
Macrophage-hitchhiked arsenic/AB bionic preparations for liver cancer†
Ce
Li
a,
Ji Ping
Zhang
 a,
Yi Chao
Yuan
a,
Yong Qin
Zhao
*a,
Hang Sheng
Zheng
*a and
Zhi Hong
Zhu
*ab
a,
Yi Chao
Yuan
a,
Yong Qin
Zhao
*a,
Hang Sheng
Zheng
*a and
Zhi Hong
Zhu
*ab
aCollege of Pharmaceutical Science, Zhejiang Chinese Medical University, Hangzhou, China. E-mail: zzhjanny@163.com
bAcademy of Chinese Medical Science, Zhejiang Chinese Medical University, Hangzhou, China
First published on 8th November 2023
Abstract
Macrophage-hitchhiked arsenic/AB bionic preparations were developed to improve the therapeutic effect on liver cancer by means of the tumor-targeting ability of macrophages in vivo. In vitro and in vivo cellular uptake assays demonstrated that arsenic/AB, with negatively charged particles of around 100–200 nm size, could hitchhike to macrophages. Dissolution experiments of arsenic/AB showed that arsenic/AB could delay the release of arsenic and ensure the safety of macrophages during its transport. Histological examination confirmed the safety of the preparations for major organs. In vivo distribution experiment showed that the arsenic/AB bionic preparations could rapidly accumulate in tumors, and in vivo treatment experiment showed a significant tumor inhibition of arsenic/AB. The therapeutic mechanism of liver cancer might be that the arsenic/AB bionic preparations could inhibit tumor growth by reducing inflammatory response and inhibiting CSF1 secretion to block CSF1R activation to induce more differentiation of tumor-associated macrophages (TAMs) towards the anti-tumor M1 phenotype. Therefore, we concluded that the arsenic/AB bionic preparations could improve the distribution of arsenic in vivo by hitchhiking on macrophages as well as make it have tumor targeting and deep penetration abilities, thus increasing the therapeutic effect of arsenic on liver cancer with reduced side effects.
Introduction
Liver cancer is the sixth most frequent cancer and the third leading cause of cancer death worldwide according to the Global Cancer Statistics 2020 report.1 The high mortality rate of liver cancer is mainly due to the high incidence and the lack of effective treatment.2 The current preferred treatment for liver cancer is surgical resection or chemotherapy drugs.3 However, recurrence and metastasis can easily occur due to non-sensitivity to chemotherapy drugs or failure to remove the entire cancer.4,5 Indeed, therapeutics with a new concept of liver cancer would be essential to achieve long-term remission.As recorded in the traditional Chinese Herbal Classics “Ben Cao Meng Sheng”, the clinical symptoms of liver cancer such as right epigastric pain and abdominal mass are consistent with the theory of “Wei Jia Ji Ju”. Arsenic has been widely used for eliminating lumps according to the theory. Research now indicates that arsenic, whose main precursor is As2O3, can induce tumor cell cytotoxicity or apoptosis6 and suppress liver cancer stem cells or metastasis.7 However, the use of arsenic is limited by its toxicity to normal tissue and the inability to achieve deep tumor penetration.8 Therefore, we hypothesized that the therapeutic effect of arsenic on liver cancer could be improved by optimizing the tumor distribution in vivo and enhancing tumor deep penetration.
Tumor-derived extracellular vesicular preparations can achieve deep tumor penetration by membrane fusion cell internalization as they can use markers that are similar to those on the surface of tumor cells to facilitate transport between tumors,9–12 but poor targeting is the drawback of extracellular vesicular preparations.13,14 Macrophages, with their ability of tumor homing, may be the solution for improving the tumor targeting of extracellular vesicular preparations.15–17 Therefore, giving full play to the advantages of tumor-derived extracellular vesicular preparations and macrophages is the key to improving the therapeutic effect of arsenic on tumors. Extracellular vesicles are broadly classified into three main subtypes based on their triggering mechanisms and biophysiological properties: microvesicles (MVs), exosomes (EVs), and apoptotic bodies (ABs).18,19 Compared to microvesicles and exosomes, ABs have a larger volume and specific surface area, increasing their ability to load more drugs and to better interact with the microenvironment or different cells.20–22 What's more, the surface of ABs also had an “eat me” signal that promoted its uptake by macrophages.23 Inspired by this, macrophages hitchhiked tumor cell-derived ABs may be a good drug delivery system based on their ability to load drugs and achieve deep tumor penetration. More importantly, the tumor targeting of tumor cell-derived ABs could be achieved by hitchhiking on macrophages in vivo according to the ability of macrophages to accurately target tumors.
In this work, an in vitro–in vivo targeted delivery system for arsenic was developed to improve the in vivo distribution and tumor targeting of arsenic. In vitro (Scheme 1), H22 cell-derived ABs were first obtained by incubating sodium arsenite with H22 cells to promote the secretion of ABs, and then arsenic-loaded ABs (arsenic/ABs) were obtained by the ultrasonic method. In vivo, arsenic/ABs could be rapidly hitchhiked to macrophages and delivered more effectively to the tumor based on the tumor-homing behavior of macrophages.
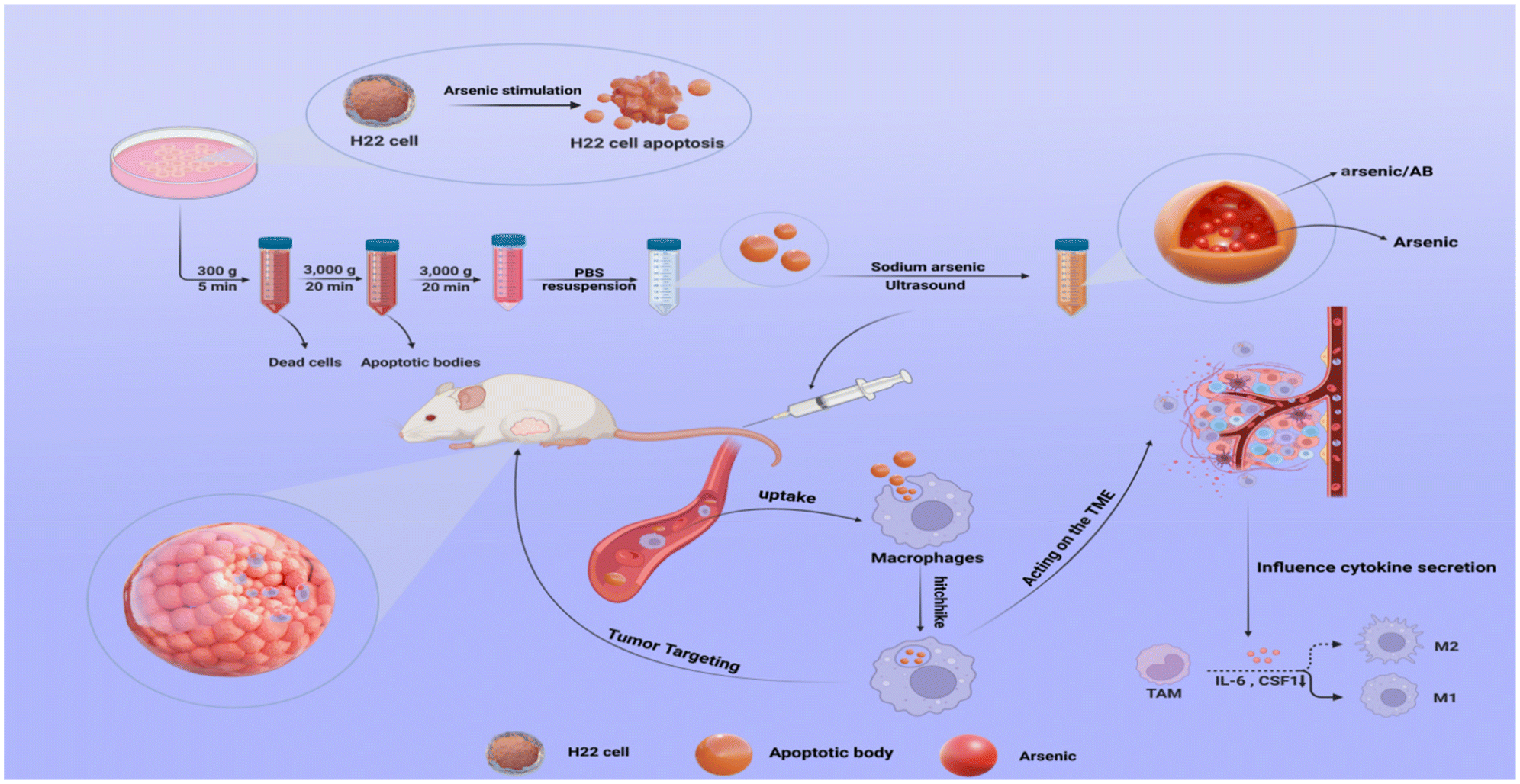 | ||
| Scheme 1 Schematic diagram of arsenic/AB bionic preparations for the enhancement of liver cancer therapy. H22 cells were first stimulated with sodium arsenite to produce ABs. arsenic/AB was obtained by ultrasound and was rapidly phagocytosed by circulating macrophages after tail vein injection in tumor-bearing KM mice. Using the natural tumor-homing tendency of macrophages, arsenic/AB could be effectively delivered into the inner regions of the tumor. In addition, arsenic/AB could affect the tumor microenvironment and induce TAM differentiation into an anti-tumor M1 phenotype by inhibiting the secretion of CSF1 and IL-6. | ||
Experimental
Materials and methods
Fluorescein isothiocyanate (FITC) (cat. no. F299199) was purchased from Aladdin Biochemical Technology (Shanghai, China). DIR (cat. no. Y18318-5 mg) was purchased from Shanghai Yuanye Bio-Technology Co., Ltd (Shanghai, China). 4,6-Diamidino-2-phenylindole dihydrochloride (DAPI) (cat. no. BS097) was purchased from Gosun Biotechnology Co., Ltd (Hangzhou, China). CD11b antibody (cat. no. 10R-CD11bgMS) was purchased from BioDee Biotechnology Co., Ltd (Beijing, China). PerCP/Cyanine5.5 anti-mouse Ly-6G/Ly-6C (Gr-1) (cat. no. 108427) and APC anti-mouse F4/80 (cat. no. 123115) were purchased from Dakewe Biotech Co., Ltd (Shenzhen, China). All chemicals are of analytical grade.
