
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDNA encoded peptide library for SARS-CoV-2 3CL protease covalent inhibitor discovery and profiling†
Yuyu
Xing
ab,
Huiya
Zhang
a,
Yanhui
Wang
a,
Zhaoyun
Zong
a,
Matthew
Bogyo
 c and
Shiyu
Chen
*ab
c and
Shiyu
Chen
*ab
aBiotech Drug Research Center, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China. E-mail: chenshiyu@simm.ac.cn
bUniversity of Chinese Academy of Sciences, No. 19A Yuquan Road, Beijing 100049, China
cDepartment of Pathology, Stanford University School of Medicine, Stanford, CA, USA
First published on 11th June 2024
Abstract
Covalent protease inhibitors serve as valuable tools for modulating protease activity and are essential for investigating the functions of protease targets. These inhibitors typically consist of a recognition motif and a covalently reactive electrophile. Substrate peptides, featuring residues capable of fitting into the substrate pockets of proteases, undergo chemical modification at the carbonyl carbon of the P1 residue with an electrophile and have been widely applied in the development of covalent inhibitors. In this study, we utilized a DNA-encoded peptide library to replicate peptide binder sequences and introduced a vinyl sulfone warhead at the C-termini to construct the DNA-encoded peptide covalent inhibitor library (DEPCIL) for targeting cysteine proteases. Screening results toward 3CL protease demonstrated the efficacy of this library, not only in identifying protease inhibitors, but also in discovering amino acids that can conform to aligned protease pockets. The identified peptide sequences provide valuable insight into the amino acid preferences within substrate binding pockets, and our novel technology is indicative of the potential for similar strategies to discover covalent inhibitors and profile binding preferences of other proteases.
Introduction
Proteases play pivotal roles in a myriad of cellular processes, ranging from protein degradation and immune response modulation to signal transduction and cell cycle regulation.1,2 Dysregulation of protease activity has been implicated in various pathological conditions, including cancer,3 inflammation,4 and neurodegenerative disorders.5 Covalent inhibitors that can permanently bind to proteases can be used to modulate their activities and be applied in agent development for tracing protease targets. Covalent inhibitors of proteases have emerged as a compelling avenue in therapeutic intervention, offering unique advantages that address challenges associated with conventional reversible inhibitors.6,7The screening of synthetic compound libraries has been the conventional method applied in the discovery of covalent protease inhibitors.8,9 Numerous covalent inhibitors of proteases have been discovered using this approach, providing useful tools for protease research.10,11 In order to generate more selective protease inhibitors, reactive electrophiles have been purposely incorporated at the C-terminus to convert substrates into inhibitors.12 Several covalent inhibitors were developed using this method and have proven useful in studying protease functions in various applications.13–15
For enabling large-scale screening of covalent libraries, protease reactive warheads have been incorporated in both phage-displayed peptide libraries16 and DNA-encoded small molecular and peptide libraries.17,18 The subsequent discovery of covalent inhibitors from these libraries has demonstrated the effectiveness of ligands obtained from combinatorial libraries. GSK has validated the principle of irreversible screening by specifically enriching a positive on-DNA Rupintrivir compound spiked into a random library.19 WuXi AppTec has introduced the acrylamide warhead to the N-termini of a peptidic DEL library and discovered a covalent inhibitor with an IC50 of 1.9 μM after 16 hours of preincubation.20 X-Chem has established the DELvalent platform and validated the selection of irreversible ligands against several challenging protease targets.21
In this work, based on reported peptide DEL extension technologies, we developed a novel format of DNA-encoded peptide covalent inhibitor library (DEPCIL) for discovering protease covalent inhibitors more efficiently and profiling amino acid binding preferences simultaneously. DEPCIL peptides not only possess a format of inhibitors that can potentially fit better into the active sites of proteases, but also the increased diversity resulting from the incorporation of both canonical and non-canonical amino acids can improve the binding affinity of peptides to the target and lead to the discovery of more potent inhibitors. Furthermore, screening with such libraries can also unveil information on the defined preferences of all substrate residues of the target protease, which is crucial for understanding the substrate recognition pockets of the target protease.
We demonstrated the DEPCIL strategy for screening the SARS-CoV-2 3CL protease (Mpro) by using an in-solution covalent inhibition and His-Tag-facilitated immobilization approach. The main 3C-like protease (3CLpro) plays an essential role in cleaving viral polyproteins to generate functional non-structural proteins and plays a crucial role in the assembly, replication, and transcription of the virus.22 The 3CL protease has no homologous proteins in humans, and it is a valid drug target for inhibiting viral replication and developing anti-COVID-19 drugs.23,24 Several reported covalent inhibitors of 3CL protease exclusively bind to the catalytic residue Cys145 located at the substrate pocket with a covalent warhead. Nirmatrelvir is an orally active 3C-like protease inhibitor developed by inserting a nitrile at the C-terminus of a non-canonical peptide binder.25 Bofutrelvir is a covalent inhibitor of 3CL protease carrying an aldehyde warhead at the C-terminus.26 In this study, screening using DEPCIL resulted in the discovery of P3C-1, a covalent inhibitor exhibiting an IC50 of 172.7 ± 4.12 nM, and unveiled the substrate binding characteristics of the 3CL protease. P3C-1 holds promise for the development of crucial therapeutics to combat SARS-CoV-2 and potential future pandemics. The screening results demonstrated the efficiency of DEPCIL technology for discovering protease inhibitors compared to related methods. DEPCIL also provides hints for developing other formats of covalent peptide libraries targeting other proteases.
