
Fluorogenic detection of cyanide ions in pure aqueous media through an intramolecular crossed-benzoin reaction: limitations unveiled and possible solutions†‡
Vincent
Gaumerd
 ab,
Yoan
Capello
a,
Quentin
Bonnin
a,
Pierre-Yves
Renard
c and
Anthony
Romieu
*a
ab,
Yoan
Capello
a,
Quentin
Bonnin
a,
Pierre-Yves
Renard
c and
Anthony
Romieu
*a
aInstitut de Chimie Moléculaire de l'Université de Bourgogne, UMR 6302, CNRS, Université de Bourgogne, 9, Avenue Alain Savary, 21000 Dijon, France. E-mail: anthony.romieu@u-bourgogne.fr; Web: https://www.icmub.com
bFrench Environment and Energy Management Agency, 20, avenue du Grésillé – BP 90406, 49004 Angers Cedex 01, France
cUniv Rouen Normandie, INSA Rouen Normandie, CNRS, Normandie Univ, COBRA UMR 6014, INC3M FR 3038, 76000 Rouen, France
First published on 15th November 2024
Abstract
Reaction-based fluorogenic sensing of lethal cyanide anions in aqueous matrices remains a big challenge. We have revisited the reported approach about an intramolecular crossed-benzoin reaction leading to the release of a phenol-based fluorophore. Fluorescence assays and RP-HPLC-MS analyses have helped us to highlight its limitations related to poor aqueous stability of probes and impossibility to achieve molecular amplification despite the assumed catalytic activation mechanism. Traceless cleavable linker strategies were considered to obtain usable cyanide-responsive chemodosimeters and statistical analyses of fluorescence data have been conducted in depth to accurately delineate their sensing performances, especially the limit of detection (LOD).
Introduction
Environmental pollution by cyanides (CN−) and related cyano compounds is clearly a topical issue of major importance. Indeed, a number of industries or industrial processes (e.g., gold mining, petrochemistry, steel manufacturing, …) have widely used and continue to use cyanides, and thus they are largely responsible for persistent pollution by this lethal anion, especially in aqueous matrices.1 This fact is exacerbated by deviant behaviours (e.g., deliberate waste dumping or chemical terrorism)2 that constantly pose heavy threats to humans and the environment. Consequently, detection and quantification of such a toxic contaminant is routinely conducted using different analytical methods (e.g., chromatographic, electrochemical, spectrophotometric techniques,…)3 in order to check that national or international legislations (e.g., the maximum acceptable concentration (MAC) of CN− in drinking water is fixed at 2.7 μM by the World Health Organization (WHO)4) are complied with. However, the high cost of these analytical instruments, tedious sample preparation steps, and requirement of trained professionals and dedicated laboratories have led to a quest for cheaper and easily usable analytical sensing technologies.5 Owing to its positive attributes including high sensitivity, signal output in a short time scale, and facile implementation within miniaturized, low-cost and portable devices, fluorescence spectroscopy quickly became a valuable alternative. The development of a cyanide-responsive molecular fluorescent probe is needed to convert the molecular recognition event into a measurable signal according to the intensiometric (ideally “OFF–ON”) or ratiometric mode.6 Depending on the reversibility or not of the reaction between the probe and analyte, either a fluorescent chemosensor or a fluorescent chemodosimeter is rationally designed.7 Over the past 25 years, a myriad of cyanide-responsive fluorescent probes have been reported and the most widely used sensing mechanisms are compiled in some comprehensive reviews.8 Due to the difficulties associated with the selectivity towards cyanide ions in complex aqueous matrices, the reaction-based approach is preferred to minimize interactions with interfering analytes. In this context, the nucleophilicity of CN− anions is often exploited in carbonyl or Michael-type addition reactions that lead to an hypsochromic shift of excitation/emission spectra of the probe, thus producing a ratiometric fluorogenic response.8c,g However, a majority of these fluorescent chemodosimeters are used in the presence of a large amount of organic co-solvent (often DMSO) due to their poor water solubility and their susceptibility to aggregation-caused quenching (ACQ) effects in aqueous solutions, or to enhance the nucleophilic reactivity of