Mouse liver cancer cells (H22) and mouse monocyte macrophage leukemia cells (RAW264.7) were obtained from the Laboratory Animal Research Center, Zhejiang Chinese Medical University (Hangzhou, China).
Male Kunming (KM) mice (weight 25 ± 10 g) were provided by the Laboratory Animal Research Center, Zhejiang Chinese Medical University (Hangzhou, China). All experiments were conducted by following the guidelines for the care and use of animals as framed and supervised by Zhejiang Chinese Medical University. The ethical number was IACUC-20190527-02.
Methods
The yield of ABs. ABs were stained with Annexin V-FITC and then quantified by CytoFlex flow cytometry (Beckmann, USA) for determining the yield of ABs. The yield of ABs was calculated using the following equation: the number of ABs/the number of cells required to secrete ABs.
Transmission electron microscopy. The morphological characteristics of ABs were observed using a transmission electron microscope (TEM) (Hitachi H-7650, Japan).
NTA. We measured the Abs’ particle size and concentration using nanoparticle tracking analysis (NTA) at VivaCell Biosciences with ZetaView PMX 110 (Particle Metrix, Meerbusch, Germany) and the corresponding software ZetaView 8.04.02. Isolated AB samples were appropriately diluted using 1× PBS buffer (Biological Industries, Israel) (5.9 × 107 particles per mL) to measure the particle size and concentration. NTA measurement was recorded and analyzed at 11 positions. The ZetaView system was calibrated using 110 nm polystyrene particles. The temperature was maintained at around 23 °C and 30 °C.
Flow cytometry. Apoptotic markers on the surface of ABs were detected by AnnexinV-FITC and PI. The precipitates centrifuged at 300g and 3000g were stained with PI after resuspension with AnnexinV-FITC and detected by flow cytometry, respectively.
Western blot. Whole lysates of H22 cells or ABs isolated from H22 cells were prepared using the lysis buffer (Servicebio, G2002-100 ML) supplemented with protease and phosphatase inhibitors (Servicebio, G2006-250UL). Protein quantification was performed using a BCA assay (Servicebio, G2026). Extracted proteins (6 μL) were separated by SDS-polyacrylamide gel electrophoresis (PAGE) and transferred onto polyvinylidene fluoride (PVDF) membranes (Servicebio, WGPVDF45). The membranes were blocked with 5% skimmed milk (Servicebio, GC310001-100 g) for 30 min at room temperature, followed by incubation with primary antibodies overnight at 4 °C. After washing 3 times with Tris-buffered saline-Tween (Servicebio, GC204002-100 ml), the membranes were incubated with secondary antibodies for 30 min at room temperature. Subsequently, by further washing with TBST, protein bands were visualized with a chemiluminescence system (CLINX, 6100). The following primary antibodies were used: Caspase-3 (1
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000; Servicebio, GB115600), and GAPDH (1:1000; Servicebio, GB15004).
:1000; Servicebio, GB115600), and GAPDH (1:1000; Servicebio, GB15004).
Preparation of arsenic/AB
1300 μg (amount of protein) of ABs isolated above were first mixed with 3 mL of 30 mg mL−1 manganese chloride and then sonicated in an ice water bath for 5 min using an ultrasonic cell crusher (JY92-IID, Ningbo Haishu Yiheng Instrument Co., Ningbo, China) (2 s “on”, 4 s “off” at 200 W). After centrifugation at 13000 rpm for 10 min, the precipitate was washed with distilled water and centrifuged for another 10 min under the same conditions. The precipitate was resuspended with 3 mL of 10 mg mL−1 of sodium arsenite and stirred for 30 min in a 37 °C water bath. Arsenic/AB was obtained by centrifugation under the same conditions and then washed with distilled water, followed by resuspension in 1.2 mL of PBS (250 μg mL−1) (see Appendix for details of the formulation process study for arsenic/AB).
Characterization of arsenic/AB
NTA. We measured the arsenic/AB particle size and zeta potential using nanoparticle tracking analysis (NTA) at VivaCell Biosciences with ZetaView PMX 110 (Particle Metrix, Meerbusch, Germany) and the corresponding software ZetaView 8.04.02. Isolated arsenic/AB samples were appropriately diluted using 1× PBS buffer (Biological Industries, Israel) (1.1 × 107 particles per mL) to measure their particle size and zeta potential. NTA measurement was recorded and analyzed at 11 positions. The ZetaView system was calibrated using 110 nm polystyrene particles. The temperature was maintained at around 23 °C and 30 °C.
Determination of arsenic/AB loadings. 500 μL of arsenic/AB was digested at 150 °C for 30 min with 2 mL of H2O2 and HNO3. After complete digestion, distilled water was added and diluted to 10 mL. The arsenic content of arsenic/AB was determined by ICAP 7400 inductively coupled plasma emission spectrometry (ICP-OES) (Thermo, USA). The loading of arsenic/AB was calculated from the measured amount of arsenic and the mass of arsenic/AB as follows
| LE (%) = We/Wm × 100% |
Dissolution test. The dissolution test for arsenic/AB was carried out in PBS (pH 7.4) at 37 °C using the paddle method at a paddle speed of 100 rpm. 1 mL of arsenic/AB was placed in a dialysis bag and then into a centrifuge tube containing 10 mL of dissolution medium tied to the paddle. The release medium was replaced at 5 min, 10 min, 15 min, 30 min, 1 h, 2 h, 4 h, 8 h, 12 h, and 24 h with 10 mL of blank release medium. The dissolution test for sodium arsenite was performed in PBS (pH 7.4) at 37 °C according to the Chinese Pharmacopoeia 2020 edition (General 0931 2nd method) at a paddle speed of 100 rpm. The arsenic content of the release medium at each time point was determined using ICP-OES, and the cumulative release rate of arsenic/AB and sodium arsenic after 24 h was calculated.
Macrophage hitchhiking experiments
The cells were cultured in DMEM medium supplemented with 10% FBS and 1% penicillin–streptomycin. In this study, ABs were used as a drug carrier to improve the distribution of arsenic in vivo and enhance the tumor targeting and deep penetration of arsenic in order to enhance the therapeutic effect and reduce the side effects. Therefore, FITC was used to replace arsenic to better track the location of the AB in vivo.Macrophage uptake assay
Qualitative analysis. RAW264.7 cells (2 × 105) were inoculated in 6-well plates, followed by the addition of ABs labelled with FITC (FITC-AB) (see Appendix for details of the preparation of FITC-AB and the stability examination of FITC-AB) and the same concentration of FITC solution (1 μg mL−1). After incubating for 1 h at 37 °C and 5% CO2, the cells were then fixed in 4% formaldehyde for 30 min and stained with 4′,6-diamidino-2-phenylindole (DAPI) for 5 min, followed by three washes with sterile PBS at the end of each step. Then, the cells were imaged using an LSM880 laser confocal scanning microscope (Zeiss, Germany). The intensity of fluorescent images was quantified using ImageJ by converting the images to grayscale and calculating the average grayscale intensity level.
Quantitative analysis. To further investigate the uptake of arsenic/AB by macrophages, RAW264.7 cells (5 × 106) were inoculated in 6-well plates, followed by the addition of sodium arsenite solution or arsenic/AB (the amount of sodium arsenite was 100 μg). After incubation at 37 °C and 5% CO2 for 1 h, the cells were collected and centrifuged at 800 rpm for 5 min at 4 °C. The precipitate was then washed thrice with sterile PBS and the arsenic content was determined by ICP. The uptake rate of arsenic/AB by macrophages was calculated by dividing the arsenic content by the number of cells.
Ex vivo cellular uptake assay. To investigate the amount of AB uptake by macrophages, male KM mice were first subcutaneously inoculated with H22 cells (5 × 106), and the blood was collected when the tumor volume reached ∼100 mm3. The red blood cells were removed using erythrocyte lysate and the remaining cells were centrifuged at 2000 rpm for 5 min, and then resuspended in 6-well plates at a density of 2 × 106. Then, 0.5 mL of FITC-AB (11.48 μg mL−1) or FITC solution (11.48 μg mL−1) was added and incubated for 2 h at 37 °C in 5% CO2. Then, the cells were labeled with CD11b, Ly-6G and F4/80 antibodies according to the instructions and analyzed with a flow cytometer. Cells expressing high levels of CD11b and F4/80 were the target cells—macrophages.25,26 The uptake of ABs by macrophages was determined by comparing the fluorescence intensity of target cells after treatment with FITC-AB and FITC solution.
In vivo cellular uptake assay. Male KM mice were first inoculated subcutaneously with H22 cells (5 × 106). 0.1 mL of FITC-AB (13.80 μg mL−1) or FITC solution (13.80 μg mL−1) was injected by tail vein when the tumor volume reached ∼100 mm3. Then, the whole blood was collected, the red blood cells were removed using erythrocyte lysate and the remaining cells were centrifuged at 2000 rpm for 5 min, and resuspended at a density of 2 × 106 per 200 μL. Then, the cells were labeled with CD11b, Ly-6G and F4/80 antibodies according to the instructions and analyzed with a flow cytometer.
In vitro effectiveness experiments
Laser confocal scanning microscope. H22 cells (5 × 106) were inoculated in a 6-well plate, followed by the addition of FITC-AB or the same concentration of FITC solution (6 μg mL−1) and incubated at 37 °C, 5% CO2 for 1 h. The cells were then fixed in 4% formaldehyde and stained with DAPI, followed by washing thrice with sterile PBS at the end of each step. Then, the cells were imaged using a laser confocal scanning microscope. The intensity of fluorescent images was quantified using ImageJ by converting the images to grayscale and calculating the average grayscale intensity level.
