE NDD (Q10–I29)) yielded RMSD values of 0.56 ± 0.05 Å, 0.74 ± 0.06 Å and 0.56 ± 0.07 Å, respectively. Furthermore, the structural and sequence alignment of VirA CDD with TobC CDD show that hydrophobic residues are well conserved at seven positions, and thus are likely to play common roles in both CDD folding and interaction with the NDD partners. Three of the hydrophobic residues are located on helix α1, one is located in the loop between helices α1 and α2, and the last three are located on helix α2 (ESI,† Fig. S6).The strong similarity of the structures is reinforced by the calculation of the TM-scores56,57 between TobC CDD (T2252–L2275) and VirFG NDD (D2–A25), VirA CDD (D6938–T6961) and MlnE NDD (Q7–K30), with values of 0.66 ± 0.04 (min = 0.55, max = 0.77), 0.62 ± 0.04 (min = 0.49, max = 0.71) and 0.57 ± 0.03 (min = 0.53, max = 0.63), respectively. (Note: TM scores vary between 0 and 1, with scores <0.17 corresponding to random, unrelated proteins, and scores >0.5 indicating the same fold56). By comparison, within the type 4HB DD family, the structural similarity is lower (RMSD for MlnE NDD vs. VirA CDD and VirFG NDD = 0.88 ± 0.06 Å and 0.90 ± 0.06 Å, respectively, with TM-scores of 0.49 ± 0.01 (min = 0.47, max = 0.52) and 0.51 ± 0.02 (min = 0.47, max = 0.54)). Globally, this analysis argues that TobC CDD adopts a type 4HB CDD fold, despite interacting with only a single α-helix from TobE NDD to form an overall 3HBb docking complex.
Evaluation of previously classified CDDs reveals unrecognized similarities
The behavior of TobC CDD prompted us to try to identify analogous situations in which individual DDs of shared structure are deployed to form docking complexes of divergent architecture, and found this to also occur with type 2 CDDs. In the prototypical type 2 docking complex, both the C- and N-terminal DDs comprise 2 α-helices.18 The second α-helix of the NDDs forms a coiled-coiled motif, and both it and the first α-helix associate with the two α-helices of the CDD to form an overall 8 α-helical bundle (Fig. 2).18 In contrast, while the two α-helix PaxB CDD (ESI,† Fig. S7) adopts a type 2 CDD fold (RMSD for PaxB CDD vs. CurG CDD = 0.70 ± 0.10 Å (ESI,† Table S5 for TM scores)), it forms a type 3HBa complex with its partner (PDB ID: 6TRP21). The type 2 character of PaxB CDD is reinforced by sequence comparison, which shows that L3304, L3307, L3312 and L3316 align with hydrophobic residues present in other type 2(-related) CDDs, including I1562, S1565, L1570 and I1574 of CurG CDD, I2625, L2628, L2633 and L2637 of Bam_5925 CDD, and I2209, L2212, L2217 and V2221 of CurK CDD. These key amino acids are distributed similarly to those in 4HB-type CDDs, with the first located on helix α1, the second in the loop between helices α1 and α2, and the last two on helix α2 (ESI,† Fig. S7).As for PaxB CDD, the type 2 CurK CDD interacts with only one α-helix furnished by the partner CurL NDD (PDB ID: 4MYZ18) and its structure resembles the PikAIII CDD (PDB ID: 3F5H17) from the pikromycin PKS, although the latter was classified as a type 1b CDD (PDB ID: 4MYZ18), as they are both composed of a one-turn α-helix associated with a longer α-helix. The measured angles between the α-helices are 125 ± 1° and 128 ± 5° for CurK CDD and PikAIII CDD, respectively, and structure superposition yielded a RMSD value of 0.53 ± 0.14 Å and a TM-score of 0.58 ± 0.03 (ESI,† Table S5 for TM scores). Furthermore, the structure and sequence comparisons show that I2209, L2212, L2217, L2221 and L2222 of CurK CDD are located at equivalent positions to those of I1544, L1547, L1552, A1556 and L1557 of PikAIII CDD (ESI,† Fig. S7), a placement they share with aliphatic residues in PaxB and CurG CDDs.
In another variation on a theme, paired DDs from the enacyloxin hybrid cis-AT/trans-AT PKS (Bamb_5925 CDD/Bamb_5924 NDD) adopt a type 2 fold but the structure of the resulting docked complex differs from canonical type 2 architectures (and consequently was designated ‘type 2-related’).14 Globally, this analysis reveals that individual CDDs of equivalent topology can be combined to yield multiple complexes of distinctive structure, a phenomenon that must depend on their structures and the interaction surface presented to the partner NDDs.