CN− anions. In order to overcome these limitations and to obtain a high-performance fluorogenic “OFF–ON” probe, a cutting-edge reaction-based approach was proposed by the Kim group in 2015.9 A strategy of protection–deprotection of the resorufin phenol moiety was established using a cyanide-assisted intramolecular crossed-benzoin reaction. IND-1 was studied in PBS (pH 7.4, containing 1% DMSO, v/v) and a limit of detection (LOD) of 4 nM was claimed. The sensing ability in complex biological systems was also evaluated in cyanide-doped living cells (HeLa and A549 cell lines). As part of a research program devoted to the development of a novel strategy for quick on-site detection of cyanide ions through a simple “mix and read” approach using a portable fluorometer, we have reconsidered the chemistry, aqueous stability and sensing performances of fluorescent chemodosimeter IND-1. Thanks to the implementation of a robust analytical methodology based on fluorescence assays and RP-HPLC-MS analyses, we have highlighted some major limitations associated with the fluorogenic intramolecular crossed-benzoin reaction, not disclosed in the Communication published by the Kim group. The most critical is the marked propensity of IND-1 to undergo undesired hydrolytic activation at physiological pH and in the absence of CN− ions, thus negatively impacting its sensing performances. In order to overcome this major difficulty and to obtain usable cyanide-responsive fluorescent probes based on this water-compatible cascade process, we have considered another phenol-based fluorophore, namely umbelliferone (or 7-hydroxycoumarin),10 known to be a worse leaving group than resorufin (the pKa value for its phenol moiety = 7.8 vs. 5.9 for resorufin).10,11 For the same purpose, structural diversification of the chemical linkage between the cyanide triggering unit and phenolic reporter (i.e., carboxamide linkage and traceless cleavable spacers12) has also been conducted to obtain three additional probes assumed as being more stable than IND-1. Herein, we report the synthesis of four novel fluorescent probes (Fig. 1) and their ability to detect CN− ions in PBS was examined in detail. Key results of this comprehensive study and recommendations for in-depth validation and evaluation of sensing performances (especially LOD) of fluorescent cyanide chemodosimeters intended to be used in pure aqueous media are highlighted.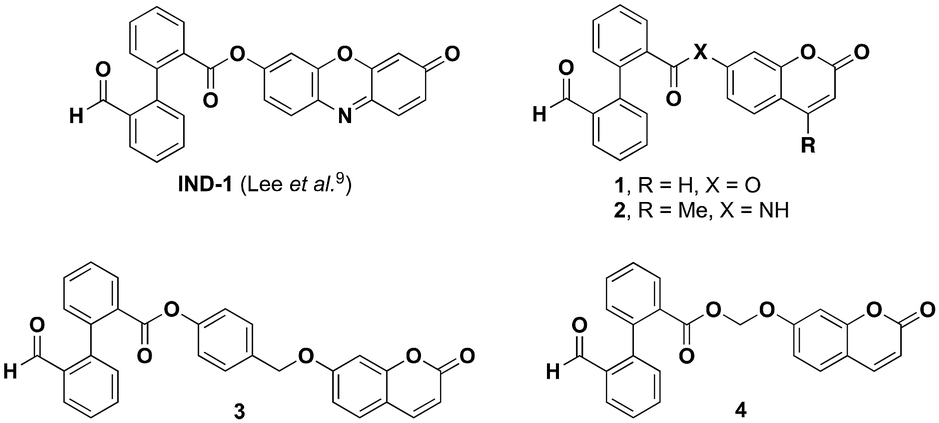 | ||
| Fig. 1 Structures of cyanide-responsive fluorogenic probes studied in this work. | ||
Experimental section
For all experimental details about general methods, instruments, chromatographic systems used for analytical and semi-preparative HPLC operations, Tables S1–S4,‡ and protocols for the synthesis of probes IND-1 and 1–4, and intermediates S1–S7, see the ESI.‡Safety note: all samples containing aq. solutions of KCN, from fluorescence assays and RP-HPLC-MS analyses, and rinsing waters (cleaning of quartz cells and HPLC glass vials) were pooled and incubated with bleach (2.6% active chlorine) for several days, and then transferred into a special tank devoted to the sole storage of cyanide wastes. Thereafter, this tank will be collected and treated by a specialist firm (SUEZ France).
Experimental details about stock solutions of probes and buffer used in fluorescence-based assays and study of the activation mechanism
- 1.0 mg mL−1 stock solutions of cyanide-responsive fluorogenic probes IND-1, 1–4, commercial umbelliferone (98%, Alfa Aesar, #L04082) and 9,10-phenanthrenequinone (>99%, Sigma-Aldrich, #156507) were prepared in DMSO (UV-spectroscopy grade, Honeywell Riedel-de-Haën, #41641).- 1.0 mg mL−1 stock solution of resorufin (sodium salt, Sigma-Aldrich, #230154-1G) was prepared in ultrapure H2O.
- 1.0 mg mL−1 stock solution of pig liver esterase (PLE, lyophilized powder, 27 units per mg solid, Sigma-Aldrich, #E3019-3.5 kU).
- PBS buffer (10 mM phosphate + 2.7 mM KCl + 137 mM NaCl, pH 7.5). This buffer was prepared by dissolving one PBS tablet (Fisher BioReagents™, #BP2944-100,) in 200 mL of ultrapure H2O. The pH value was checked using a pH-meter.
- Phosphate buffer (PB, 0.1 M, pH 7.4) was prepared by mixing aqueous solutions of Na2HPO4 (0.1 M) and KH2PO4 (0.1 M) prepared in ultrapure H2O (805 mL and 195 mL respectively for preparing 1 L). The pH value was adjusted to 7.4 by partial dissolution of a NaOH pellet.