Flow cytometry. To examine the amount of AB uptake by H22 cells, H22 cells were inoculated in 6-well plates at a density of 5 × 106 cells per well, 500 μL of FITC-AB (6 μg mL−1) or the same concentration of FITC solution was added and incubated in an incubator. After 2 h, the cell suspension was centrifuged at 1000 rpm for 5 min at 4 °C. The precipitate was washed with PBS and the uptake of H22 cells was detected by flow cytometry.
H22 cytotoxicity assay. H22 cells were inoculated in 96-well plates at a density of 1 × 104 cells per well and incubated with different concentrations of arsenic/AB or sodium arsenite for 24 h at 37 °C and 5% CO2. MTT was added to the reaction for 4 h and the absorbance was measured at 570 nm using Synergy H1 Enzyme Labeler (Burton, USA) to probe the cytotoxicity of arsenic/AB.
H22 apoptosis assay. H22 cells were inoculated in 6-well plates at a density of 5 × 106 cells per well and incubated with different concentrations of arsenic/AB or sodium arsenite solutions for 24 h at 37 °C and 5% CO2. Then, the cells were treated with the apoptosis Kit and analyzed with a flow cytometer.
In vivo effectiveness experiments
All animal experiments have been approved by the Animal Ethics Committee of Zhejiang Chinese Medical University. The ethical number was IACUC-20190527-02.Statistical analysis
Experimental data were expressed as mean ± standard deviation (mean ± SD). Comparisons between two groups were made by independent t-test using GraphPad Prism 9.0 or between three or more groups by one-way analysis of variance (ANOVA). p < 0.05 was considered a statistically significant difference (ns: p > 0.05, *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.001).Results and discussion
Generation and characterization of ABs
ABs were obtained according to differential centrifugation and characterized by shape, size, protein markers, apoptotic markers and yield. TEM assay showed that ABs had the morphological characteristics of a cup-shaped membranous vesicle (Fig. 1A).23,27 The NTA particle size distribution data for ABs showed a diameter range of 50–600 nm, with a main peak of 100–200 nm (Fig. 1B). The protein marker Caspase-3 was detected by western blotting. Western blot analysis showed that the isolated ABs expressed the specific marker Caspase-3 (Fig. 1C). The detection of apoptotic markers on the surface of ABs was performed by staining live cells, cells from centrifugation at 300g, and ABs from centrifugation at 3000g with Annexin V-FITC and PI, respectively.28 As shown in Fig. 1D, the apoptosis rate of cells obtained by centrifugation at 300g was 70.39%, which was significantly higher than that of the live cell group (15.97%). The positive rate for Annexin V-FITC of the ABs obtained by centrifugation at 3000g was 35.84%. This result demonstrated that the stimulation accelerated the apoptosis and promoted the production of ABs, and the surface of isolated ABs had apoptotic markers. Together, the above results demonstrated that the product obtained by differential centrifugation was ABs. In addition, the yield of ABs was 0.2% according to the PS-specific fluorescent antibody Annexin V-FITC.29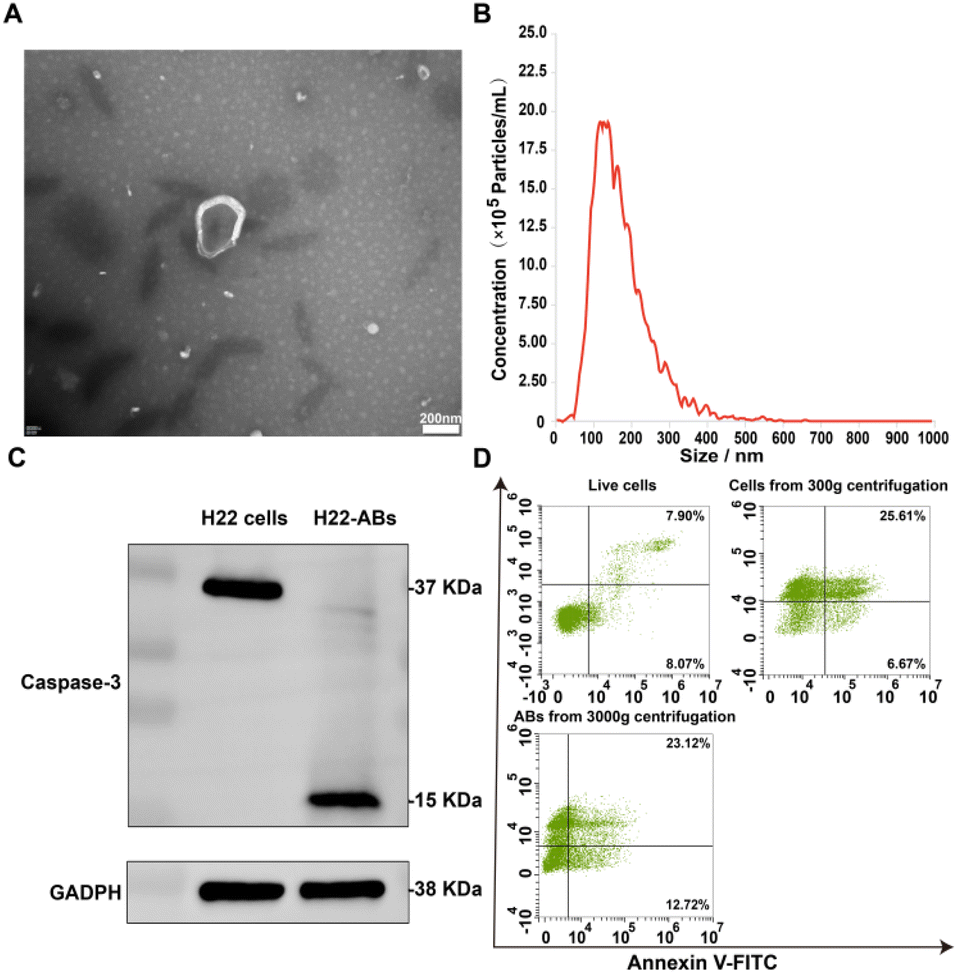 | ||
| Fig. 1 (A) TEM image of ABs, scale bar: 200 nm, (B) particle size distribution of ABs, (C) western blots of the apoptotic marker protein caspase-3 in the H22 cells and H22 cell-derived ABs, (D) live cells, cells from 300g centrifugation and ABs from 3000g centrifugation were analyzed with Annexin V-FITC and PI dot plots. Quadrant gates were set up on the corresponding unstained control populations. | ||
Preparation and characterization of arsenic/AB
Arsenic/AB was characterized and the results showed that it had the suitable particle size and potential to hitchhike on macrophages. In addition, arsenic/AB exhibited delayed release properties, allowing for the safety of macrophages during transport. Fig. 2A and B showed that arsenic/AB had a spherical or sphere-like shape with a particle size of 136.7 ± 25.8 nm. The uptake of nanoparticles by macrophages was size-dependent, and most nano-preparations were designed to be in the 10–200 nm diameter range.30,31Fig. 2C demonstrated the ζ value of (−) 13.27 ± 5.16 mV for arsenic/AB. Zeta potential (ζ potential) values influenced the physical stability of nanoparticles, and nanoparticles with a negative surface charge were readily phagocytosed by macrophages.32–34 The ζ potential and particle size of arsenic/AB made it readily available for uptake by macrophages in vivo, which facilitated its improved distribution in vivo and penetration deeply into the tumor. Fig. 2D demonstrated the delayed release of arsenic/AB compared to sodium arsenite, allowing for the safety of macrophages during transport. The drug loading of arsenic/AB was calculated to be 0.48%. In conclusion, these results demonstrated that arsenic/AB had an appropriate size and potential with delayed release characteristics, creating favorable conditions for its hitchhiking on macrophages.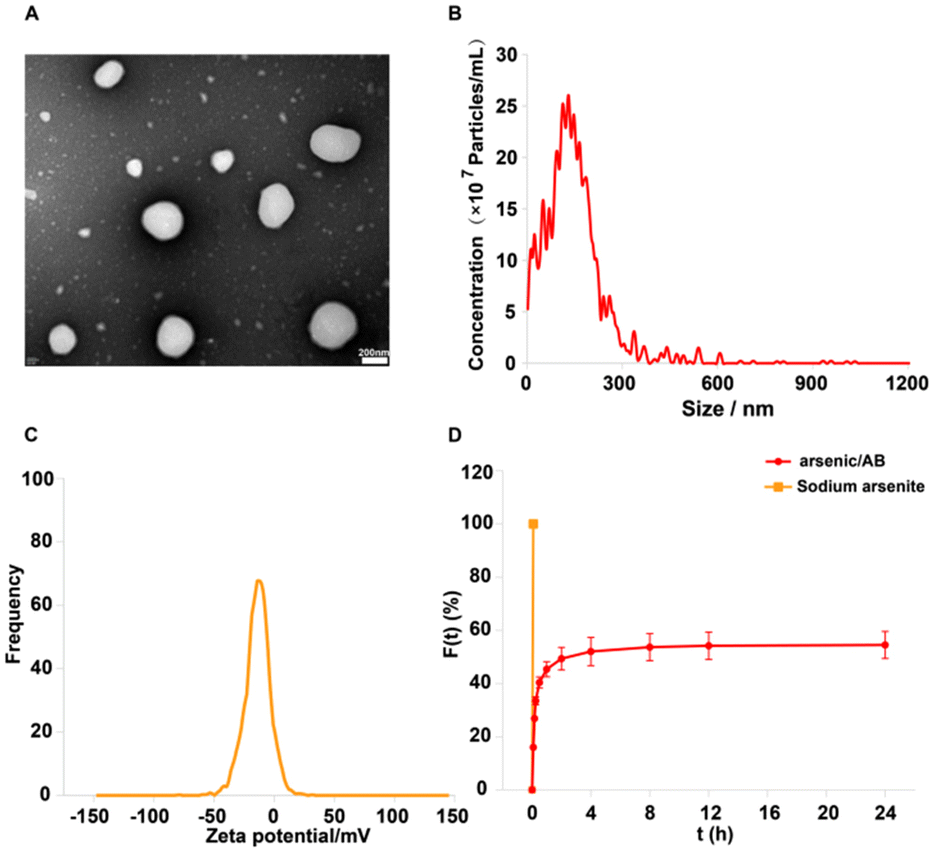 | ||