Results and discussion
Design of DEPCIL
The hydrolysis of a peptide substrate by a protease is initiated with the nucleophilic attack from the active site residue of the protease to the carbonyl carbon of the P1 residue of the substrate peptide. The substrate peptide can be transformed into a covalent inhibitor by introducing an electrophile to replace the P1 carbonyl carbon (Fig. 1A). This substitution facilitates a covalent attachment to the active site residue, achieving protease inhibition. Currently, this chemical modification strategy is the most commonly employed method for developing covalent inhibitors of protease and for studying the positional preferences of individual substrate pockets.27,28 However, to achieve a relatively potent inhibitor often requires a substantial amount of medicinal chemistry efforts to perform a series of modifications to find sequences where all residues that can fit the pockets of the target.29,30 In this study, we employed a DNA-encoded peptide library for the discovery of covalent inhibiting peptides against cysteine proteases. | ||
| Fig. 1 The principle of covalent inhibition of cysteine protease and application of DEPCIL library screening. (A) The mechanism of covalent inhibition involves coupling reaction between the active site cysteine residue (yellow shadow) and a peptide-based inhibitor containing multiple residues binding to substrate recognition pockets and a vinyl sulfone warhead conjugated at the C-terminus. The nucleophilic attach between the active site cysteine and the vinyl sulfone group quenched the activity of the cysteine protease. (B) Chemical structure of the designed inhibitor library comprising a vinyl sulfone, a (3S)-γ-lactam alanine amino acid, and a peptide library of four random amino acid lengths. (C) Schematic representation of the procedure for discovering inhibitors of cysteine protease by applying the vinyl sulfone peptide inhibitor library. Here, peptide inhibitors that react with the target protease can be recovered and sequenced to unveil the sequence information of the peptide. Following DNA tag sequencing, peptide synthesis, and inhibition testing, the inhibitor can be discovered. (D) Procedure for constructing the DNA-encoded library of covalent inhibitors of cysteine proteases by incorporating the vinyl sulfone warhead and extending peptide residues. | ||
A number of previous studies have reported building DNA-encode small molecular and peptide based covalent inhibitor libraries of proteases.17,18 In contrast to incorporating the warhead at the sidechain of an amino acid or the N-terminus of the DEL peptide library, we chose to insert the warhead at the C-terminus of the peptide library to develop a novel format of DNA-encoded peptide covalent inhibitor library (Fig. 1B). Previous studies have indicated that potent peptide binders of protease generally adopt a substrate-like binding pattern, with individual residues occupying the pockets of the protease.31,32 In the complex structures of peptide inhibitors with protease targets, the atom closest to the active site residue is the carboxylic terminal carbon. Incorporating the warhead at the carboxylic group of a P1 residue allows individual residues in the substrate peptide to align with the substrate recognition pockets of the target protease. This strategy offers several advantages. Firstly, it closely mimics the process of substrate binding and the nucleophilic attack from the catalytic residue. Aligned peptide sequences can bind to all of the target protease substrate sites, positioning the C-terminal at the center of the active site. This methodology is expected to reveal substrate binding pocket preference properties that may be challenging to study using other methods. Secondly, the incorporation of non-canonical amino acids supplements the limited natural amino acids. Natural amino acids generally lack the ability to surpass the improvements inherent in sequences shaped by natural evolution. Non-canonical amino acids, however, have demonstrated greater potential for expanding the scope of further enhancements.33 HATU-activated amide coupling and T4 ligations are compatible with various chemical moieties, enabling the incorporation of diverse noncanonical amino acids. Consequently, this approach generates a broader structural and spatial diversity, facilitating the discovery of more potent binders and reducing the likelihood of binding to off-target proteins (Fig. 1C).
Generation of P1-vinyl sulfone conjugate
Cysteine 145 is the key catalytic residue for 3CL protease to exert hydrolytic activity. The most commonly used covalent functional groups of cysteine proteases are acrylamide, vinyl sulfone and aldehyde, among others.12 The olefin part of vinyl sulfone can react with the sulfhydryl group in cysteine to form a covalent bond and has been applied in a large number of bioactive molecules and drug candidates.34 The Wittig reaction was employed to couple aldehyde with sulfide phosphate, for the generation of a vinyl sulfone (VS) electrophile. Recognizing the significance of the P1 residue in the binding of the target protease, we sequentially reduced protected (3S)-γ-lactam alanine,35 which was then reacted with diethyl iodomethyl phosphate to produce (3S)-γ-lactam vinyl sulfone (P1-VS). Additionally, we coupled a hydrophilic and flexible polyethylene glycol trimer (PEG3) linker to the C-terminus of the lactam vinyl sulfone warhead. This modification allows the generated peptide library to freely adopt the proper position in space to fit into the active site of the screened protease. Finally, an Fmoc protection group was introduced at the N-terminus of the P1-VS-PEG for being able to be used as a building block in the DNA-encoded library synthesis (Fig. S1, ESI†).Construction of DEL Library bearing a VS at the C-terminus
With the synthesized Fmoc-P1-VS-PEG-COOH in hand, we applied it in the synthesis of the DEPCIL library following the general DNA-encoded library (DEL) peptide library preparation procedure. First, the stability of P1-VS in subsequent DEL was examined. We subjected compound 9 to 95 °C incubation for 16 hours in the BOC deprotection buffer utilized for DEL construction and subsequently analyzed the samples using LCMS. The results revealed that the integrity of the vinyl sulfone moiety was preserved (Fig. S2, ESI†). To efficiently incorporate the VS warhead into the DEPCIL, the Fmoc-protected P1-VS amino acid was reacted with the headpiece to generate a universal starting building block. For ensuring purity, the product underwent additional purification through precipitation and ultrafiltration. LCMS analysis of the Fmoc-P1-VS-PEG3-HP product confirmed a purity of >95% (Fig. S3, ESI†), and quantification using UV absorption indicated a yield of 94.2%.Considering that most proteases recognize substrates through short central recognition sequences, we decided to construct the DEPCIL library with a length of four residues (Fig. 1D and Fig. S4, ESI†).36 Amino acids with sidechain protection suitable for DEL construction and exhibiting diverse properties, such as hydrophilic/hydrophobic, positive/negative charge, varying sizes, aromatic/aliphatic, were selected for library construction (Table S1, ESI†). These building blocks were assessed for coupling efficiency by reacting them with the headpiece, and only those demonstrating a coupling efficiency greater than 80% were selected for use (unpublished data). The presence of the VS warhead at the N-terminus of the headpiece did not affect the efficiency to couple amino acid to the amino group. The remaining steps of library construction followed standard DEL library synthesis procedures. Firstly, in each cycle, we selected 37 amino acids with various properties that exhibited promising coupling efficiency. Following acylation reaction to conjugate amino acids onto the primary amine, 37 unique DNA tags were ligated to headpiece. After pooling, deprotection of Fmoc, and purification, the cycle-1 product was split into 37 wells for carrying out the next round of synthesis. After four rounds of synthesis, a covalent DEL with a library size of 1.87 million was achieved.
We also optimized barcode tags to ensure sufficient distance, preventing overlap due to PCR and NGS errors. DNA tags (Table S2, ESI†), 9 bases in length, were designed with a minimum of 3-base difference, using a previously reported Python script (reference: https://github.com/pelinakan/UBD). The DNA tags were customized and synthesized with 5′ phosphorylation and a two-base overhang at 3′ end for enabling T4 ligation. During the extension reaction steps of library construction, each coupling reaction was monitored with PAGE analysis until complete reaction achievement. The expected sizes of molecular upper shifts could be observed for each round of DNA tag attachment (Fig. S5A, ESI†).
To minimize the impact of DNA damage during the final step of DEPCIL library deprotection, which aimed to remove side chain protection groups, a mild condition of heated sodium acetate solution was applied to remove the t-butyloxycarbonyl (BOC) from the side chains of free amine side chains.37 Followed by another precipitation purification, a final DNA sequence for the PCR primer binding was incorporated to achieve the expected DEPCIL library (Fig. S5B, ESI†).
Screening of 3CL inhibitors
The defined S1 to S5 pockets of 3CL cysteine protease fit the format of the designed DEPCIL library. The covalent linkage between inhibitors and the target protease provided an opportunity to selectively identify active inhibitors through the enrichment of the target protein, subsequently leading to the identification of the carried DNA tag. Conversely, weak noncovalent interactions with nonspecific peptides allowed for the elimination of peptides that did not form stable covalent interactions with the target.We designed the target 3CL protease carrying a His-tag for both purification and inhibitor immobilization with Ni NTA resin. Following codon optimization, gene synthesis, and expression construct building in the pET28b vector, the 3CL protease was well expressed in BL21(DE3) cells. A sufficient amount of protease (1 mg, 500 mL 2YT expression media) was achieved through affinity purification and size-exclusive chromatography. The obtained 3CL protease exhibited potent efficiency in hydrolyzing a fluorogenic substrate based on the native substrate protein sequence (Fig. S6, ESI†). Due to the decreased hydrolysis efficiency of the 3CL protease after immobilization on solid support, we decided to conduct the screening process through in-solution inhibition followed by enrichment with target protein immobilization.