Towards understanding the basis for alternative type 2 NDD folding
To extend this analysis to the NDDs, we first compared the sequences of NDDs that alternatively fold into one or two α-helices. Two α-helices CurH (PDB ID: 4MYY18), Bam_5924 (PDB ID: 6TDN14), and PaxB NDDs (PDB ID: 7B2B20) share similar structural features (ESI,† Fig. S7 and Table S6). The structure and sequence alignments of the domains identify three conserved aliphatic residues (I18, L21, and L29 for CurH NDD; L18, L21, and L29 for Bam_5924 NDD; and L6, L9 and L17 for PaxB NDD), the first of which is located on helix α1, the second on the turn in the interhelical region, and the third on helix α2 (ESI,† Fig. S7). These residues are clustered together in the structure of the NDDs, creating a hydrophobic core that likely contributes to the overall V-shaped structure of the DDs, as well as stabilizes the resulting DD complex (ESI,† Fig. S7).In contrast, the NDDs of CurL (PDB ID: 4MYZ18) and PaxC (PDB ID: 6TRP20) from the same two systems contain only one α-helix, although the PaxC NDD is long enough to accommodate an additional, upstream α-helix. It is also notable that in the absence of its partner, the CurL NDD can fold into a longer α-helix (PDB ID: 4MZ018) (ESI,† Fig. S7). The sequence alignment of one α-helix CurL and PaxC NDDs with the NDDs comprising two α-helices (CurH, Bam_5924 and PaxB NDDs), shows that the aliphatic residues L13 and I21 of CurL and L8 and L16 of PaxC, align with L9 and L17 of PaxB, L21 and L29 of Bam_5924, and L21 and L29 of CurH NDDs. Structural alignment confirms that L13 and I21 of CurL superimpose with L9 and L17 of PaxB, L21 and L29 of Bam_5924, and L21 and L29 of CurH NDDs. However, only L16 of PaxC superimposes with L17 of PaxB, L29 of Bam_5924, and L29 of CurH NDDs, because L8 of PaxC is located within the continuity of the same α-helix, and not on a loop as in the other complexes (ESI,† Fig. S7). Nonetheless, L8 and L16 align structurally with L13 and L21 of the alternative extended α-helical form adopted by isolated CurL NDD18 (ESI,† Fig. S7). A potential additional explanation for the absence of the first α-helix in CurL and PaxC NDDs is the presence of a negatively charged residue (E10 in CurL and E5 in PaxC (ESI,† Fig. S7)) in place of an aliphatic residue in the extreme N-terminal region which may disrupt its folding and/or destabilize it. Thus, although PaxC NDD contains a single α-helix, it closely resembles CurL NDD and the second α-helix of type 2 NDDs.
Key architectural and specificity features of type 2(-related) complexes
To further explore the similarity between the type 3HBa PaxB CDD/PaxC NDD20 pair and type 2 DDs, we calculated backbone RMSDs for the complex structure (PDB ID: 6TRP20) (V3300–L3316/D9–I20) vs. the complexes of type 2 CurG CDD/CurH NDD (PDB ID: 4MYY18) (L1558–I1574/S22–K33) and type 2-related Bam_5925 CDD/Bam_5924 NDD (PDB ID: 6TDN14) (E2621–L2637/S22–L33), yielding values of 1.13 ± 0.07 Å and 1.18 ± 0.07 Å, respectively (note, 1.13 represents an average for the two distinct reported conformations of CurG CDD/CurH NDD) and TM-score of 0.51 and 0.49 respectively. Therefore, despite PaxC NDD comprising a single α-helix, the docked PaxB CDD/PaxC NDD interaction is closely similar to these type 2(-related) complexes. Notably, both PKS and purely NRPS systems employ similar types of docking domains but the PaxB CDD/PaxC NDD complex is monomeric instead of dimeric, reflecting the likely monomeric state of the NRPS subunits58 from which it derives.Among the type 2 and type 2-related complexes, selectivity is proposed to be promoted by an electrostatic interaction involving an acidic residue present in the first α-helix of the CDD and one conserved basic residue in the second α-helix of the NDD (e.g., CurG CDD E1561/CurH NDD K32 and Bam_5925 CDD E2624/Bam_5924 NDD K32 (ESI,† Fig. S7)).14,18 In the case of CurK CDD, as the first α-helix of the CDD is shorter than in typical type 2 CDDs, the acidic residue involved (E2224) is located in the second helix of the CDD, and interacts with CurL NDD K24 and also R27.18 Similarly, within the PaxB CDD/PaxC NDD complex, K19, which is located at a similar position in the NDD, forms a salt bridge with E3303 situated in helix α1 of the CDD. This interaction in the PaxB CDD/PaxC NDD complex complements a salt bridge formed by the couple R23/D3296 (ESI,† Fig. S7). Thus, key charge:charge interactions are conserved among all of these DD pairs, consistent with their relatedness.
Concerning the alternative association modes of type 2 and type 2-related DDs, the main difference lies in the way the V-shaped NDD monomers homodimerize and so contact their CDD partners (Fig. 2). The presence of heptad repeats in classical type 2 complexes results in coiled-coil formation by the NDD, and each CDD contacts both NDDs18 (Fig. 2, ESI,† Fig. S7). In contrast, in the type 2-related complexes which lack heptad repeats, the NDDs interleave and each NDD contacts principally one CDD14 (Fig. 2, ESI,† Fig. S7).
Interestingly, the PaxA-T1 CDD/PaxB NDD interaction seems to be related to type 2(-related) complexes, even though the first α-helix of the CDD is contributed by the upstream PCP domain (T1). Moreover, PaxB NDD is monomeric, as the canonical heptad repeat needed for coil–coil formation is not present (ESI,† Fig. S7). Nonetheless, the lengths of the α-helices and the inter-α-helical angles are similar (RMSD between the backbones of PaxA CDD (I1064–Y1080) and Bam_5925 (E2621–L2637), CurG CDD (L1558–I1574) and PaxB CDD (V3300–L3316) = 1.41 ± 0.09 Å, 1.30 ± 0.10 Å and 1.40 ± 0.10 Å, respectively). As mentioned earlier, the type 2(-related) Bam_5925 and PaxB CDDs share four conserved hydrophobic residues that are implicated both in CDD assembly and the docking interaction. CurG only incorporates three of these residues (I1562, L1570, and I1574), as it lacks the hydrophobic residue within the loop region, and this amino acid is not well-conserved among type 2 DDs.18 Notably, the corresponding residues in PaxA T1-CDD are L1068, F1071, L1076, and Y1080. Although the presence of aromatic residues at these 4 positions is uncommon, such substitutions have already been observed in other PKS type 2 CDDs.18 Thus, the type 2(-related) DD folding and mode of association are tolerant to these sorts of variations. Further arguing for the type 2(-related) character of the PaxA-T1 CDD/PaxB NDD complex, it incorporates a polar interaction resembling the salt bridge implicated in docking specificity. This involves Q1067 in the first α-helix of the CDD and K22 in the second α-helix of the NDD, although other residue couples are present in the complex, which also contribute to complex formation21 (ESI,† Fig. S7). Nonetheless, as the complex is unique in incorporating an α-helix contributed by the PCP domain and having perpendicularly interlaced DDs, it would be prudent to hold off on its classification, pending the identification of additional, analogous interactions.