| Fig. 2 (A) TEM image of arsenic/AB, scale bar: 200 nm, (B) particle size distribution of arsenic/AB, (C) Zeta potential of arsenic/AB, and (D) in vitro release of sodium arsenite and arsenic/AB, ***p < 0.001. | ||
Macrophage hitchhiking experiments
In vitro, ex vivo and in vivo cellular uptake experiments were used to demonstrate that arsenic/AB could be taken up by macrophages for tumor targeting. In vitro cell uptake assay showed that the amount of FITC uptake by macrophages was significantly higher in the FITC-AB group than in the FITC solution group (Fig. 3A and B). ICP assay was used to further determine the amount of arsenic taken up by macrophages. The uptake amount of arsenic/AB by macrophages was calculated to be 1.18 μg/(1 × 106) cells, which was 40 times compared to the arsenic solution (Fig. 3C). This was consistent with the results of ex vivo and in vivo cellular uptake. As shown in Fig. 3E, cells from the blood were first analyzed by fluorescence-activated cell sorting. CD11b+Ly6G+ were neutrophils and CD11b+F4/80+ were macrophages. The analysis of flow cytometry showed that the fluorescence intensity of FITC was significantly higher in CD11b+F4/80+ macrophages in the FITC-AB group than in the FITC solution group. Fig. 3D shows that at an administration concentration of 6.25 μg mL−1, the survival rate of macrophages in the arsenic/AB group (89.80%) was still greater than 85.0%, which was significantly higher than the sodium arsenite solution group (34.95%), indicating that arsenic/AB had no significant toxicity to macrophages. The results of the cytotoxicity assay for arsenic/AB were consistent with the results of the dissolution assay that demonstrated the safety of arsenic/AB on macrophages. In conclusion, the results provided further evidence that arsenic/AB could hitchhike on macrophages and had little effect on the biological behavior of macrophages. This provided favorable conditions for arsenic/AB to hitchhike on macrophages and enhance the therapeutic effect of arsenic on liver cancer.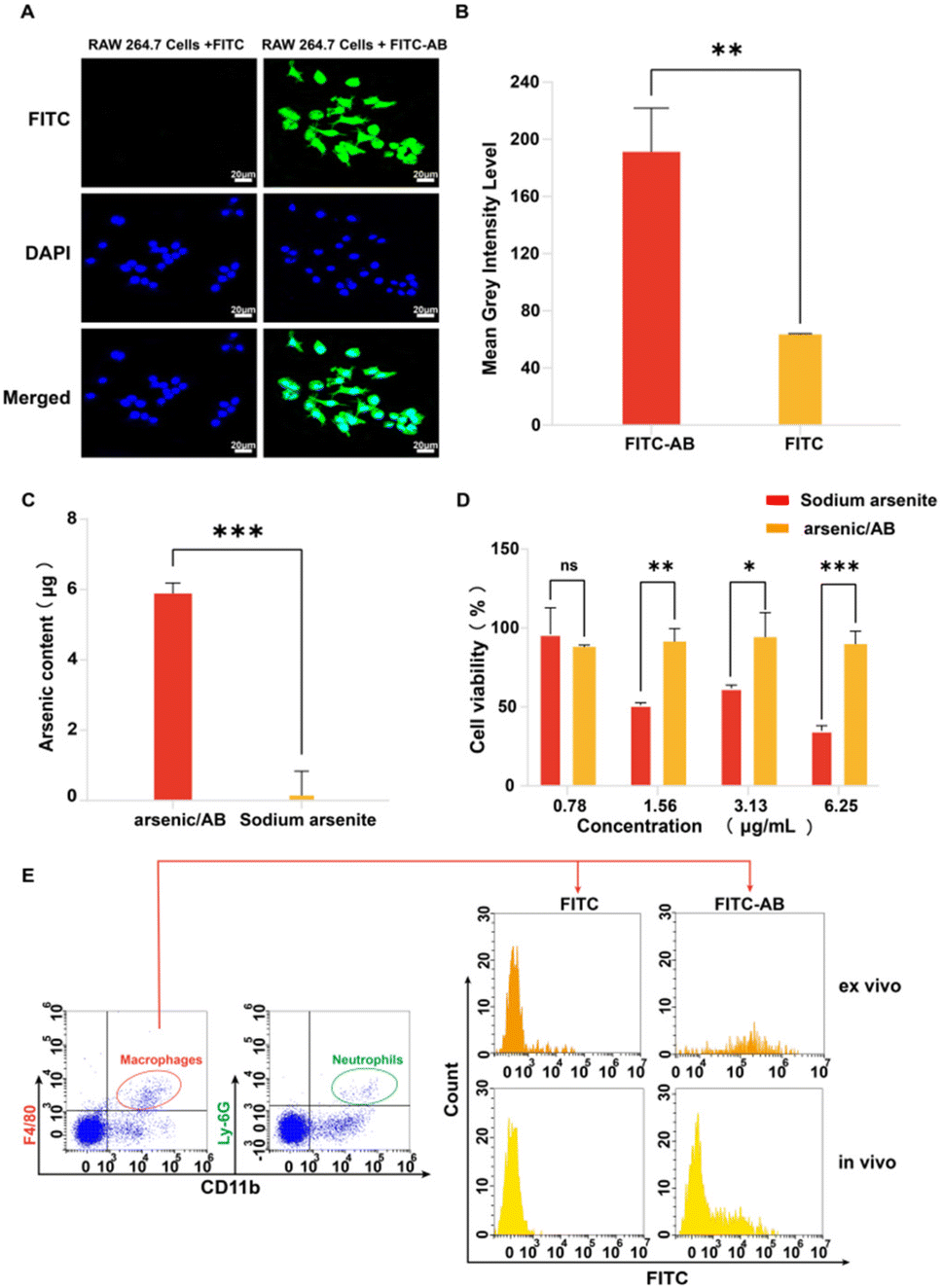 | ||
| Fig. 3 (A) Fluorescence imaging of cellular uptake of FITC-AB or FITC in macrophages after 1 h incubation, (B) the fluorescence intensity of cellular uptake of FITC-AB or FITC after 1 h incubation (quantified using ImageJ), (C) arsenic content in the treatment of RAW 264.7 cells with arsenic/AB and sodium arsenite solution after 1 h incubation, (D) determination of the toxicity of arsenic/AB and sodium arsenite to macrophages by MTT, and (E) ex vivo and in vivo macrophage uptake of FITC-AB or FITC, macrophages were identified by CD11b and F4/80 staining gating. Scale bar = 20 μm, ns: p > 0.05, *p < 0.05, **p < 0.01, and ***p < 0.001. | ||
In vitro effectiveness experiments
The results of in vitro cellular uptake, cytotoxicity, and apoptosis assay showed that arsenic/AB had a positive therapeutic effect on liver cancer. Firstly, in vitro cellular uptake results showed that compared to the FITC solution, there was more FITC-AB taken up by H22 cells (Fig. 4A–C). This might be attributed to the presence of markers on the surface of tumor-derived ABs that were similar to tumor cell membranes, facilitating the passage of arsenic/AB through tumor cell membranes.35 This allowed arsenic/AB for deep tumor penetration and enhanced the therapeutic effect of arsenic on liver cancer. Next, MTT was used to study the in vitro cytotoxicity of arsenic/AB and sodium arsenite solutions. Arsenic/AB was significantly more toxic to H22 cells than sodium arsenite. When the concentration of arsenic/AB was as low as 0.78 μg mL−1, the cell viability of H22 was only about 40% (Fig. 4D).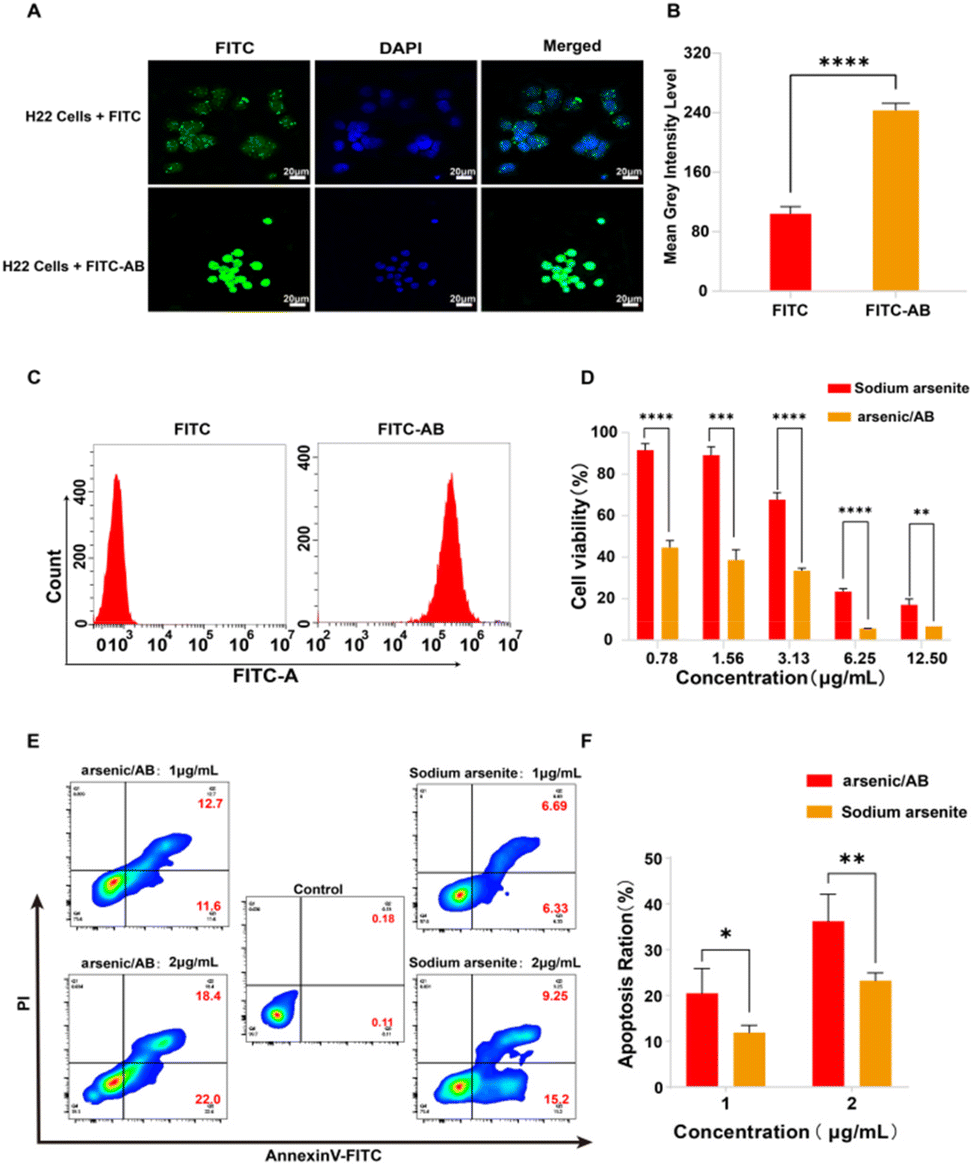 | ||
| Fig. 4 (A) Fluorescence imaging of cellular uptake of FITC-AB or FITC in H22 cells after 1 h incubation, (B) the fluorescence intensity of cellular uptake of FITC-AB or FITC in H22 cells after 1 h incubation(quantified using ImageJ), (C) the cellular uptake of FITC-AB or FITC in H22 cells after 2 h incubation by flow cytometry, (D) determination of the toxicity of arsenic/AB and sodium arsenite to H22 cells by MTT, (E) the apoptosis rate of transfected H22 cells was analysed with Annexin V-FITC and PI dot plots. Quadrant gates were set up on the corresponding unstained control populations, and (F) the apoptosis rate of H22 cells after treatment with different concentrations of arsenic/AB or sodium arsenite solution. Scale bar = 20 μm, *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. | ||
Then, the IC50 values for arsenic/AB and sodium arsenite are summarized in Table 1. It could be seen that the IC50 values of arsenic/AB for H22 cells (0.7546 ± 0.1674 μg mL−1) were lower than those of sodium arsenite (4.149 ± 0.3064 μg mL−1). The results demonstrated that arsenic/AB had a high inhibitory effect on tumor cells.