To increase protease activity and enhance the binding and selection of inhibitors that can covalently react with the target protease, 10 pmol of the DEPCIL library was mixed with 140 pmol of 3CL protease in a buffer displaying maximum 3CL activity. The mixture was then incubated for an extended time of 1 h at 37 °C. Additionally, to minimize nonspecific binding to Ni NTA resin, sheared salmon sperm DNA was applied to the resin to block unspecific interactions before being utilized to immobilize the inhibitor-bound target 3CL protease. By imidazole elution, the active DEPCIL members binding to 3CL protease were recovered and quantified using qPCR. This quantification was achieved by comparing with a standard curve generated by plotting the Cq value against a series of concentrations of DNA (Fig. S7A, ESI†). For the sake of comparison, negative selection without 3CL incubation showed a dramatic decrease of DEPCIL in binding to Ni NTA resin. The recovered DNA with and without 3CL incubation was 4.11 pg μL−1 and 1.94 pg μL−1, respectively, representing a 2.1-fold enrichment for 3CL covalent binding (Fig. S7B, ESI†).
To unveil the sequence of the selected peptide, we applied Illumina NGS to sequence the selected peptides as many as possible. The recovered DNA of DEPCIL was PCR amplified, and a pair of universal sequencing adaptors for Illumina NGS sequencing were attached to both ends. Following PAGE analysis (Fig. S8A and B, ESI†), the desired PCR product was sequenced by an NGS service provider with sample pooling. Upon receiving the acquired data, a quality filter with an error cutoff of 1% was applied to retain accurate data, and this process led to the discovery of 2 million independent DNA sequences. A minimum redundancy of 2 for the discovered peptides also indicated a high sequence recovery of the selected peptides. Due to a minimum of 3 base differences designed in the tags, the error of peptide sequences due to sequencing could be as low as 0.05%. The discovered DNA sequences were found to be in the expected format of the DEPCIL library (Fig. S8C, ESI†) and were decoded with tools published on GitHub (https://github.com/sunghunbae/decode). Duplication statistics were analyzed and sorted according to the discovered population to indicate the enrichment of the discovered peptides. Details of the data analysis procedure was reported previously. Highly populated peptides showed enrichment compared with the corresponding population in the native library, indicating a strong selection enforcement (Fig. S8D and E, ESI†).
Characterizing discovered inhibitors
The most highly enriched peptides were selected for synthesis and tested for in vitro potency to inhibit the 3CL protease. Multiple synthesis routines were performed to discover one that could achieve the desired peptides in high yield. Fmoc-protected P1-VS was initially used as an amino acid and loaded onto Wang resin. However, due to the low reactivity of the phenol hydroxyl group, the yield of loading did not reach a level sufficient for peptide synthesis. Consequently, we decided to couple the P1-VS motif to the peptide in solution. Peptide sequences comprising P2 to P5 residues were synthesized on chlorotrityl resin and cleaved with N terminus BOC protection and fully-protected side chains. The free C-terminal carboxyl acid was then activated for coupling with the P1-VS warhead to achieve the full-length inhibitor. After complete deprotection with a cleavage cocktail K, the crude peptide was precipitated with ethyl ether, and the precipitate was purified by HPLC to obtain the final covalent inhibitor peptides (Fig. S9, ESI†). Following lyophilization, the peptides were analyzed with LCMS to confirm purity and molecular weight, ensuring suitability for the activity test (Table S3, ESI†). To assess the potency in inhibiting the 3CL protease, peptide solutions were diluted to 10 μM and incubated with the 3CL protease, allowing covalent binding. After coincubation at 37 °C for 10 min, the fluorogenic substrate Dabcyl-KTSAVLQ↓SGFRKM-Glu(Edans) of 3CL was added, and the fluorescence signal was monitored to indicate the residual activity of hydrolyzing the native sequence. The top 10 most enriched peptides were tested for inhibitory activity at a concentration of 10 μM, with the majority showing weak inhibition. The most enriched peptide sequence, P3C-1, inhibited at 98% (Fig. 2A). We suspect that the prevalence of weak binders may stem from bias resulting from library distribution and PCR amplification of DNA tags. This bias issue is less pronounced in other selection technologies such as phage display, mRNA display, and yeast display, which entail amplification and competitive binding across multiple selection rounds. A correlation trend of selection frequency and inhibition rate could be observed (Fig. 2B and C). To determine the half-inhibition potency, the inhibiting peptide was further subjected to kinetic test, and the half inhibitory ratio (IC50) was calculated. Peptide P3C-1, with a sequence of Gln-Val-Orn-Leu-(3S)-γ-lactam alanine-VS, exhibited the highest potency with an IC50 of 1.60 μM after 10 min of pre-incubatio. | ||
| Fig. 2 The sequence of enriched peptides and the characterization of discovered inhibitors. (A) Top 10 most enriched peptide sequences discovered in the 3CL screening. Amino acid names used in each building blocks (BBs) were indicated. (B) Selection frequencies of the top 10 discovered peptides. (C) Inhibition rate of the inhibitors carrying the sequence of the top 10 sequences. | ||