Having identified similarities between isolated type 2 CurK CDD and type 1b PikAIII CDD, we evaluated whether their respective complexes with their NDD partners were also related. As noted earlier, in the absence of CurK CDD, CurL NDD can fold into a longer α-helix (PDB ID: 4MZ018), and the two different conformations of the NDD adopt a coiled-coil configuration.18 This coiled-coil superimposes well with the type 1b PikAIV NDD coiled-coil within the PikAIII CDD/PikAIV NDD complex (PDB ID: 3F5H) with a RMSD value 1.93 Å and a TM-score of 0.74 (ESI,† Fig. S7 and Table S5). While direct comparison of the overall docked CDDs/NDDs complexes of PikAIII/PikAIV and CurK/CurL yields a high RMSD (2.05 Å) (ESI,† Fig. S7 and Table S5), the TM-score is 0.58, showing that the overall topologies are similar.
A systematic comparison of the structures of available type 2(-related) and type 1 CDD/NDD complexes gives RMSD values between 1.71 and 2.54 Å, with TM scores higher than 0.5. Higher RMSD values (>4 Å) for the type 2-related Bam_5925 CDD/Bam_5924 NDD complex relative to the other CDD/NDD complexes (ESI,† Table S5) are explained by the fact that its NDDs are not folded as a coiled-coil, but are interleaved. As a result, compared to the other CDDs, the binding of Bam_5925 CDD is offset (ESI,† Fig. S7). Thus, overall, the conclusion that emerges is that type 1 and type 2 DD complexes are structurally closely related, even though their sequences have often been differentiated in previous studies.12,14,17 Such similarity rarely provokes specificity issues in their native contexts, as type 1a/1b DDs are typically found in actinomycetes, while type 2 DDs are present principally in myxobacteria and cyanobacteria.59 In cases such as the unusual, hybrid cis-AT/trans-AT enacyloxin IIa PKS of Burkholderia ambifaria where both type 1- and type 2-related domains are present, they are intrinsically orthogonal,14 demonstrating that the key interaction residues in each case are sufficient to confer specificity.
Conclusions
We aimed to investigate potential DDs from the recently discovered toblerol (Tob) trans-AT PKS system.11 Both the origin of the PKS from a methylotroph Methylorubrum extorquens AM1, and its atypical organization, suggested that it might contain DDs exhibiting novel characteristics. We have shown herein that both DDs operating at the central TobC/TobE interface are intrinsically disordered (IDRs) as discrete domains, only adopting stable α-helical structures in the presence of their partner. Neither standard secondary structure prediction programs nor AlphaFold230 were able to confidently predict the number and extent of the α-helices in TobE NDD, thus illustrating the importance of experimental investigations. The finding that both DDs are IDRs further strengthens earlier observations14 concerning the utility of such elements in modular PKS systems for achieving both high specificity and low-to-medium affinity interactions. Furthermore, while folded TobC CDD resembles the so-called 4 α-helix bundle (4HB) CDDs from other trans-AT PKSs,15 it induces its partner NDD to adopt only a single α-helix, giving rise to a novel 3HBb docking complex with a measured interaction strength (KD = 4 μM) on par with other pairs of 4HB domains.14,16 Taken together with previous work, these data show that even within trans-AT PKSs, equivalent DD elements can be deployed in several different ways (i.e. to form 4- or 3- α-helix bundles) to meet common affinity and specificity imperatives.The fact that individual 4HB fold DDs can interface productively with both mono- and bi-helical partners prompted us to reexamine other known types of DD complexes for the re-use of common elements. This analysis revealed that type 2 DDs also participate in assemblies of several different configurations. When the CDDs adopt the canonical type 2 fold, they form 8 α-helix bundles of varying topology (canonical type 2 and type 2-related), along with 3 α-helix bundles (type 3HBa) of geometry distinct from that of the TobC CDD/TobE NDD complex (type 3HBb). In each case, a limited set of conserved hydrophobic and/or charged residues appears to play a decisive role in defining the overall architecture of the complexes and the specificity of the interactions, while ensuring similar binding affinities.