| Cell | H22 |
|---|---|
| Arsenic/AB | 0.7546 ± 0.1674 |
| Sodium arsenite | 4.149 ± 0.3064 |
Finally, the efficiency of arsenic/AB was further confirmed by the Annexin V FITC/PI double staining assay. As illustrated in Fig. 4E and F, the apoptosis rate of H22 cells in the arsenic/AB group (24.30%) was higher than that in the sodium arsenite solution group (13.02%) after treatment with the same drug concentration (1 μg mL−1). The apoptosis rate was also significantly higher in the arsenic/AB group (40.40%) than in the sodium arsenite (24.45%) group when administered at a concentration of 2 μg mL−1. This was consistent with the results of cytotoxicity. This allowed arsenic/AB to suppress the tumor by inducing apoptosis in tumor cells, achieving a therapeutic effect on liver cancer.
In vivo bio-distribution and tumor penetration
In vivo distribution assays were used to demonstrate the ability of ABs to improve in vivo distribution and tumor penetration. Firstly, FITC solution and FITC-AB of the same fluorescence intensity were injected into the tumor-bearing mice through the tail vein. The tumor-bearing mice were dissected 1 h later and the isolated tissues were imaged. As shown in Fig. 5A and B, the fluorescence intensity of the tumor tissues in the FITC-AB group was significantly higher than that in the FITC solution group. The results showed that ABs could rapidly target tumor sites and achieve improved drug distribution in vivo. To further explore the tumor-targeting ability of arsenic/AB, we examined its in vivo distribution after 24 h (see Appendix for details of in vivo bio-distribution of arsenic/AB at 24 h). The results showed that there was more arsenic/AB accumulating at the tumor site after 24 h compared to 1 h (Fig. S5†). This was mainly attributed to the tumor-homing ability of macrophages. Rapid tumor targeting could reduce the release of arsenic/AB during transport by macrophages, which improved the safety of arsenic/AB to macrophages and allowed more drug release at the tumor site to enhance the efficacy of treating liver cancer. The improved in vivo distribution allowed arsenic/AB to accumulate at the tumor site, further enhancing the efficacy of liver cancer and reducing the systemic toxicity of arsenic/AB.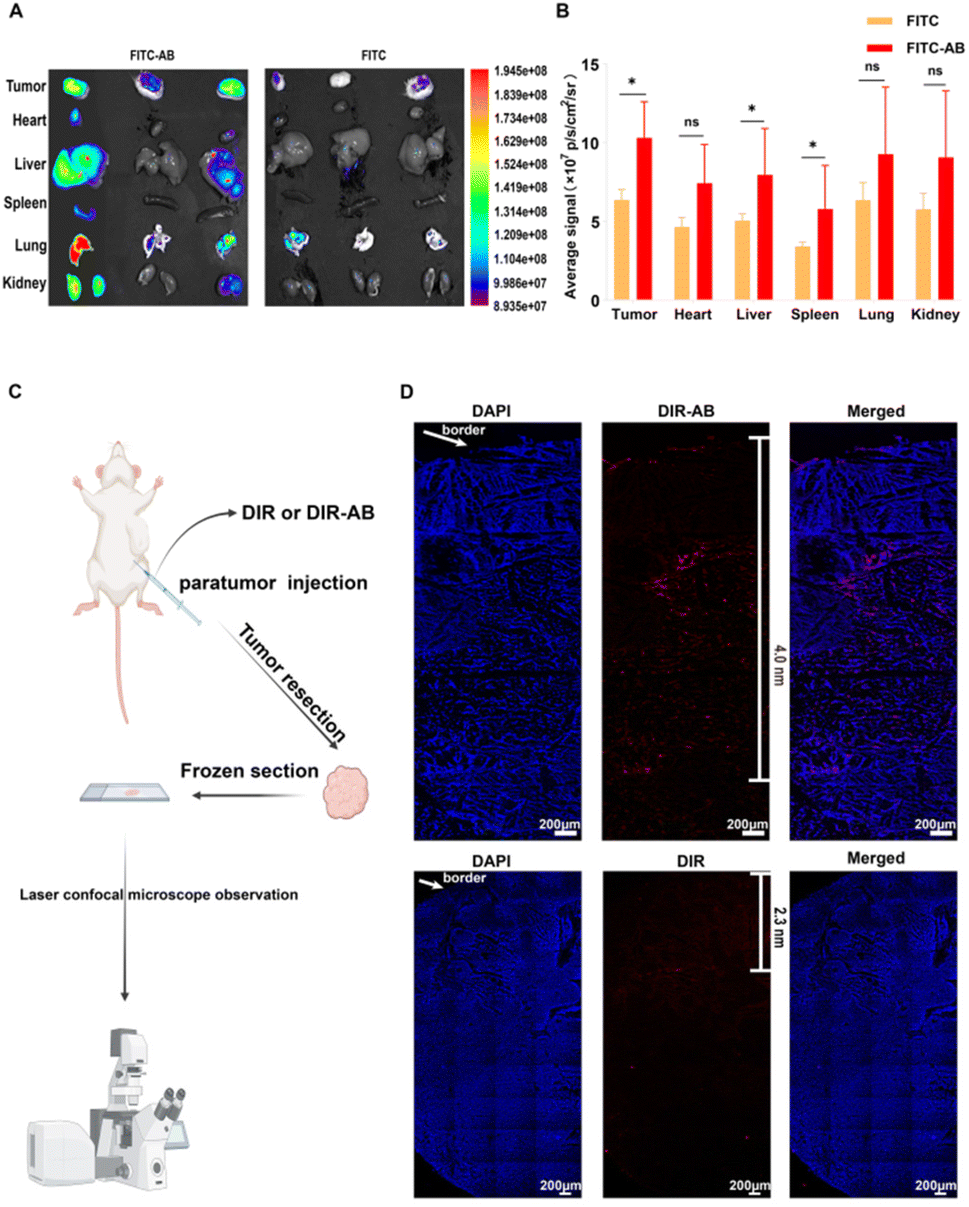 | ||
| Fig. 5 (A) Ex vivo fluorescent images of tissue samples collected 1 h after injection, (B) quantitative fluorescence intensity and statistical analysis of tissue samples collected 1 h after injection, (C) flow diagram of deep tumor penetration assay, and (D) laser confocal image of a frozen tumor section, scale bar = 200 μm. | ||
The tumor penetration ability of arsenic/AB was then examined and the experimental results showed that arsenic/AB was able to penetrate better deep into the tumor. As shown in Fig. 5C, the DIR-AB group (the depth of penetration was 4.0 nm) penetrated significantly deeper into the tumor compared to the DIR group (the depth of penetration was 2.3 nm). This might be due to the fact that tumor-derived ABs had markers on their surface that are similar to the tumor cell membrane, making it easier for ABs to penetrate through the cell membrane to the depths of the tumor. This also confirmed the suspicion that arsenic/AB could penetrate deep into the tumor to effectively inhibit tumor growth.