Positional preference of 3CL
In addition to the inhibitor discovered by DEPCIL screening, the properties of the substrate binding pockets were even more important to uncover. Since the increased binding of peptides could be a contributor to facilitating the rapid reaction between DEPCIL members and the target protease, we analyzed inhibition correlated with mutations at individual residues, for studying protease substrate sites individually. Based on the sequence of peptide P3C-1, single mutation peptides with variations at P2 to P5 were selected from the discovered peptides. Each series of peptides were calculated for discovery frequency, and the inhibition rate was tested for the synthesized peptide inhibitors (Fig. S10, ESI†). As expected, the results indicated that the P2 amino acid (corresponding to building block1, BB1) is crucial for the binding of peptides to 3CL, and the replacement of leucine with other amino acids almost eliminated inhibitor activity (Fig. 3A and B). In contrast, the S3 pocket (corresponding to BB2) can adapt to quite a range of amino acids; the replacement with amino acids of different properties did not dramatically change the activity of the mutant peptides (Fig. 3C and D). The S4 pocket (corresponding to BB3) of 3CL is also quite conservative, and valine was the only potent peptide discovered in the selections. The discovery rate of the valine substituent also dominated (Fig. 3E and F). The S5 pocket (corresponding to BB4) also accepts a quite range of amino acids with different properties (Fig. 3G and H), due to its remote location from the active site and open position to solvent exposure.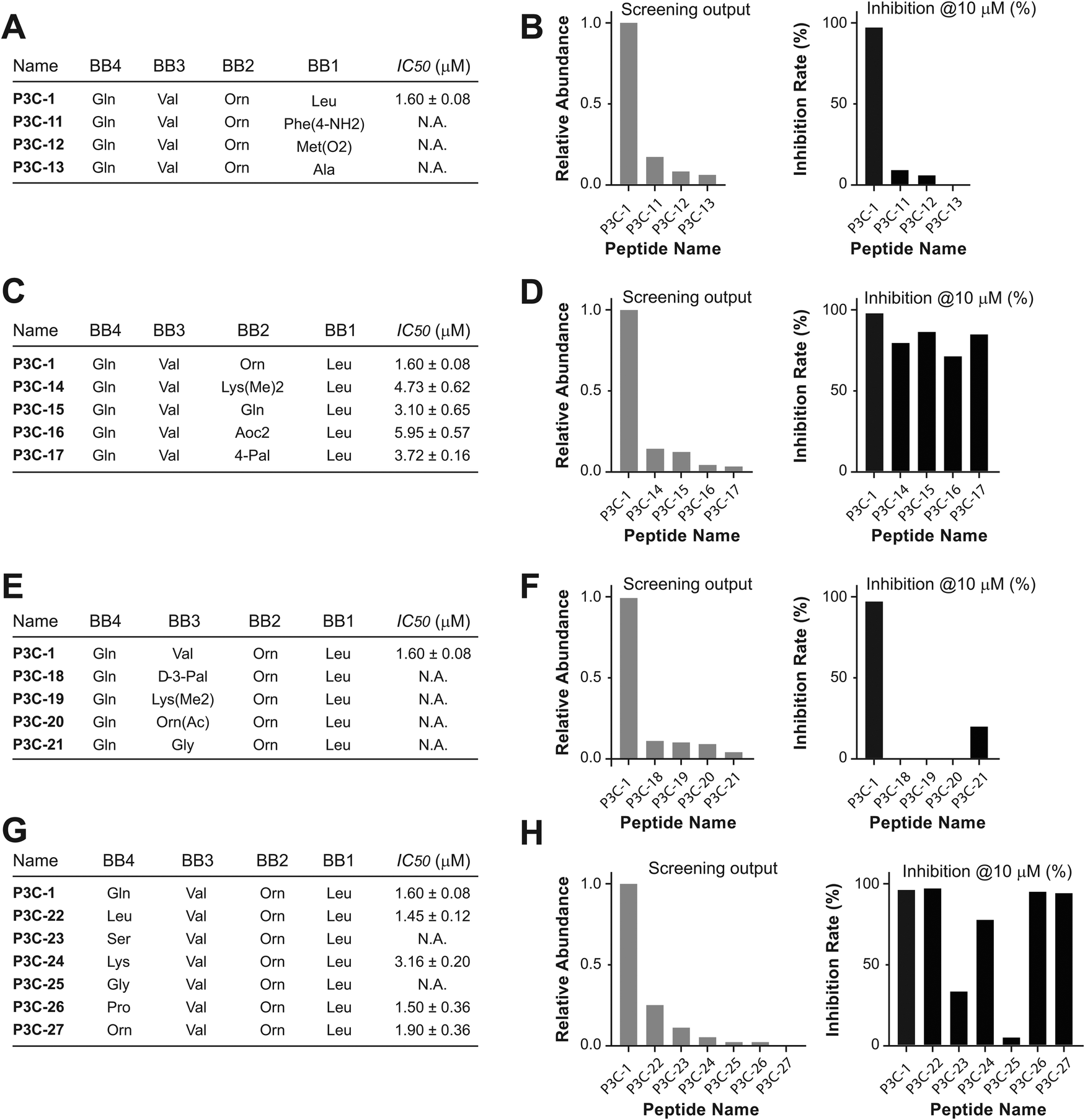 | ||
| Fig. 3 Analysis of subsite characteristics by testing inhibitors with variants at P2–P5 positions. (A), (C), (E) and (G) Discovered peptides with variations in the P2, P3, P4, and P5 residues for comparing their positional preferences. (B), (D), (F) and (H) Comparison of the discovery (left) and inhibition (right) rates of peptide inhibitors with corresponding mutations. | ||
Comparison with native peptide
To better understand the efficiency of DEPCIL for discovering better inhibitors, the discovered inhibitors were compared with naturally evolved peptide sequence. Although there are a number of covalent inhibitors of 3CL protease, we chose the naturally evolved peptide sequence due to its well-established structure activity relationship (SAR). The native substrate sequence of the 3CL peptide, the peptides composing P1–P5 of the 3CL native substrate protein, were conjugated with VS for the generation of a control peptide. To minimize the difference caused by S1 site binding, the P1 glutamine from the native peptide was also converted to (3S)-γ-lactam alanine for a comparison of the binding properties of peptide sequences selected from the developed DECPIL library (Fig. 4A). P3C-1 presented a conserved P2 leucine as the native sequence. The replacement of native alanine with noncanonical Orn at the P3 position, alanine with valine at the P4 position, and serine with glutamine at the P5 position enhanced the inhibition 1.5-fold (Fig. 4B). The most potent inhibitor showed a similar sequence as the natural substrate with critical residue conserved. These data suggested that the DEPCIL technology can identify conformations that can present both key anchor residues and less critical residue for binding to the target.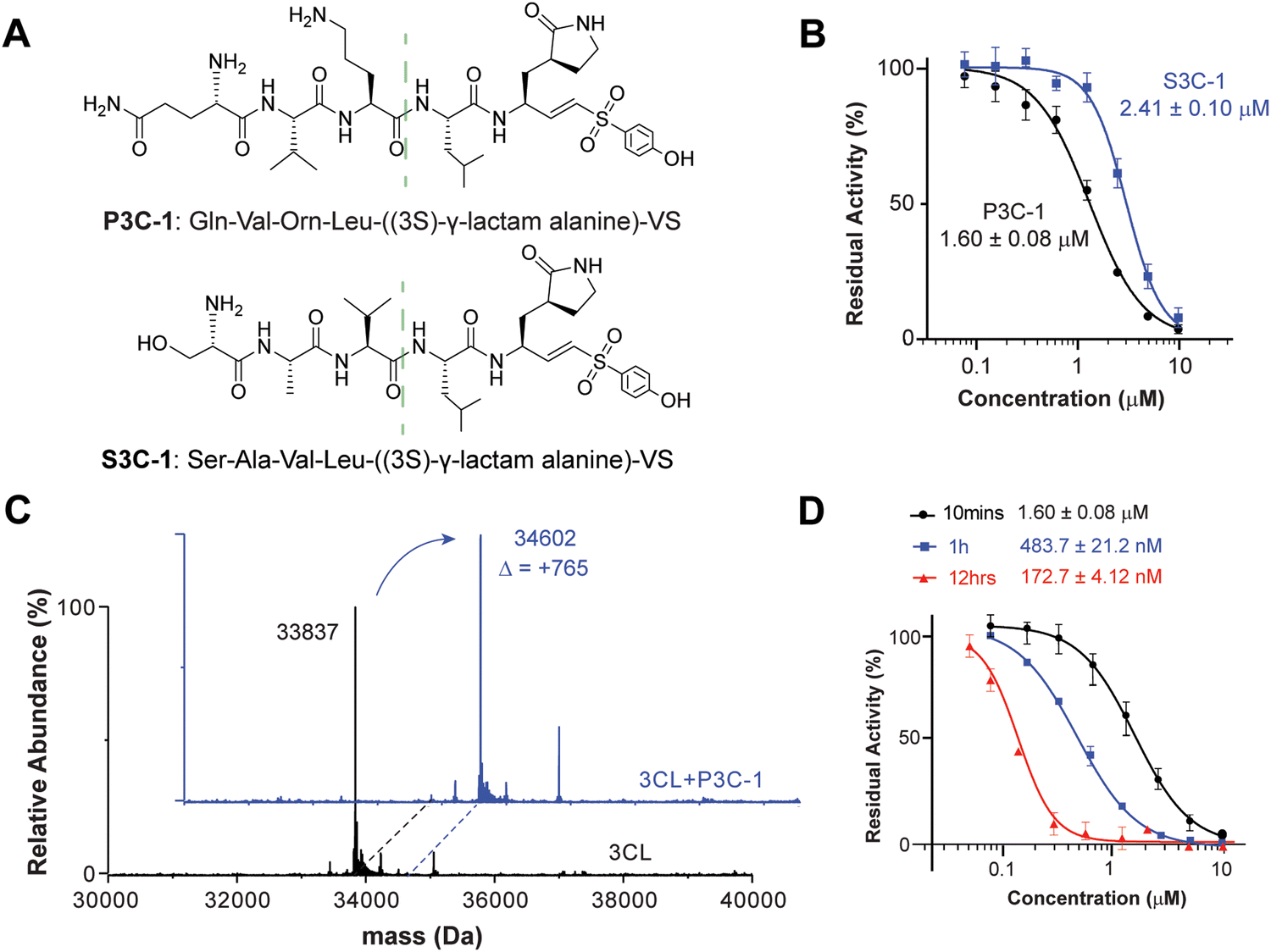 | ||
| Fig. 4 Comparison with native 3CL substrate-based inhibitor. (A) Chemical structure of the inhibitor discovered by DEPCIL screening (top) and the native sequence inhibitor (bottom), comprising the natural substrate sequence with substitution of the P1 residue for (3S)-γ-lactam alanine and the incorporation of vinyl sulfone at the C-terminus. The structures shared a common C-terminus structure, separated by a green dashed line. (B) Inhibition curves for both inhibitors. (C) Analysis of the binding properties of the developed covalent 3CL protease inhibitor, showing the inactive mass of 3CL protease before (black) and after (blue) the incubation with developed covalent inhibitors. (D) Time-dependent inhibition of the 3CL protease by compound P3C-1. The kinetics of residual protease activity of 3CL treated with a series of concentrations of P3C-1 for pre-incubation times of 10 minutes, 1 hour, and 16 hours were plotted, and the IC50 values were calculated to be 1.60 ± 0.08 μM, 483.7 ± 21.2 nM, and 172.7 ± 4.12 nM, respectively. | ||
Interactions between noncanonical peptide and 3CL protease
To determine the covalent binding ability of P3C-1 with 3CL protease, mass spectrometry of intact protein was used to determine the relative molecular weight of the target. By comparing the molecular weight changes of the protein before and after inhibition, it was possible to determine whether covalent binding had occurred. The experimental results revealed that in the Tris–HCl buffer (pH 7.4), the covalent functional group VS of P3C-1 reacted with active site cysteine to form the C–S bond (Fig. 4C). Upon pre-incubation of P3C-1 with 3CL for 10 minutes, 1 hour, and 12 hours, the residual protease activity significantly decreased, indicating an IC50 of 172.7 ± 4.12 nM after 12 hours of pre-incubation. The time-dependent inhibition further confirmed the covalent binding between P3C-1 and the 3CL protease (Fig. 4D).MD study of interactions between inhibitor and the 3CL protease