Globally, these observations illustrate the high versatility and adaptability of α-helical DDs as docking elements. However, the fact that individual DDs that exhibit the same folds can form divergent complexes further complicates efforts to reliably predict DD types. The classification of DDs into types should thus not be based on the unpaired elements but on the experimentally determined topologies of the native complexes. We anticipate, however, that additional progress in elucidating DD structures coupled with ongoing improvements in AlphaFold30-based structure prediction will ultimately allow for DD characterization based on sequence analysis alone. In the meantime, the results reported here further expand the toolbox of DDs available for PKS synthetic biology.24–26
Materials and methods
Sequence analysis
The sequences of the analyzed docking domains were retrieved from UniProt.60 To identify putative docking domains at the extremities of the toblerol PKS subunits, the boundaries of their adjacent domains were determined by multiple sequence alignments of known PKS domains using Clustal Omega61 and HHpred.62 Secondary structure prediction of the putative docking domains was performed with PSIPRED,36,63 and disorder and interaction propensity were predicted with IUPred3/ANCHOR2.37 Since no confident secondary structure prediction emerged, the regions directly down-/upstream of the flanking domains were expressed as TobC CDD and TobE NDD, respectively. In terms of the TobE NDD-ACP construct, the C-terminal boundary was set to two residues after the last α-helix of the ACP domain (Table S1, ESI†).Materials, DNA manipulation, and sequencing
The toblerol PKS gene cluster from Methylorubrum extorquens AM1 was provided by J. Piel's laboratory (Institute of Microbiology, ETH Zürich, CH) on a fosmid. Biochemicals and media were purchased from ThermoFisher Scientific (Tris, EDTA), Merck (NaPi), CARLO ERBA (NaCl), BD (peptone, yeast extract), VWR (glycerol), Sigma-Aldrich (imidazole, IPTG) and Eurisotop (15NH4Cl, 13C6-glucose). The enzymes for genetic manipulation were purchased from ThermoFisher Scientific. DNA isolation and manipulation in Escherichia coli were carried out using standard methods.30 The purification of PCR products and digested plasmids and mini-preparation of plasmid DNA were performed using the NucleoSpin® Gel and PCR Clean-up or NucleoSpin® Plasmid DNA kits (Macherey Nagel). PCR amplifications were carried out on a Mastercycler Pro (Eppendorf) using Phusion Hot Start II High-Fidelity DNA polymerase. Primers were purchased from Sigma-Aldrich (France), and DNA sequencing was performed by Eurofins Genomics (Köln, DE).Cloning, expression, and purification of recombinant proteins
Three constructs (TobC CDD, TobE NDD, and TobE NDD-ACP) were amplified from the fosmid encoding the toblerol PKS cluster using forward and reverse primers incorporating EcoRI and HindIII sites for TobC CDD and BamH1 and NheI restriction sites for TobE NDD and TobE NDD-ACP (ESI,† Table S2). Amplicons were ligated into the equivalent sites of vector pBG102 (Center for Structural Biology, Vanderbilt University). This vector allows the expression of proteins with an N-terminal His6-SUMO tag. Cleavage of the tag leaves a non-native N-terminal Gly–Pro–Gly–Ser sequence when the BamHI site is used and a Gly–Pro–Gly–Ser–Pro–Asn–Ser sequence when EcoRI is used. The vectors were used to transform E. coli Rosetta 2 (DE3) (Novagen) and constructs were expressed by growth in LB medium at 37 °C to an A600 of 0.6, followed by induction with IPTG (0.5 mM) and incubation at 20 °C for a further 15–18 h.The E. coli cells were collected by centrifugation (8000 g) and resuspended in buffer A (50 mM Tris, 500 mM NaCl, pH 8.5). The cells were lysed by sonication and the cell debris was removed by centrifugation (48000 g for 30 min). The supernatant was filtered (0.22 μm) and loaded onto a HisTrap HP 5 mL column (GE Healthcare), equilibrated in buffer A. The stationary phase was washed extensively with buffer A containing 50 mM imidazole, and His-tagged proteins were eluted using a one-step elution with buffer A containing 300 mM imidazole. The obtained fusion proteins were then incubated with His-tagged human rhinovirus 3C protease (1 μM) for 14–18 h at 4 °C to cleave the His6-SUMO tag. The target proteins were separated from the remaining His-tagged proteins by loading onto a HisTrap HP 5 mL column (GE Healthcare), followed by elution in buffer A containing 20 mM imidazole. Flow-through containing the proteins was concentrated for a final purification step by size-exclusion chromatography using a HiLoad 16/600 Superdex 75 PG (GE Healthcare) equilibrated with 100 mM sodium phosphate (pH 6.5). The purification resulted in a homogeneous preparation of each protein. The production of 15N- and 13C, 15N-enriched TobC CDD and TobE NDD for NMR structure elucidation was carried out by growth in M9 minimal medium. The minimal medium was supplemented with 15NH4Cl (0.5 g L−1) and 13C-glucose (2 g L−1) as the sole sources of nitrogen and carbon. The isotopically labeled constructs were purified to homogeneity using the same protocol as for the unlabeled proteins. For the stereospecific NMR assignment of the methyl groups of valine and leucine,51 TobC CDD and TobE NDD were expressed in M9 minimal medium containing 0.2 g of 13C-glucose and 1.8 g of 12C-glucose as the sole carbon sources to generate 10% 13C-enriched samples.51
Analytical gel filtration
The interaction between the DDs was assessed by size exclusion chromatography using a Superdex 75 10/300 GL column (GE Healthcare) equilibrated with 100 mM sodium phosphate (pH 6.5) buffer. SEC was carried out on isolated TobC CDD and TobE NDD (400 μM of each protein) and an equimolar mixture of TobC CDD and TobE NDD (both at 400 μM). Elution of the proteins was followed via the absorbance at 280 nm.DD analysis by circular dichroism
Circular dichroism (CD) was performed on a Chirascan CD from Applied Photophysics (UMS2008/US40 Ingénierie Biologie Santé en Lorraine (IBSLor), Université de Lorraine-CNRS-INSERM). Data were collected using a 0.1 mm path-length cuvette containing 30 μL of 100 mM protein sample, at 0.5 nm intervals in the wavelength range of 180–260 nm at 20 °C. Measurements were made in triplicate, and sample spectra were corrected for buffer background by subtracting the average spectrum of buffer alone. To estimate the protein secondary structure and evaluate the extent of induced folding when the two DDs were combined,14,15 TobC CDD (100 μM) was placed in one cuvette and TobE NDD (100 μM) into another cuvette, and a combined spectrum of the two cuvettes was then recorded. The two DD samples were subsequently combined, and the mixture (100 μM of each DD) was placed into both cuvettes and then the signal was recorded from both cuvettes simultaneously. Deconvolution of the CD spectra was carried out using the Pro-Data Viewer software (Applied Photophysics, Ltd, Leatherhead, UK).DD analysis by isothermal titration calorimetry