In vivo antitumor efficacy
To verify the therapeutic effect of the arsenic/AB bionic preparation on liver cancer, tumor-bearing mice were given sodium arsenite solution or arsenic/AB through tail vein injection every three days, and the tumor volume and mouse body weight were recorded. As shown in Fig. 6A, sodium arsenite solution or arsenic/AB inhibited tumor growth to varying degrees compared to the rapid tumor growth in the control group. The mice in the arsenic/AB group had smaller tumor volumes compared to the other two groups (p < 0.01), indicating that arsenic/AB had the greatest inhibition of tumor growth. This may be attributed to the fact that ABs derived from tumor cells could penetrate deeply through the cell membrane of tumor cells when they reach the tumor site. This was also due to the fact that arsenic/AB could reach tumor sites more efficiently by hitchhiking on macrophages for tumor targeting.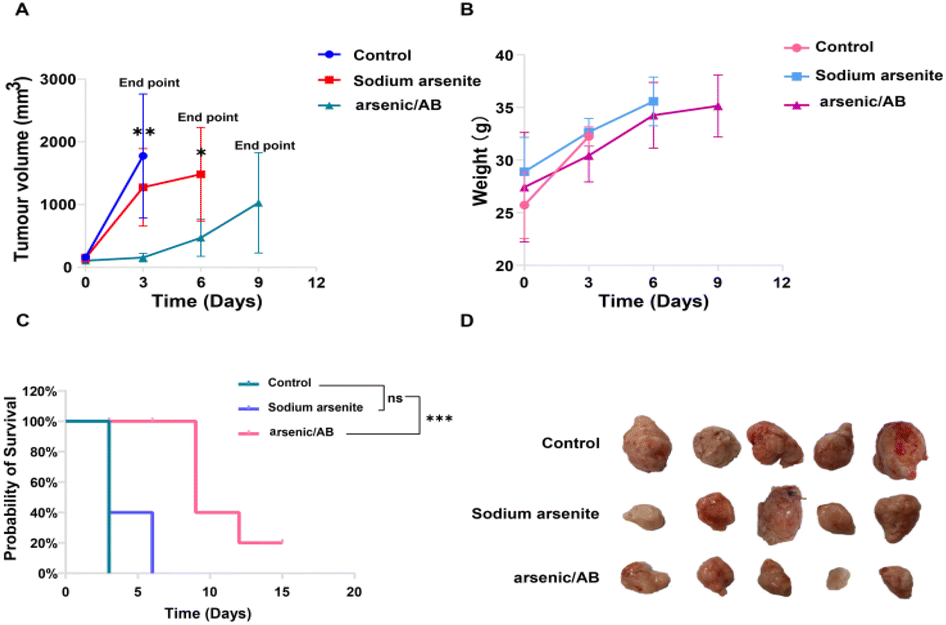 | ||
| Fig. 6 (A) Tumor volumes at different time points, shown as mean and standard error bars of repeated measurements (n = 5 per group), (B) body weights of mice at different time points, expressed as mean and standard error bars of repeated measurements (n = 5 per group), (C) survival curves of mice under different administration methods (n = 5 per group), and (D) the photograph of the tumor after biopsy. *p < 0.05, **p < 0.01, and ***p < 0.001. | ||
Body weight changes in the mice were used to assess the in vivo safety of drugs.36 As shown in Fig. 6B, the change in the body weight of mice in the arsenic/AB group was not significantly lower compared to that in the control groups, indicating the weak systemic toxicity of arsenic/AB. This was consistent with the results shown in Fig. 6C, although the final survival rate of arsenic/AB was only 20%, the experimental results showed that we significantly improved the survival time compared with sodium arsenite solution. All the mice in the sodium arsenite solution group reached the endpoint at 6 days, while the arsenic/AB group had a 20% survival rate at 15 days.
In addition, the final volume of the tumor of arsenic/AB reached about 1000 mm3 and there was no difference in the weight loss of the mice between the three groups, this might be due to the fact that the tumor exhibited rapid growth on modelling mice. Considering the high dose of arsenic, more adverse effects occurred. Therefore, we explored a smaller dose (1 mg kg−1) compared with the highest dose of 2 mg kg−1.37–39 In this case, arsenic/AB still showed some effect of tumor inhibition, which better reflects the advantages of arsenic/AB for the treatment of liver cancer. Tumor inhibitory effects of arsenic/AB might be attributed to the fact that arsenic/AB could be taken up by macrophages and targeted to the tumor by the homing ability of macrophages, improving the in vivo distribution of arsenic/AB, allowing arsenic/AB to accumulate at the tumor site, and reducing the toxicity to other tissues and organs.
Histological examination
The strong toxicity of arsenic was a major reason for limiting its use, so it was necessary to examine the safety of arsenic/AB. H&E was used to assess pathological changes in major organs to verify the therapeutic effect of arsenic/AB on tumor as well as their safety. Compared to the control group and the sodium arsenite solution group, there were no significant inflammatory reactions or necrosis in the major organs of the arsenic/AB group, indicating the reduced toxicity of the arsenic/AB preparation compared to the sodium arsenite solution (Fig. 7). However, significant necrotic and apoptotic cells could be seen in the tumor tissue sections of the arsenic/AB group. Together, these results demonstrated that arsenic/AB produced satisfactory therapeutic effects with reduced side effects.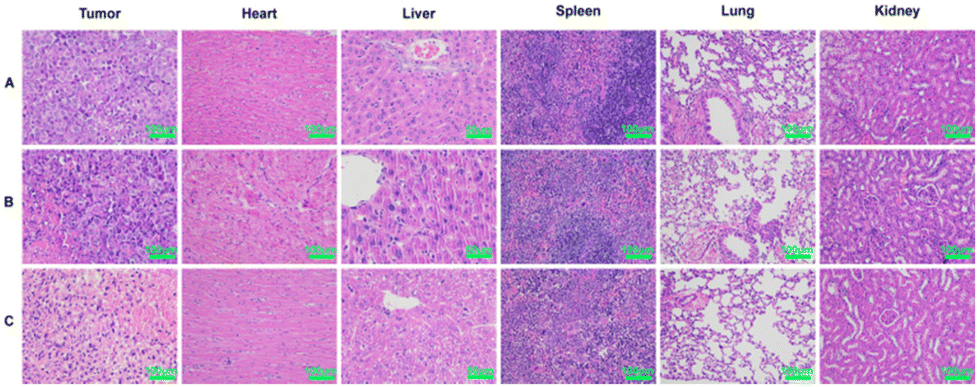 | ||
| Fig. 7 Histopathological analysis after 15 days of administration of different drug delivery methods. (A) Control, (B) sodium arsenite solution, and (C) arsenic/AB preparations. | ||
Immunohistochemistry
The tumor microenvironment (TME) plays a crucial role in the progression, metastasis, and treatment of cancer.40 Macrophages, often referred to as tumor-associated macrophages (TAMs), which were clustered around tumor cells, play a key role in tumor progression as an important component of the TME. TAMs could be divided into two phenotypes: M1 and M2. In general, M1 macrophages promoted an inflammatory response against tumor cells and inhibited tumor growth, while M2 macrophages tended to play an immune suppressive phenotype and favor tumor progression.41 Within the TME, many factors influenced the differentiation of the TAMs. For example, the overproduction of interleukin 6 (IL-6), which is closely associated with the inflammatory response, promoted the differentiation of TAMs into the M2 phenotype.42 In addition, colony-stimulating factor 1 (CSF1), a classical monocyte inducer, could activate the colony-stimulating factor 1 receptor (CSF1R), causing CSF1R overexpression, which favors the differentiation of macrophages into the tumor-promoting M2 phenotype.43–45 To investigate the reason for the tumor suppressive effect of arsenic/AB, immunohistochemical (IHC) examination of the tumor tissue was carried out. After IHC examination, we found that arsenic/AB could promote TAM differentiation towards the M1 phenotype with anti-tumor effects (Fig. 8A), which effectively inhibit tumor growth. CD86 was highly expressed on the surface of M1 phenotype macrophages, whereas CD206 was highly expressed on the surface of M2 phenotype macrophages.46 As shown in Fig. 8E, the positive area rate for CD86 was significantly higher in the arsenic/AB group than in the other two groups. At the same time, the positive area rate for CD206 was significantly lower than in the other two groups. This result demonstrated that arsenic/AB could promote more differentiation of TAMs towards the M1 phenotype. To investigate the mechanism by which arsenic/AB affects TAM differentiation, we further analyzed IL-6, CSF1 secretion and CSF1R expression by IHC (Fig. 8B–D). The results showed that the positive area rates for IL-6, CSF1, and CSF1R were all significantly lower in the arsenic/AB group than in the other two groups (Fig. 8F–H). The above results suggest that arsenic/AB influenced TAM differentiation in two ways. Firstly, arsenic/AB inhibited the secretion of IL-6 and avoided the development of excessive inflammatory responses. Secondly, arsenic/AB could inhibit the secretion of CSF1, blocking CSF1R activation and avoiding CSF1 overexpression. Together, these two actions promoted the differentiation of macrophages into the anti-tumor M1 phenotype. On one hand, it could allow macrophage-hitchhiked arsenic/AB to reach the tumor with little differentiation into the tumor-promoting M2 phenotype, leading to reduced efficacy of arsenic/AB in liver cancer. On the other hand, it could lead to a greater differentiation of TAMs towards the M1 phenotype, enhancing the therapeutic effect of arsenic/AB on liver cancer.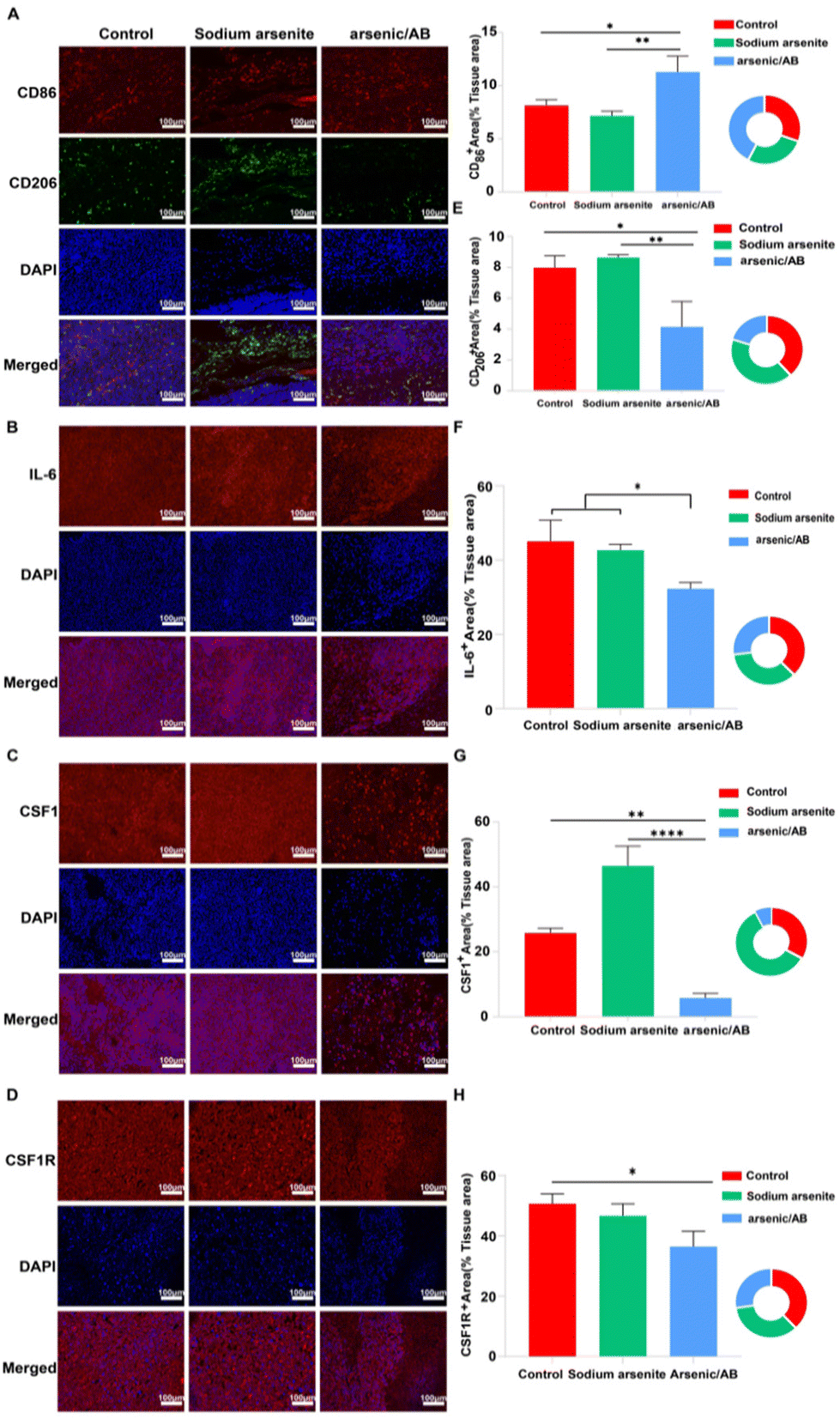 | ||