To determine how the discovered noncanonical peptide can better bond to the 3CL protease, we analyzed this peptide-protein interaction using structural information. We applied molecular dynamics (MD) to simulate the interactions between P3C-1 and the 3CL protease, using the high-resolution structure of 3CL protease bound to a five-residue substrate peptide as a template (7T70, chain B). While the power of MD calculations for the de novo prediction of ligand-protein interactions are limited, there are several reasons why MD simulations are nonetheless informative for predicting the interactions between P3C-1 and 3CL protease. First, the covalent linkage between P3C-1 and 3CL protease largely defines the orientation in which the peptide can bind to the target protein, thus narrowing the chemical space to explore and improving the probability of discovering relevant interactions. Second, the critical residues in P3C-1 are similar to the native 3CL substrate sequence of SAVLQ, allowing us to generate a more reliable initial conformation of P3C-1. To perform the MD simulations, we covalently linked the VS of P3C-1 to the active-site cysteine of 3CL protease to generate input files that were loaded into the Assisted Model Building with Energy Refinement (AMBER) biomolecular simulation package. The complex structure was subjected to MD simulation in explicit TIP3P solvent models with ff14SB and general AMBER force field (GAFF) parameter sets. After 100 ns of production simulation, the binding of P3C-1 to 3CL protease reached a constant and stable state. The energy of the system remained constant, and the structure converged to a single low-energy state for the complex. This predicted structure showed P3C-1 stably bound to the 3CL protease in the primary substrate-binding pocket (Fig. 5A). The side chains of residues besides the N-terminal Gln1 adopted a conformation similar to that of the native substrate peptide. The side chain of P1 (3S)-γ-lactam ring adopted the S1 pocket of 3CL protease and aligned with the native substrate P1 glutamine (Fig. 5B). Leu in P3C-1 matches the positioning of the P2 Leucine in the substrate peptide, forming a hydrophobic center with H41, Y54 and M165 that forms the S2 pocket of the 3CL protease. Val replaced the P4 Alanine in the substrate peptide by forming multiple hydrophobic interactions with M165, L167, and F185 in the S4 pocket. This could explain the preference for hydrophobic residues at P2 and P4 positions. The substitution of native Ala in the P3 position with Orn introduced extra hydrogen binding with E166, explaining the increased efficiency of P3C-1 binding. The flexible end of P3C-1 appeared to have a weak interaction with Q189, which could elucidate the relatively broad tolerance profile observed for the P5 amino acids. However, the optimized MD structure supports the finding of the contribution of noncanonical amino acids to strengthening the interaction with 3CL protease and enhancing the binding of P3C-1 to the active site (Fig. 5C and D). Overall, the MD simulations confirmed that stable and low-energy conformations can be found in which the binding of selected P3C-1 peptide aligns the C-terminal at the center of the active site and enforces the covalent modification by the active-site cysteine residue. | ||
| Fig. 5 MD analysis of the binding of the P3C-1 inhibitor at the active site of 3CL protease, compared with the binding of native substrate sequences. (A) AMBER simulated structure of P3C-1 bound to the 3CL protease active site, with a covalent bond formed between VS warhead and active site cysteine 145. (B) Aligned structures of simulated P3C-1 and 3CL substrate peptide bound at the active site. (C) Diagram representing the interaction between P3C-1 and 3CL protease by formed hydrophobic (red radiations), H-bonding (green dashed lines), and electrostatic interactions (red dashed lines). (D) Diagram representing the interaction between 3CL protease with the natural substrate bound by formed various interactions. | ||
Conclusions
In summary, we believe that our DEPCIL screening approach could be used to construct highly diverse non-canonical covalent inhibitor libraries and applied to discovering peptides for inhibiting proteases and unveiling substrate pocket preferences. Our results suggest that this DEPCIL approach works well for 3CL protease by applying the vinyl sulfone warhead, and it is likely that the same library could be used for screening against other targets that contains a reactive cysteine. For example, a number of proteins were found to have mutation with free cysteine on surface and DEPCIL can also be applied for screening for covalent inhibitors.38,39 This DEPCIL strategy could be expanded to similar methods for constructing covalent inhibitor libraries for profiling a broad range of proteases by substituting the vinyl sulfone warhead with corresponding reactive electrophiles. Thus, this DEPCIL screening approach has a high potential for enhancing the discovery and screening of non-canonical covalent ligands in various chemical biology research fields.Materials and methods