ITC was performed using a MicroCal ITC 200 (GE Healthcare) at 25 °C (UMS2008/US40 IBSLor, Université de Lorraine-CNRS-INSERM). A 300 μL aliquot of TobC CDD or TobE NDD at 60 μM was placed in the calorimeter cell, and TobE NDD or TobC CDD at 600 μM was added as follows: 0.5 μL over 1.0 s for the first injection, followed by 19 injections of 2 μL over 4.0 s with 120 s spacing time. The heat of reaction per injection (μcal s−1) was determined by integration of the peak areas using NITPIC64 and the data were fitted with Origin software (MicroCal, LLC, USA) to determine the heat of binding (ΔH), the stoichiometry of binding (N) and the dissociation constant (KD). The heats of dilution were determined by injecting the DDs (initial concentration of 600 μM) into the cell containing only the buffer, and these data were subtracted from the titration data before curve fitting. The thermodynamic parameters of the interaction were determined using the relationship ΔG = ΔH − TΔS, where ΔG = −RT*ln(KD) and ΔH is experimentally determined.NMR data acquisition
NMR data were acquired on four samples: (i) 0.6 mM 15N/13C-labeled TobC CDD; (ii) 0.5 mM 15N/13C-labeled TobE NDD; (iii) 1 mM 15N/13C-labeled TobC CDD and 2 mM unlabeled TobE NDD; (iv) 1 mM 15N/13C-labeled TobE NDD and 2 mM unlabeled TobC CDD. To minimize the amount of protein needed, 300 μL of protein solution was loaded into a 4 mm NMR tube. All NMR spectra were recorded at 298K on a Brucker DRX600 spectrometer equipped with a cryoprobe (UMS2008/US40 IBSLor, Université de Lorraine-CNRS-INSERM) using standard NMR experiments (reviewed in ref. 40). Backbone and sequential resonance assignments were obtained by the combined use of 2D 15N–1H and 13C–1H HSQC spectra and 3D HNCA, HNCACB, HNCO, HN(CA)CO, HNHA, and CBCA(CO)NH experiments. Sidechain atoms were assigned using 2D aromatic 13C–1H HSQC, 3D (H)CC(CO)NH, H(CC)(CO)NH, (H)CCH-TOCSY, and H(C)CH-TOCSY experiments. Stereospecific assignments of valine and leucine methyl groups were determined by recording 1H–13C constant-time HSQCs on the 10% fractionally 13C-labeled samples.51 To collect NOE-based distance restraints for the TobC CDD/TobE NDD structure calculation, 3D 15N NOESY-HSQC and 13C NOESY-HSQC were recorded using a 120 ms mixing time. Intermolecular NOE were recorded using a 3D 13C/15N X-filtered NOESY experiment on the [12C,14N]TobE NDD/[13C,15N] TobC CDD sample.65 {1H}-15N-heteronuclear nuclear Overhauser effect experiments66 were carried out using standard Bruker pulse sequences on 15N-labeled DD alone and in complex with its unlabeled DD partner. Experiments were recorded twice in an interleaved fashion with and without proton saturation using a 7 s recovery delay. {1H}–15N-heteronuclear NOE was defined as NOE = Ssat/S0 and errors using signal-to-noise ratios (SNR) as δNOE = NOE*((SNRsat)−2 + (SNRunsat)−2)½.67 Chemical shift perturbations were calculated using the following formula: CSP = (Δδ2HN + (ΔδN/6.5)2)½.68Z-scores were calculated as (CSP-μ)/σ with μ and σ being the average and the standard deviation of the titration CSPs, respectively.
NMR data analysis
NMR data were processed using Topspin 3.1 (Bruker) and were analyzed with CcpNMR 3.0.469 and NMRFAM-Sparky.70 Secondary structure prediction and random coil index analysis71 were carried out using TALOS-N.41NMR structure calculation and analysis
Initial structures were generated using CYANA 3.0 software.48 Starting from a set of manually assigned NOEs (Nuclear Overhauser Effect), the standard CYANA protocol of seven iterative cycles of calculation was performed with NOE assignments by the embedded CANDID routine combined with torsion angle dynamics structure calculation.72 In each of the 7 cycles, 100 structures starting from random torsion angle values were calculated using 15000 steps of torsion angle dynamics-driven simulated annealing. A total of 1349 NOE-based distances and 68 backbone angle restraints were used for the final calculations. The angle restraints were obtained from 13Cα, 13Cβ, 13C′, and 15N chemical shifts using TALOS-N41 with an assigned minimum range of ±20°. No hydrogen-bond restraints were used for structure calculation. The second stage consisted of refinement of the 50 lowest CYANA target function conformers by restrained MD simulations in Amber 22,49 using previously published procedures.14,15 The representative ensemble corresponds to the 20 conformers with the lowest restraint energy terms. The quality of the structure was assessed using the Protein Structure Validation Software Suite (PSVS),47 PROCHECK-NMR,53 and MolProbity54 servers. Complex analysis was performed by visual inspection using PyMOL73 and PDBsum1.74 Root-mean-square-deviation calculation, structure superimposition, analyses, and illustrations were carried out using PyMOL.73 TM scores were computed using US-align.56,57 Solvent accessible surface areas were calculated using Naccess V2.1.1.75 3D docking domain models were generated using AlphaFold2.301H, 13C, and 15N chemical shift assignments for isolated TobC CDD and TobE NDD, and the TobC CDD/TobE NDD complex have been deposited in the Biological Magnetic Resonance Data Bank under accession numbers 52224, 52225 and 34886, respectively, and the structure coordinates and NMR restraints for the TobC CDD/TobE NDD complex in the Protein Data Bank under accession number 8RAJ.
Author contributions
S. S.: formal analysis, investigation, methodology, validation, visualization, writing – original draft, review and editing. K. J. W.: conceptualization, funding acquisition, project administration, supervision, writing – original draft, review and editing. B. C.: conceptualization, formal analysis, investigation, methodology, project administration, supervision, validation, visualization, writing – original draft, review and editing.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We are grateful for funding of this work by the Agence Nationale de la Recherche (ANR-20-CE93-0002-01 PKSOx to K. J. W.), the Université de Lorraine, and the Centre National de la Recherche Scientifique (CNRS). We also acknowledge J. Piel, ETH Zurich, CH, for the kind gift of the toblerol gene cluster. The NMR, ITC and CD analysis were carried out on the Plateforme de Biophysique et Biologie Structurale (B2S) of the UMS2008/US40 IBSLor (Université de Lorraine-CNRS-INSERM).Notes and references
- A. L. Demain, J. Ind. Microbiol. Biotechnol., 2014, 41, 185–201 CrossRef CAS PubMed.