| Fig. 8 Immunohistochemical detection of tumor tissue sections stained with different antibodies after reaching the endpoint of the experiment by different administration methods of drug delivery. (A) Stained with CD86 and CD206 antibodies, (B) stained with the IL-6 antibody, (C) stained with the CSF1 antibody, and (D) stained with the CSF1R antibody. Calculation of the positive area for different cytokines in the percentage of tissue area using ImageJ. (E) CD86 and CD206, (F) IL-6, (G) CSF1, and (H) CSF1R. The results are shown as group means ± SEM (standard error of the mean). *p < 0.05, **p < 0.01, and ****p < 0.0001. | ||
Conclusions
In this study, we designed macrophage-hitchhiked arsenic/AB bionic preparations to enhance the therapeutic efficacy in liver cancer while improving the safety of the clinical use of arsenic. Firstly, in vitro release and in vitro macrophage toxicity assay demonstrated that due to the delayed release properties of arsenic/AB, it had a high safety profile on macrophages. Next, we demonstrated the specific uptake of arsenic/AB by macrophages through in vitro, ex vivo and in vivo cellular uptake assays. This was attributed to the arsenic/AB with the negatively charged particles of approximately 100–200 nm size and the “eat-me” signal of the AB surface. Macrophages had strong phagocytic activity and tumor-homing ability, therefore arsenic/AB hitchhiked macrophages could efficiently accumulate at the tumor site and then gradually penetrate the tumor, effectively enhancing its tumor targeting and deep tumor penetration abilities. However, the results of macrophage uptake stability in vitro (Fig. S4†) suggested that arsenic/AB might be present in vivo in more than just a macrophage-carrying form, thus arsenic/AB may be targeted to tumors by other pathways. Firstly, tumor targeting may be achieved through the enhanced permeation and penetration (EPR) effect. Due to the rapid proliferation of tumor cells and the abnormal tumor vascular system, the EPR effect has become a commonly used strategy for nanoparticle targeting of tumors.47 The effectiveness of the EPR effect depends on the nanoparticle particle size and surface charge. Nanoparticles with particle sizes of 10–100 nm and a neutral or slightly negative surface charge exhibit a stronger EPR effect.48,49 Arsenic/AB had a particle size of 136.7 ± 25.8 nm and a slightly negative surface charge, allowing it to target tumors via the EPR effect. However, relying solely on the EPR effect for tumor targeting has limited efficiency, as studies have shown that only 0.7% of nanoparticles accumulated at the tumor site through the EPR effect.50 Secondly, since the carrier of arsenic/AB was tumor cell-derived ABs, it may also target the tumor site through homologous targeting ability.48 However, the number of nanoparticles targeted to the tumor in this way is also limited. The tumor targeting by hitchhiking on macrophages is more efficient than other ways, which is the main mechanism of arsenic/AB tumor targeting. Then in vitro effectiveness tests showed that arsenic/AB could be taken up by tumor cells and produce anti-tumor effects by inducing apoptosis. This was made possible by the fact that the surface of tumor cell-derived ABs had markers similar to tumor cell membranes, allowing further deep tumor penetration of arsenic/AB. In vivo experiments further demonstrated the ability of arsenic/AB to rapidly target tumors and produce strong tumor suppression with low side effects. The results of the in vivo and in vitro experiments together supported the suspicion that arsenic/AB could improve the therapeutic effect on liver cancer by hitchhiking on macrophages to improve distribution in vivo and enhance deep tumor penetration. Finally, through the IHC assay, we found that arsenic/AB produced strong tumor suppression by reducing the inflammatory response and inhibiting CSF1 secretion to block CSF1R activation, thereby inducing TAM differentiation towards the M1 phenotype with tumor-suppressive effects. Finally, through the detection of cytokines, we found that arsenic/AB induced the differentiation of TAMs towards the M1 phenotype with tumor-suppressive effects by inhibiting the secretion of IL-6 and CSF1, thus producing a strong tumor suppressive effect. In summary, the advantage of arsenic/AB was that it could be targeted for drug delivery through spontaneous physiological activity in vivo, which enhanced the targeting of arsenic/AB while effectively avoiding its elimination as a foreign body during tumor targeting and reducing its systemic toxicity. This indicated that arsenic/AB had high potential for the treatment of liver cancer and was therefore worthwhile to further optimize for addressing some of the remaining issues. Firstly, the yield of ABs was low, which made the extraction of ABs more expensive and unfavorable for the application of arsenic/AB. Then, the low loading rate and high pre-target release rate of arsenic/AB reduced the effective dose when it reached the tumor site and weakened the tumor inhibition effect. Finally, the short retention time of arsenic/AB at the tumor site could result in more frequent dosing to maintain a good therapeutic effect. As arsenic/AB was administered intravenously, frequent administration became more difficult, which further limited the use of arsenic/AB bionic preparations. In future studies, we will optimize the arsenic/AB bionic preparations to address these issues and provide a more reliable solution for the treatment of liver cancer.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This study was supported by the National Natural Science Foundation of China (82003954, 82174096), the Zhejiang Provincial Natural Science Foundation of China (LY23H280010), and the Research Project of Zhejiang Chinese Medical University (2023JKZKTS23). We appreciate the experimental support from the Public Platform of Pharmaceutical Research Center, Academy of Medical Sciences, Zhejiang Chinese Medical University. The graphical abstract was created with BioRender.com.References
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, CA Cancer J. Clin., 2021, 71(3), 209–249 CrossRef PubMed.
- D. Anwanwan, S. K. Singh, S. Singh, V. Saikam and R. Singh, Biochim. Biophys. Acta, Rev. Cancer, 2020, 1873(1), 188314 CrossRef CAS PubMed.
- S. Man, C. Luo, M. Yan, G. Zhao, L. Ma and W. Gao, Eur. J. Med. Chem., 2021, 224, 113690 CrossRef CAS.
- Z. Cheng, J. Wei-Qi and D. Jin, Biochim. Biophys. Acta, Rev. Cancer, 2020, 1874(1), 188382 CrossRef CAS PubMed.
- X. D. Zhu, Z. Y. Tang and H. C. Sun, Genes Dis., 2020, 7(3), 328–335 CrossRef CAS.
- N. Sadaf, N. Kumar, M. Ali, V. Ali, S. Bimal and R. Haque, Life Sci., 2018, 205, 9–17 CrossRef CAS PubMed.
- H. Y. Wang, B. Zhang, J. N. Zhou, D. X. Wang, Y. C. Xu, Q. Zeng, Y. L. Jia, J. F. Xi, X. Nan, L. J. He, W. Yue and X. T. Pei, Cell Death Dis., 2019, 10(6), 453 CrossRef PubMed.