Chromatographic separations were performed by automated flash chromatography using SepaBean T equipped with SepaFlash UltraPure irregular silica columns (40–64 μm, 60 Å, Santai, Science Inc., Shanghai). Commercial plates (HTPLC Silica Gel 6-GF254, Leyan, Qingdao) were used for analytical thin-layer chromatography to follow the progress of reactions. Unless otherwise specified, 1H NMR spectra and 13C NMR spectra were obtained on a Varian Mercury 400-MHz console connected with an Oxford NMR AS400 actively shielded magnet spectrometer at room temperature. Chemical shifts for 1H or 13C are given in ppm (δ) relative to tetramethylsilane as an internal standard. Mass spectra (m/z) of chemical compounds, peptides, and proteins were recorded on a 1260 Infinity II LC 6230 TOF MS system (Agilent Technologies, America) or an ACQUITY Arc SQ Detector 2 system (Waters, America). Reverse-phase HPLC (RP-HPLC) purifications were performed using a 1260 Infinity II preparative HPLC equipped with a semi-prep C18 column (5 μm, C18, 100 Å, 250 × 10 mm liquid chromatography column; Agilent Technologies, America) eluted over a linear gradient from 95% solvent A (water and 0.1% trifluoroacetic acid) to 100% solvent B (acetonitrile and 0.1% trifluoroacetic acid).Unless otherwise noted, chemicals were purchased from Sigma Aldrich, J&K Chemical, or Sinopharm and were used without further purification. Protected Fmoc-amino acids and coupling reagents were purchased from Bidepharm and GL Biochem (Shanghai, China) Ltd. DPAL were purchased from Generalbio (Anhui, China). RinkAmide-MBHA resin (0.458 mmol g−1) was purchased from GL Biochem (Shanghai) Ltd (Lot. Nr. GLS220524-4910).
DEPCIL construction
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 30 minutes to reduce the volume to less than 1 mL. This wash step was repeated three additional times to remove impurities. Finally, the remaining solution was recovered, and quantification was performed based on the DNA absorbance using a Nanodrop spectrophotometer.
000 rpm for 30 minutes to reduce the volume to less than 1 mL. This wash step was repeated three additional times to remove impurities. Finally, the remaining solution was recovered, and quantification was performed based on the DNA absorbance using a Nanodrop spectrophotometer.
Recombinant 3CL protein purification
A gene encoding SARS-CoV-2 Mpro was synthesized and subcloned into the vector pET28b. To obtain the main protease with authentic N- and C-termini, the construct designed for SARS-CoV-2 Mpro followed a similar approach as previously describe,31 and the gene sequence was verified by Sanger sequencing. The sequence-verified expression vector was then transformed into E. coli strain BL21 (DE3). A transformed single clone was cultured at 37 °C in 10 mL LB medium with kanamycin (50 μg mL−1) for 8 h and diluted 1:100 into 500 mL fresh LB medium supplied with 50 μg mL−1 kanamycin. Induction of the overexpression of the Mpro gene was initiated by adding 0.5 mM isopropyl-D-thiogalactoside (IPTG) at 1 °C when the OD600 reached 0.6. After 16 h, cells were harvested by centrifugation at 5000g, 4 °C for 8 min. The pellets were resuspended in 50 mL buffer R (20 mM Tris, pH 7.8, 150 mM NaCl) and lysed by sonication on ice. The lysate was clarified by centrifugation at 10000g at 4 °C for 30 min, and the supernatant was loaded onto a HisTrap column. The column was washed with 15 mL buffer A (buffer R with 50 mM imidazole) to remove unspecific binding proteins, followed by elution using 5 mL buffer B (buffer R with 500 mM imidazole). HiPrep Sephacryl S-100 HR gel-filtration chromatography (GE Healthcare) was employed to further purify the fractions with a running condition of buffer R. Fractions containing the target protein were pooled and mixed with PreScission protease (P2302, Beyotime) following the manufacturer's instructions, resulting in the target protein with authentic N- and C-termini. The PreScission-treated Mpro was applied to BeyoGold™GST-tag Purification Resin (P2251, Beyotime) and nickel columns to remove the GST-tagged PreScission protease, the His-tag, and proteins with uncleaved His-tag. Fractions containing the target protein were pooled and concentrated using Amicon Ultra 15 centrifugal filters (10 kD, Merck Millipore) at 2300g, 4 °C. The purified protein concentration was determined via BCA protein assay and was >90% pure, as estimated by a 12% SDS-PAGE.
Covalent peptide synthesis
Measurement of SARS-CoV-2 3CL protease inhibition
The potency of peptides to inhibit SARS-CoV-2 3CL protease was quantified by measuring its residual activity to hydrolyze a fluorogenic substrate peptide designed from its native substrate protein. The Dabcyl-KTSAVLQ↓SGFRKM-Glu(Edans) substrate was synthesized according to a previous publication,32 and the increase of Edans fluorescence signal was recorded to reflect the activity of 3CL protease. The half inhibition constants IC50 were determined by incubating different concentrations of peptides with fixed concentration of 3CL protease (25 nM), and the enzymatic assays were performed at 25 °C in 50 mM Tris, pH 7.3, 1 mM EDTA, and 1 mM DTT buffer. In a 384-well black plate was placed 20 μL protease and 20 μL of different concentrations of peptides were added (0–10 μM final concentration at 2-fold serial dilutions). After being incubated at 37 °C for 10 min, 20 μL of 150 μM substrate was added to allow hydrolysis by the residually active 3CL protease. Fluorescent signal (excitation 340 nm, emission 490 nm) was read in 10-min intervals, which were plotted against time to calculate the rate of 3CL hydrolysis in the presence of different peptide inhibitors. The acquired data were analyzed in the GraphPad Prism software 9.0 and fit according to Y = bottom + (top–bottom)/(1 + 10^((logIC50 − X) × HillSlope)) equation to calculate the IC50 values.