- A. L. Demain and P. Vaishnav, Microb. Biotechnol., 2011, 4, 687–699 CrossRef PubMed.
- J. Staunton and K. J. Weissman, Nat. Prod. Rep., 2001, 18, 380–416 RSC.
- M. A. Fischbach and C. T. Walsh, Chem. Rev., 2006, 106, 3468–3496 CrossRef CAS PubMed.
- T. Nguyen, K. Ishida, H. Jenke-Kodama, E. Dittmann, C. Gurgui, T. Hochmuth, S. Taudien, M. Platzer, C. Hertweck and J. Piel, Nat. Biotechnol., 2008, 26, 225–233 CrossRef CAS PubMed.
- E. J. N. Helfrich, R. Ueoka, M. G. Chevrette, F. Hemmerling, X. Lu, S. Leopold-Messer, H. A. Minas, A. Y. Burch, S. E. Lindow, J. Piel and M. H. Medema, Nat. Commun., 2021, 12, 1422 CrossRef CAS PubMed.
- H. Jenke-Kodama and E. Dittmann, Phytochemistry, 2009, 70, 1858–1866 CrossRef CAS PubMed.
- H. Jenke-Kodama, A. Sandmann, R. Müller and E. Dittmann, Mol. Biol. Evol., 2005, 22, 2027–2039 CrossRef CAS PubMed.
- J. Piel, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14002–14007 CrossRef CAS PubMed.
- Y.-Q. Cheng, G.-L. Tang and B. Shen, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3149–3154 CrossRef CAS PubMed.
- R. Ueoka, M. Bortfeld-Miller, B. I. Morinaka, J. A. Vorholt and J. Piel, Angew. Chem., Int. Ed., 2018, 57, 977–981 CrossRef CAS PubMed.
- R. W. Broadhurst, D. Nietlispach, M. P. Wheatcroft, P. F. Leadlay and K. J. Weissman, Chem. Biol., 2003, 10, 723–731 CrossRef CAS PubMed.
- C. D. Richter, D. Nietlispach, R. W. Broadhurst and K. J. Weissman, Nat. Chem. Biol., 2008, 4, 75–81 CrossRef CAS PubMed.
- F. Risser, S. Collin, R. Dos Santos-Morais, A. Gruez, B. Chagot and K. J. Weissman, J. Struct. Biol., 2020, 212, 107581 CrossRef CAS PubMed.
- J. Dorival, T. Annaval, F. Risser, S. Collin, P. Roblin, C. Jacob, A. Gruez, B. Chagot and K. J. Weissman, J. Am. Chem. Soc., 2016, 138, 4155–4167 CrossRef CAS PubMed.
- J. Zeng, D. T. Wagner, Z. Zhang, L. Moretto, J. D. Addison and A. T. Keatinge-Clay, ACS Chem. Biol., 2016, 11, 2466–2474 CrossRef CAS PubMed.
- T. J. Buchholz, T. W. Geders, F. E. Bartley, K. A. Reynolds, J. L. Smith and D. H. Sherman, ACS Chem. Biol., 2009, 4, 41–52 CrossRef CAS PubMed.
- J. R. Whicher, S. S. Smaga, D. A. Hansen, W. C. Brown, W. H. Gerwick, D. H. Sherman and J. L. Smith, Chem. Biol., 2013, 20, 1340–1351 CrossRef CAS PubMed.
- C. Hacker, X. Cai, C. Kegler, L. Zhao, A. K. Weickhmann, J. P. Wurm, H. B. Bode and J. Wöhnert, Nat. Commun., 2018, 9, 4366 CrossRef PubMed.
- J. Watzel, C. Hacker, E. Duchardt-Ferner, H. B. Bode and J. Wöhnert, ACS Chem. Biol., 2020, 15, 982–989 CrossRef CAS PubMed.
- J. Watzel, E. Duchardt-Ferner, S. Sarawi, H. B. Bode and J. Wöhnert, Angew. Chem., Int. Ed., 2021, 60, 14171–14178 CrossRef CAS PubMed.
- M. Jenner, S. Kosol, D. Griffiths, P. Prasongpholchai, L. Manzi, A. S. Barrow, J. E. Moses, N. J. Oldham, J. R. Lewandowski and G. L. Challis, Nat. Chem. Biol., 2018, 14, 270–275 CrossRef CAS PubMed.
- J. Yan, S. Gupta, D. H. Sherman and K. A. Reynolds, ChemBioChem, 2009, 10, 1537–1543 CrossRef CAS PubMed.
- T. Miyazawa, M. Hirsch, Z. Zhang and A. T. Keatinge-Clay, Nat. Commun., 2020, 11, 80 CrossRef CAS PubMed.
- T. Miyazawa, B. J. Fitzgerald and A. T. Keatinge-Clay, Chem. Commun., 2021, 57, 8762–8765 RSC.
- L. Su, L. Hôtel, C. Paris, C. Chepkirui, A. O. Brachmann, J. Piel, C. Jacob, B. Aigle and K. J. Weissman, Nat. Commun., 2022, 13, 515 CrossRef CAS PubMed.
- X. Sun, Y. Yuan, Q. Chen, S. Nie, J. Guo, Z. Ou, M. Huang, Z. Deng, T. Liu and T. Ma, Nat. Commun., 2022, 13, 5541 CrossRef CAS PubMed.
- J. L. Meinke, A. J. Simon, D. T. Wagner, B. R. Morrow, S. You, A. D. Ellington and A. T. Keatinge-Clay, ACS Synth. Biol., 2019, 8, 2017–2024 CrossRef CAS PubMed.