- A. Mishra, P. Oliinyk, R. Lysiuk, L. Lenchyk, S. S. S. Rathod, H. Antonyak, R. Darmohray, N. Dub, O. Antoniv, O. Tsal and T. Upyr, Environ. Toxicol. Pharmacol., 2022, 95, 103970 CrossRef CAS PubMed.
- T. Yong, X. Zhang, N. Bie, H. Zhang, X. Zhang, F. Li, A. Hakeem, J. Hu, L. Gan, H. A. Santos and X. Yang, Nat. Commun., 2019, 10(1), 3838 CrossRef PubMed.
- D. Kim, H. K. Woo, C. Lee, Y. Min, S. Kumar, V. Sunkara, H. G. Jo, Y. J. Lee, J. Kim, H. K. Ha and Y. K. Cho, Anal. Chem., 2020, 92(8), 6010–6018 CrossRef CAS.
- S. Li, Y. Qu, L. Liu, C. Wang, L. Yuan, H. Bai and J. Wang, Cell Proliferation, 2023, 20 DOI:10.1111/cpr.13452.
- D. Wang, Y. Yao, J. He, X. Zhong, B. Li, S. Rao, H. Yu, S. He, X. Feng, T. Xu, B. Yang, T. Yong, L. Gan, J. Hu and X. Yang, Adv. Sci., 2019, 7(3), 1901293 CrossRef.
- M. Yang, X. Wang, F. Pu, Y. Liu, J. Guo, S. Chang, G. Sun and Y. Peng, Pharmaceutics, 2021, 13(10), 1593 CrossRef CAS.
- X. Zhao, D. Wu, X. Ma, J. Wang, W. Hou and W. Zhang, Biomed. Pharmacother., 2020, 128, 110237 CrossRef CAS PubMed.
- Y. Wu, S. Wan, S. Yang, H. Hu, C. Zhang, J. Lai, J. Zhou, W. Chen, X. Tang, J. Luo, X. Zhou, L. Yu, L. Wang, A. Wu, Q. Fan and J. Wu, J. Nanobiotechnol., 2022, 20(1), 542 CrossRef CAS PubMed.
- T. Xiao, W. Hu, Y. Fan, M. Shen and X. Shi, Theranostics, 2021, 11(14), 7057–7071 CrossRef CAS PubMed.
- C. Chen, M. Song, Y. Du, Y. Yu, C. Li, Y. Han, F. Yan, Z. Shi and S. Feng, Nano Lett., 2021, 21(13), 5522–5531 CrossRef CAS.
- S. Wang, C. Li, Y. Yuan, Y. Xiong, H. Xu, W. Pan and H. Pan, J. Drug Delivery Sci. Technol., 2022, 104088 Search PubMed.
- F. Urabe, N. Kosaka, K. Ito, T. Kimura, S. Egawa and T. Ochiya, Am. J. Physiol.: Cell Physiol., 2020, 318(1), C29–C39 CrossRef CAS.
- M. Zhou, Y. J. Li, Y. C. Tang, X. Y. Hao, W. J. Xu, D. X. Xiang and J. Y. Wu, J. Controlled Release, 2022, 351, 394–406 CrossRef CAS PubMed.
- E. I. Buzás, E.Á Tóth, B. W. Sódar and K.É Szabó-Taylor, Semin. Immunopathol., 2018, 40(5), 453–464 CrossRef.
- H. Liu, S. Liu, X. Qiu, X. Yang, L. Bao, F. Pu, X. Liu, C. Li, K. Xuan, J. Zhou, Z. Deng, S. Liu and Y. Jin, Autophagy, 2020, 16(12), 2140–2155 CrossRef CAS PubMed.
- C. Zheng, B. Sui, X. Zhang, J. Hu, J. Chen, J. Liu, D. Wu, Q. Ye, L. Xiang, X. Qiu, S. Liu, Z. Deng, J. Zhou, S. Liu, S. Shi and Y. Jin, J. Extracell. Vesicles, 2021, 10(7) DOI:10.1002/jev2.12109.
- J. Hahm, J. Kim and J. Park, Tissue Eng. Regener. Med., 2021, 18(4), 513–524 CrossRef CAS.
- L. Zheng, X. Hu, H. Wu, L. Mo, S. Xie, J. Li, C. Peng, S. Xu, L. Qiu and W. Tan, J. Am. Chem. Soc., 2020, 142(1), 382–391 CrossRef CAS PubMed.
- A. E. Shin, Y. Tesfagiorgis, F. Larsen, M. Derouet, P. Y. F. Zeng, H. J. Good, L. Zhang, M. R. Rubinstein, Y. W. Han, S. M. Kerfoot, A. C. Nichols, Y. Hayakawa, C. J. Howlett, T. C. Wang and S. Asfaha, Gastroenterology, 2023, 164(4), 593–609 CrossRef CAS.
- D. Chen, Z. Zhao, K. Zhang, F. Jin, C. Zheng and Y. Jin, STAR Protoc, 2022, 3(4), 101695 CrossRef CAS.
- Q. Ma, M. Liang, N. Limjunyawong, Y. Dan, J. Xing, J. Li, J. Xu and C. Dou, Theranostics, 2020, 10(15), 6825–6838 CrossRef CAS PubMed.
- H. W. Shin and H. Takatsu, Crit. Rev. Biochem. Mol. Biol., 2020, 55(2), 166–178 CrossRef CAS PubMed.
- V. Ejigah, O. Owoseni, P. Bataille-Backer, O. D. Ogundipe, F. A. Fisusi and S. K. Adesina, Polymers, 2022, 14(13), 2601 CrossRef CAS.
- W. Yu, R. Liu, Y. Zhou and H. Gao, ACS Cent. Sci., 2020, 6(2), 100–116 CrossRef CAS.
- J. Fang, W. Islam and H. Maeda, Adv. Drug Delivery Rev., 2020, 157, 142–160 CrossRef CAS PubMed.
- B. Yadav, M. Chauhan, S. Shekhar, A. Kumar, A. K. Mehata, A. K. Nayak, R. Dutt, V. Garg, V. Kailashiya, M. S. Muthu, S. Sonali and R. P. Singh, Int. J. Pharm., 2023, 633, 122587 CrossRef CAS.
- O. M. Elsharkasy, J. Z. Nordin, D. W. Hagey, O. G. de Jong, R. M. Schiffelers, S. E. Andaloussi and P. Vader, Adv. Drug Delivery Rev., 2020, 159, 332–343 CrossRef CAS PubMed.
- T. Saleem, A. Sumrin, M. Bilal, H. Bashir and M. B. Khawar, Saudi J. Biol. Sci., 2022, 29(4), 2063–2071 CrossRef CAS PubMed.
- L. Li, D. He, Q. Guo, Z. Zhang, D. Ru, L. Wang, K. Gong, F. Liu, Y. Duan and H. Li, J. Nanobiotechnol., 2022, 20(1), 50 CrossRef CAS PubMed.
- J. Hu, Y. Dong, L. Ding, Y. Dong, Z. Wu, W. Wang, M. Shen and Y. Duan, Signal Transduction Targeted Ther., 2019, 4, 28 CrossRef PubMed.
- Y. Huang, B. Zhou, H. Luo, J. Mao, Y. Huang, K. Zhang, C. Mei, Y. Yan, H. Jin, J. Gao, Z. Su, P. Pang, D. Li and H. Shan, Theranostics, 2019, 9(15), 4391–4408 CrossRef CAS.
- C. Li, K. Zhang, A. Liu, T. Yue, Y. Wei, H. Zheng, J. G. Piao and F. Li, Int. J. Pharm., 2021, 609, 121209 CrossRef CAS PubMed.
- Y. Lin, J. Xu and H. Lan, J. Hematol. Oncol., 2019, 12(1), 76 CrossRef.
- Y. S. Weng, H. Y. Tseng, Y. A. Chen, P. C. Shen, A. T. Al Haq, L. M. Chen, Y. C. Tung and H. L. Hsu, Mol. Cancer, 2019, 18(1), 42 CrossRef.
- M. R. Galdiero, G. Marone and A. Mantovani, Cold Spring Harbor Perspect. Biol., 2018, 10(8) DOI:10.1101/cshperspect.a028662.
- K. Inamura, Y. Shigematsu, H. Ninomiya, Y. Nakashima, M. Kobayashi, H. Saito, K. Takahashi, E. Futaya, S. Okumura, Y. Ishikawa and H. Kanda, Cancers, 2018, 10(8), 252 CrossRef PubMed.
- M. Zhu, L. Bai, X. Liu, S. Peng, Y. Xie, H. Bai, H. Yu, X. Wang, P. Yuan, R. Ma, J. Lin, L. Wu, M. Huang, Y. Li and Y. Luo, J. Immunother. Cancer, 2022, 10(12) DOI:10.1136/jitc-2022-005610.
- A. J. Boutilier and S. F. Elsawa, Int. J. Mol. Sci., 2021, 22(13), 6995 CrossRef CAS.
- V. Ejigah, O. Owoseni, P. Bataille-Backer, O. D. Ogundipe, F. A. Fisusi and S. K. Adesina, Polymers, 2022, 14(13), 2601 CrossRef CAS PubMed.
- J. Fang, W. Islam and H. Maeda, Adv. Drug Delivery Rev., 2020, 157, 142–160 CrossRef CAS.
- M. Izci, C. Maksoudian, B. B. Manshian and S. J. Soenen, Chem. Rev., 2021, 121(3), 1746–1803 CrossRef CAS PubMed.
- W. Yu, R. Liu, Y. Zhou and H. Gao, ACS Cent. Sci., 2020, 6(2), 100–116 CrossRef CAS PubMed.
- G. Zhao, H. Liu, Z. Wang, H. Yang, H. Zhao, Y. Zhang, K. Ge, X. Wang, L. Luo, X. Zhou, J. Zhang and Z. Li, Acta Biomater., 2022, 153, 529–539 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3bm01311a |
| This journal is © The Royal Society of Chemistry 2024 |