Peptide inhibitors mechanism research
To assess the covalent binding ability of the synthesized peptide molecules to SARS-CoV-2 3CLpro, Electrospray Ionization Quadrupole Time-of-Flight Mass Spectrometry (ESI Q-TOF MS) was employed to determine the exact molecular mass of the protein. By comparing the relative molecular mass changes of the protein before and after inhibitor binding, the occurrence of covalent binding can be ascertained. The experimental setup involved reacting SARS-CoV-2 Mpro at a concentration of 4 μM with inhibitors at a concentration of 100 μM. The reaction was conducted in a buffer consisting of 50 mM Tris (pH = 7.3), 1 mM EDTA, and 1 mM DTT. After 12 hours of reaction time, 10 μL aliquots were extracted and subjected to electrospray mass spectrometry using a Waters Acquity ESI Q-TOF instrument. The obtained mass spectra were then analyzed to determine any changes in the molecular mass of the protein resulting from inhibitor binding. This methodology allows for the identification and confirmation of covalent binding events between the synthesized peptide inhibitors and SARS-CoV-2 3CLpro through precise mass measurements.AMBER molecular dynamics simulation
The initial structure of the peptide inhibitor was minimized with the Gaussian 16 software package and parameterized with GAFF. Tleap was used for generating MD simulation input files, and the linkages between cyan and cysteine residues in the active site of 3CLpro were manually specified. P3C-1 inhibitor adopting random positions were built. Force field parameters for the 3CLpro-inhibitor complex and surrounding atoms were taken from GAFF, and partial charges were calculated with the HF/6-31G* RESP charge method. All structures were first minimized by holding the conformation of the 3CL protease, followed by whole-system minimization. Then, the system was heated to 300 K before equilibration on the whole system. The production equilibration was run at 300 K with constant pressure and periodic boundary for 100 ns. Langevin dynamics was used to control the temperature using a collision frequency of 1.0 collision per ps. The final structures were analyzed by using the VMD package, and a common final structure was presented by using the pymol package. All the calculations were performed by the AMBER biomolecular simulation package (code available at https://ambermd.org/) and were compiled in a GPU accelerated workstation.Data availability
Supplementary information and chemical compound information are available in the online version of the paper.Author contributions
S. C., M. B. and Y. X. conceived the project and designed the experiments. Y. X., H. Z., and Y. W. performed the experiments and analyzed the data. All authors wrote the manuscript. S. C. obtained funding for the work.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by The Hundred Talents Program of the Pioneer Initiative from Chinese Academy of Sciences and the Distinguished Young Scholars of the National Natural Science Foundation of China (overseas) (to S. C.).References
- M. Egeblad and Z. Werb, Nat. Rev. Cancer, 2002, 2, 161–174 CrossRef CAS PubMed.
- N. D. Rawlings, F. R. Morton and A. J. Barrett, Nucleic Acids Res., 2006, 34, D270–D272 CrossRef CAS PubMed.
- A. Eatemadi, H. T. Aiyelabegan, B. Negahdari, M. A. Mazlomi, H. Daraee, N. Daraee, R. Eatemadi and E. Sadroddiny, Biomed. Pharmacother., 2017, 86, 221–231 CrossRef CAS PubMed.
- Z. Y. Cai, A. L. Zhang, S. Choksi, W. H. Li, T. Li, X. M. Zhang and Z. G. Liu, Cell Res., 2016, 26, 886–900 CrossRef CAS PubMed.
- M. Patron, H. G. Sprenger and T. Langer, Cell Res., 2018, 28, 296–306 CrossRef CAS PubMed.
- J. Singh, R. C. Petter, T. A. Baillie and A. Whitty, Nat. Rev. Drug Discovery, 2011, 10, 307–317 CrossRef CAS PubMed.
- R. A. Bauer, Drug Discovery Today, 2015, 20, 1061–1073 CrossRef CAS PubMed.
- M. Bon, A. Bilsland, J. Bower and K. McAulay, Mol. Oncol., 2022, 16, 3761–3777 CrossRef CAS PubMed.
- R. Lagoutte, R. Patouret and N. Winssinger, Curr. Opin. Chem. Biol., 2017, 39, 54–63 CrossRef CAS PubMed.
- J. Johansen-Leete, S. Ullrich, S. E. Fry, R. Frkic, M. J. Bedding, A. Aggarwal, A. S. Ashhurst, K. B. Ekanayake, M. C. Mahawaththa, V. M. Sasi, S. Luedtke, D. J. Ford, A. J. O'Donoghue, T. Passioura, M. Larance, G. Otting, S. Turville, C. J. Jackson, C. Nitsche and R. J. Payne, Chem. Sci., 2022, 13, 3826–3836 RSC.
- P. Lito, M. Solomon, L. S. Li, R. Hansen and N. Rosen, Science, 2016, 351, 604–608 CrossRef CAS PubMed.
- H. Kim, Y. S. Hwang, M. Kim and S. B. Park, RSC Med. Chem., 2021, 12, 1037–1045 RSC.
- D. J. Augeri, J. A. Robl, D. A. Betebenner, D. R. Magnin, A. Khanna, J. G. Robertson, A. Y. Wang, L. M. Simpkins, P. Taunk, Q. Huang, S. P. Han, B. Abboa-Offei, M. Cap, L. Xin, L. Tao, E. Tozzo, G. E. Welzel, D. M. Egan, J. Marcinkeviciene, S. Y. Chang, S. A. Biller, M. S. Kirby, R. A. Parker and L. G. Hamann, J. Med. Chem., 2005, 48, 5025–5037 CrossRef CAS PubMed.
- C. Kaya, I. Walter, S. Yahiaoui, A. Sikandar, A. Alhayek, J. Konstantinovic, A. M. Kany, J. Haupenthal, J. Köhnke, R. W. Hartmann and A. K. H. Hirsch, Angew. Chem., Int. Ed., 2022, 61, e202112295 CrossRef CAS PubMed.
- R. D. Healey, E. M. Saied, X. J. Cong, G. Karsai, L. Gabellier, J. Saint-Paul, E. Del Nero, S. Jeannot, M. Drapeau, S. Fontanel, D. Maurel, S. Basu, C. Leyrat, J. Golebiowski, G. Bossis, C. Bechara, T. Hornemann, C. Arenz and S. Granier, Angew. Chem., Int. Ed., 2022, 61, e202109967 CrossRef CAS PubMed.
- S. Y. Chen, S. Lovell, S. M. Lee, M. Fellner, P. D. Mace and M. Bogyo, Nat. Biotechnol., 2021, 39, 490–498 CrossRef CAS PubMed.
- Y. Q. Deng, J. Z. Peng, F. Xiong, Y. A. Song, Y. Zhou, J. F. Zhang, F. S. Lam, C. Xie, W. Y. Shen, Y. R. Huang, L. Meng and X. Y. Li, Angew. Chem., Int. Ed., 2020, 59, 14965–14972 CrossRef CAS PubMed.