- S. Kosol, A. Gallo, D. Griffiths, T. R. Valentic, J. Masschelein, M. Jenner, E. L. C. de los Santos, L. Manzi, P. K. Sydor, D. Rea, S. Zhou, V. Fülöp, N. J. Oldham, S.-C. Tsai, G. L. Challis and J. R. Lewandowski, Nat. Chem., 2019, 11, 913–923 CrossRef CAS PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- V. Monzon, D. H. Haft and A. Bateman, Bioinform. Adv., 2022, 2, vbab043 CrossRef PubMed.
- E. J. N. Helfrich and J. Piel, Nat. Prod. Rep., 2016, 33, 231–316 RSC.
- J. Davison, J. Dorival, H. Rabeharindranto, H. Mazon, B. Chagot, A. Gruez and K. J. Weissman, Chem. Sci., 2014, 5, 3081–3095 RSC.
- V. Y. Alekseyev, C. W. Liu, D. E. Cane, J. D. Puglisi and C. Khosla, Protein Sci., 2007, 16, 2093–2107 CrossRef CAS PubMed.
- Y. Tang, C.-Y. Kim, I. I. Mathews, D. E. Cane and C. Khosla, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11124–11129 CrossRef CAS PubMed.
- D. W. A. Buchan and D. T. Jones, Nucleic Acids Res., 2019, 47, W402–W407 CrossRef CAS PubMed.
- G. Erdős, M. Pajkos and Z. Dosztányi, Nucleic Acids Res., 2021, 49, W297–W303 CrossRef PubMed.
- D. Sahu, M. Bastidas, C. W. Lawrence, W. G. Noid and S. A. Showalter, Methods Enzymol., 2016, 567, 23–45 CAS.
- L. J. McGuffin, K. Bryson and D. T. Jones, Bioinformatics, 2000, 16, 404–405 CrossRef CAS PubMed.
- J. Cavanagh, W. J. Fairbrother, A. G. Palmer, M. Rance and N. J. Skeltonv, in Protein NMR Spectroscopy, ed. J. Cavanagh, W. J. Fairbrother, A. G. Palmer, M. Rance and N. J. Skelton, Academic Press, Burlington, 2nd edn, 2007, pp. v–vi Search PubMed.
- Y. Shen and A. Bax, J. Biomol. NMR, 2013, 56, 227–241 CrossRef CAS PubMed.
- D.-H. Kim and K.-H. Han, Mol. Cells, 2018, 41, 889–899 CAS.
- C. J. Oldfield, Y. Cheng, M. S. Cortese, P. Romero, V. N. Uversky and A. K. Dunker, Biochemistry, 2005, 44, 12454–12470 CrossRef CAS PubMed.
- P. Tompa and M. Fuxreiter, Trends Biochem. Sci., 2008, 33, 2–8 CrossRef CAS PubMed.
- R. Evans, M. O’Neill, A. Pritzel, N. Antropova, A. Senior, T. Green, A. Žídek, R. Bates, S. Blackwell, J. Yim, O. Ronneberger, S. Bodenstein, M. Zielinski, A. Bridgland, A. Potapenko, A. Cowie, K. Tunyasuvunakool, R. Jain, E. Clancy, P. Kohli, J. Jumper and D. Hassabis, bioRxiv, 2022, preprint DOI:10.1101/2021.10.04.463034.
- R. Yin, B. Y. Feng, A. Varshney and B. G. Pierce, Protein Sci., 2022, 31, e4379 CrossRef CAS PubMed.
- A. Bhattacharya, R. Tejero and G. T. Montelione, Proteins, 2006, 66, 778–795 CrossRef PubMed.
- P. Güntert, in Protein NMR Techniques, ed. A. K. Downing, Humana Press, Totowa, NJ, 2004, pp. 353–378 Search PubMed.
- D. A. Case, T. E. Cheatham III, T. Darden, H. Gohlke, R. Luo, K. M. Merz Jr., A. Onufriev, C. Simmerling, B. Wang and R. J. Woods, J. Comput. Chem., 2005, 26, 1668–1688 CrossRef CAS PubMed.
- B. Chagot and W. J. Chazin, J. Mol. Biol., 2011, 406, 106–119 CrossRef CAS PubMed.
- D. Neri, T. Szyperski, G. Otting, H. Senn and K. Wuethrich, Biochemistry, 1989, 28, 7510–7516 CrossRef CAS PubMed.
- G. T. Montelione, M. Nilges, A. Bax, P. Güntert, T. Herrmann, J. S. Richardson, C. D. Schwieters, W. F. Vranken, G. W. Vuister, D. S. Wishart, H. M. Berman, G. J. Kleywegt and J. L. Markley, Structure, 2013, 21, 1563–1570 CrossRef CAS PubMed.
- R. A. Laskowski, J. A. Rullmannn, M. W. MacArthur, R. Kaptein and J. M. Thornton, J. Biomol. NMR, 1996, 8, 477–486 CrossRef CAS PubMed.
- V. B. Chen, W. B. Arendall, J. J. Headd, D. A. Keedy, R. M. Immormino, G. J. Kapral, L. W. Murray, J. S. Richardson and D. C. Richardson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 12–21 CrossRef CAS PubMed.
- M. Schubert, D. Labudde, H. Oschkinat and P. Schmieder, J. Biomol. NMR, 2002, 24, 149–154 CrossRef CAS PubMed.
- Y. Zhang and J. Skolnick, Proteins, 2004, 57, 702–710 CrossRef CAS PubMed.
- C. Zhang, M. Shine, A. M. Pyle and Y. Zhang, Nat. Methods, 2022, 19, 1109–1115 CrossRef CAS PubMed.
- J. M. Reimer, A. S. Haque, M. J. Tarry and T. M. Schmeing, Curr. Opin. Struct. Biol., 2018, 49, 104–113 CrossRef CAS PubMed.
- M. Thattai, Y. Burak and B. Shraiman, PLoS Comput. Biol., 2018, 3, 1827–1835 Search PubMed.
- A. Bateman, M.-J. Martin, S. Orchard, M. Magrane, S. Ahmad, E. Alpi, E. H. Bowler-Barnett, R. Britto, H. Bye-A-Jee, A. Cukura, P. Denny, T. Dogan, T. Ebenezer, J. Fan, P. Garmiri, L. J. da Costa Gonzales, E. Hatton-Ellis, A. Hussein, A. Ignatchenko, G. Insana, R. Ishtiaq, V. Joshi, D. Jyothi, S. Kandasaamy, A. Lock, A. Luciani, M. Lugaric, J. Luo, Y. Lussi, A. MacDougall, F. Madeira, M. Mahmoudy, A. Mishra, K. Moulang, A. Nightingale, S. Pundir, G. Qi, S. Raj, P. Raposo, D. L. Rice, R. Saidi, R. Santos, E. Speretta, J. Stephenson, P. Totoo, E. Turner, N. Tyagi, P. Vasudev, K. Warner, X. Watkins, R. Zaru, H. Zellner, A. J. Bridge, L. Aimo, G. Argoud-Puy, A. H. Auchincloss, K. B. Axelsen, P. Bansal, D. Baratin, T. M. Batista Neto, M.-C. Blatter, J. T. Bolleman, E. Boutet, L. Breuza, B. C. Gil, C. Casals-Casas, K. C. Echioukh, E. Coudert, B. Cuche, E. de Castro, A. Estreicher, M. L. Famiglietti, M. Feuermann, E. Gasteiger, P. Gaudet, S. Gehant, V. Gerritsen, A. Gos, N. Gruaz, C. Hulo, N. Hyka-Nouspikel, F. Jungo, A. Kerhornou, P. Le Mercier, D. Lieberherr, P. Masson, A. Morgat, V. Muthukrishnan, S. Paesano, I. Pedruzzi, S. Pilbout, L. Pourcel, S. Poux, M. Pozzato, M. Pruess, N. Redaschi, C. Rivoire, C. J. A. Sigrist, K. Sonesson, S. Sundaram, C. H. Wu, C. N. Arighi, L. Arminski, C. Chen, Y. Chen, H. Huang, K. Laiho, P. McGarvey, D. A. Natale, K. Ross, C. R. Vinayaka, Q. Wang, Y. Wang and J. Zhang, Nucleic Acids Res, 2023, 51, D523–D531 CrossRef CAS PubMed.
- F. Sievers, A. Wilm, D. Dineen, T. J. Gibson, K. Karplus, W. Li, R. Lopez, H. McWilliam, M. Remmert, J. Söding, J. D. Thompson and D. G. Higgins, Mol. Syst. Biol., 2011, 7, 539 CrossRef PubMed.
- F. Gabler, S.-Z. Nam, S. Till, M. Mirdita, M. Steinegger, J. Söding, A. N. Lupas and V. Alva, Curr. Protoc. Bioinf., 2020, 72, e108 CrossRef CAS PubMed.
- D. T. Jones, J. Mol. Biol., 1999, 292, 195–202 CrossRef CAS PubMed.
- S. Keller, C. Vargas, H. Zhao, G. Piszczek, C. A. Brautigam and P. Schuck, Anal. Chem., 2012, 84, 5066–5073 CrossRef CAS PubMed.
- A. L. Breeze, Prog. Nucl. Magn. Reson. Spectrosc., 2000, 36, 323–372 CrossRef CAS.
- N. A. Farrow, O. Zhang, J. D. Forman-Kay and L. E. Kay, J. Biomol. NMR, 1994, 4, 727–734 CrossRef CAS PubMed.
- V. Kharchenko, M. Nowakowski, M. Jaremko, A. Ejchart and Ł. Jaremko, J. Biomol. NMR, 2020, 74, 707–716 CrossRef PubMed.
- F. A. A. Mulder, D. Schipper, R. Bott and R. Boelens, J. Mol. Biol., 1999, 292, 111–123 CrossRef CAS PubMed.
- S. P. Skinner, R. H. Fogh, W. Boucher, T. J. Ragan, L. G. Mureddu and G. W. Vuister, J. Biomol. NMR, 2016, 66, 111–124 CrossRef CAS PubMed.
- W. Lee, M. Tonelli and J. L. Markley, Bioinformatics, 2015, 31, 1325–1327 CrossRef PubMed.
- M. V. Berjanskii and D. S. Wishart, J. Am. Chem. Soc., 2005, 127, 14970–14971 CrossRef CAS PubMed.
- T. Herrmann, P. Güntert and K. Wüthrich, J. Mol. Biol., 2002, 319, 209–227 CrossRef CAS PubMed.
- The PyMOL Molecular Graphics System, Version 3.0, Schrödinger, LLC.
- R. A. Laskowski, Nucleic Acids Res., 2009, 37, D355–359 CrossRef CAS PubMed.
- S. J. Hubbard and J. M. Thornton, Computer Program, Department of Biochemistry and Molecular Biology, University College, London, 1993 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting tables summarizing the oligonucleotides used in this work, the structural statistics for the NMR solution structure of the individual TobC CDD and TobE NDD domains and the TobC CDD/TobE NDD complex, and the calculated RMSD and TM-scores; supporting figures showing the bioinformatics analysis of the TobC/TobE interface, the gel filtration and CD analyses, the AlphaFold2 DD structure predictions, the ITC- and CD-based analysis of the DD mutants, and the DD structural comparisons; supporting tables reporting the sequences of constructs used in this work and the primers used for amplification, NMR structural statistics, the DD interhelical angles, the calculated RMSDs between proteins/complexes, and the calculated TM scores. See DOI: https://doi.org/10.1039/d4cb00075g |
| This journal is © The Royal Society of Chemistry 2024 |