- L. J. Li, M. B. Su, W. W. Lu, H. Z. Song, J. X. Liu, X. Wen, Y. R. Suo, J. J. Qi, X. M. Luo, Y. B. Zhou, X. H. Liao, J. Li and X. J. Lu, ACS Med. Chem. Lett., 2022, 13, 1574–1581 CrossRef CAS PubMed.
- Z. R. Zhu, L. C. Grady, Y. Ding, K. E. Lind, C. P. Davie, C. B. Phelps and G. Evindar, SLAS Discovery, 2019, 24, 169–174 CrossRef CAS PubMed.
- R. Ge, Z. Shen, J. Yin, W. Chen, Q. Zhang, Y. An, D. Tang, A. L. Satz, W. Su and L. Kuai, SLAS Discovery, 2022, 27, 79–85 CrossRef CAS PubMed.
- J. P. Guilinger, A. Archana, M. Augustin, A. Bergmann, P. A. Centrella, M. A. Clark, J. W. Cuozzo, M. Däther, M. A. Guié, S. Habeshian, R. Kiefersauer, S. Krapp, A. Lammens, L. Lercher, J. L. Liu, Y. B. Liu, K. Maskos, M. Mrosek, K. Pflügler, M. Siegert, H. A. Thomson, X. Tian, Y. Zhang, D. L. K. Makino and A. D. Keefe, Bioorg. Med. Chem., 2021, 42, 116223 CrossRef CAS PubMed.
- A. Hegyi and J. Ziebuhr, J. Gen. Virol., 2002, 83, 595–599 CrossRef PubMed.
- J. Ziebuhr, E. J. Snijder and A. E. Gorbalenya, J. Gen. Virol., 2000, 81, 853–879 CrossRef CAS PubMed.
- K. Vandyck and J. Deval, Curr. Opin. Virol., 2021, 49, 36–40 CrossRef CAS PubMed.
- D. R. Owen, C. M. N. Allerton, A. S. Anderson, L. Aschenbrenner, M. Avery, S. Berritt, B. Boras, R. D. Cardin, A. Carlo, K. J. Coffman, A. Dantonio, L. Di, H. Eng, R. Ferre, K. S. Gajiwala, S. A. Gibson, S. E. Greasley, B. L. Hurst, E. P. Kadar, A. S. Kalgutkar, J. C. Lee, J. Lee, W. Liu, S. W. Mason, S. Noell, J. J. Novak, R. S. Obach, K. Ogilvie, N. C. Patel, M. Pettersson, D. K. Rai, M. R. Reese, M. F. Sammons, J. G. Sathish, R. S. P. Singh, C. M. Steppan, A. Stewart, J. B. Tuttle, L. Updyke, P. R. Verhoest, L. Q. Wei, Q. Y. Yang and Y. A. Zhu, Science, 2021, 374, 1586–1593 CrossRef CAS PubMed.
- W. H. Dai, B. Zhang, X. M. Jiang, H. X. Su, J. A. Li, Y. Zhao, X. Xie, Z. M. Jin, J. J. Peng, F. J. Liu, C. P. Li, Y. Li, F. Bai, H. F. Wang, X. Cheng, X. B. Cen, S. L. Hu, X. N. Yang, J. Wang, X. Liu, G. F. Xiao, H. L. Jiang, Z. H. Rao, L. K. Zhang, Y. C. Xu, H. T. Yang and H. Liu, Science, 2020, 368, 1331–1335 CrossRef CAS PubMed.
- L. Boike, N. J. Henning and D. K. Nomura, Nat. Rev. Drug Discovery, 2022, 21, 881–898 CrossRef CAS PubMed.
- B. J. Pinch, Z. M. Doctor, B. Nabet, C. M. Browne, H. S. Seo, M. L. Mohardt, S. Kozono, X. L. Lian, T. D. Manz, Y. J. Chun, S. Kibe, D. Zaidman, D. Daitchman, Z. C. Yeoh, N. E. Vangos, E. A. Geffken, L. Tan, S. B. Ficarro, N. London, J. A. Marto, S. Buratowski, S. Dhe-Paganon, X. Z. Zhou, K. P. Lu and N. S. Gray, Nat. Chem. Biol., 2020, 16, 979–987 CrossRef PubMed.
- W. J. Zhou, D. Ercan, L. Chen, C. H. Yun, D. N. Li, M. Capelletti, A. B. Cortot, L. Chirieac, R. E. Iacob, R. Padera, J. R. Engen, K. K. Wong, M. J. Eck, N. S. Gray and P. A. Jänne, Nature, 2009, 462, 1070–1074 CrossRef CAS PubMed.
- N. Liu, Y. C. Zhang, Y. S. Lei, R. Wang, M. M. Zhan, J. B. Liu, Y. H. An, Y. Q. Zhou, J. Zhan, F. Yin and Z. G. Li, J. Med. Chem., 2022, 65, 876–884 CrossRef CAS PubMed.
- A. Angelini, L. Cendron, S. Chen, J. Touati, G. Winter, G. Zanotti and C. Heinis, ACS Chem. Biol., 2012, 7, 817–821 CrossRef CAS PubMed.
- S. Chen, D. Bertoldo, A. Angelini, F. Pojer and C. Heinis, Angew. Chem., Int. Ed., 2014, 53, 1602–1606 CrossRef CAS PubMed.
- C. J. Vickers, G. E. González-Páez and D. W. Wolan, ACS Chem. Biol., 2013, 8, 1558–1566 CrossRef CAS PubMed.
- R. Ahmadi and S. Emami, Eur. J. Med. Chem., 2022, 234, 114255 CrossRef CAS PubMed.
- L. L. Zhang, D. Z. Lin, Y. Kusov, Y. Nian, Q. J. Ma, J. Wang, A. von Brunn, P. Leyssen, K. Lanko, J. Neyts, A. de Wilde, E. J. Snijder, H. Liu and R. Hilgenfeld, J. Med. Chem., 2020, 63, 4562–4578 CrossRef CAS PubMed.
- R. A. Houghten, C. Pinilla, S. E. Blondelle, J. R. Appel, C. T. Dooley and J. H. Cuervo, Nature, 1991, 354, 84–86 CrossRef CAS PubMed.
- T. Franch, M. D. Lundorf, S. N. Jakobsen, E. K. Olsen, A. L. Andersen, A. Holtmann, A. H. Hansen, A. M. Sørensen, A. Goldbech, D. de Leon, D. K. Kaldor, F. A. Sløk, G. N. Husemoen, J. Dolberg, K. B. Jensen, L. Petersen, M. Nørregaard-Madsen, M. A. Godskesen, S. S. Glad, S. Neve, T. Thisted, T. T. A. Kronborg, C. K. Sams, J. Felding, P. O. Freskgard, A. H. Gouliaev and H. Pedersen, US Pat., US11702652B2, 2023.
- D. Q. Zhao, Y. Liu, F. C. Yi, X. Zhao and K. Lu, Eur. J. Med. Chem., 2023, 259, 115698 CrossRef CAS PubMed.
- J. M. Ostrem, U. Peters, M. L. Sos, J. A. Wells and K. M. Shokat, Nature, 2013, 503, 548–551 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cb00097h |
| This journal is © The Royal Society of Chemistry 2024 |