Experimental details related to UV-vis absorbance measurements
UV-vis absorption spectra were obtained on a Varian Cary 50 Scan (single beam) spectrophotometer (software Cary WinUV) using rectangular quartz cells (Hellma, 100-QS, 45 × 12.5 × 12.5 mm, pathlength: 10 mm, chamber volume: 3.5 mL), at 25 °C (using a Lauda Ecoline Recirculating Chiller RE 106 combined with a temperature controller Lauda E100, connected to the spectrophotometer cell holder). The absorption spectra of 2′-formyl-[1,1′-biphenyl]-2-carboxylic acid cyanide-responsive probes IND-1 and 1–4 were recorded in PBS within the concentration range 1–30 μM (total volume = 3.0 mL, three distinct dilutions for the accurate determination of molar extinction coefficients of probes IND-1, and 1–3; five distinct dilutions for probe 4).Experimental details related to fluorescence-based assays and the methodology employed for LOD determination
Fluorescence spectroscopic studies (scan and kinetics modes) were performed with a SAFAS Flx-Xenius XC spectrofluorimeter (software: SP2000 v7.8.13.0) using specific fluorometer quartz cells (flx CELL QZ F MACRO, light pass: 10 mm, width: 10 mm, chamber volume 3.0 mL). All assays were performed at 25 °C (using a Grant instrument LT ecocool 100 combined with a temperature controller Grant instrument T ecocool 100, connected to the spectrofluorometer cell rack) and conducted with continuous mechanical stirring using small stirrer blades, in PBS + 1% (v/v) DMSO. The SAFAS Flx-Xenius XC spectrofluorimeter enables achieving until ten distinct kinetics within the same experiment. All Em spectra are automatically corrected by applying the correction file provided by the SAFAS company.Fluorescence emission time-course comparison at 10 μM
To minimize inaccuracies in the preparation of the diluted solutions, we prepared batches of 10 μM probes solution for each assay (see Table S1‡ for data related to the preparation of batches of probes). For all fluorescence emission time-course assays, we repeated each experiment thrice with three different batches of KCN dilutions, freshly prepared each time from the corresponding commercial salt (Sigma-Aldrich, #31252-100G, purity ≥ 97%). A 0.1 M KCN solution was prepared, then successive dilutions, calculated for adding 5 μL of analyte in a quartz cell, were achieved to obtain a range of 0.1–0.005 M (see Table S2‡ for conditions map of fluorescence time-course assays (concentration: 10 μM)).Fluorescence Em spectra and time-course assays were recorded after excitation at the suitable wavelength using the parameters compiled in Table S3.‡
Concentration dependence of fluorescence Em intensity for resorufin
Five distinct solutions of resorufin (sodium salt) were prepared in PBS from the stock solution and covering a range of concentrations ranging from 1.0 μM to 20 μM. Em spectra (wavelength range: 555–800 nm) were recorded in the scan mode, after excitation at 540 nm (Ex/Em bandwidths = 5/5 nm, PMT voltage = 439 V). Fluorescence Em intensity at λEmmax was plotted against concentration.Fluorescence emission time-course assay of probe IND-1 – influence of probe's concentration (1.0 μM vs. 10 μM)
To highlight the effect of resorufin fluorescence self-quenching on the fluorescence output signal measured during the time-course experiments, we conducted two distinct sets of kinetics (probe's concentration: 1.0 μM and 10 μM), with three distinct conditions: control, 10 equiv. KCN and 20 equiv. KCN. For each concentration, three independent probe's solutions were prepared in PBS + 1% (v/v) DMSO from the corresponding stock solution. KCN solutions at 0.1 and 0.01 M (entries 1 and 8 of Table S2‡) from the third replicate of “Fluorescence emission time-course comparison at 10 μM” were used. We determined the fluorescence parameters from a resorufin solution prepared in PBS + 1% (v/v) DMSO for each concentration (see Table S4‡ for the conditions map of fluorescence time-course assays conducted with 1.0 μM and 10 μM concentrations).Parameters for kinetics mode:
1.0 μM: Ex/Em = 565/595 nm; bandwidths Ex/Em = 2/2 nm; PMT voltage = 891 V,
10 μM: Ex/Em = 565/594 nm*; bandwidths Ex/Em = 2/2 nm; PMT voltage = 721 V.
(*)Please note: Em wavelength is different due to the technical impossibility to set the same Ex/Em wavelength couple with two different PMT voltage values.
Parameters for scan mode:
1.0 μM: Ex/Em range = 540/555–800 nm; bandwidths Ex/Em = 5/5 nm; PMT voltage = 515 V,
10 μM: Ex/Em range = 540/555–800 nm; bandwidths Ex/Em = 5/5 nm; PMT voltage = 439 V.
Determination of LOD
The determination of LOD was achieved using a methodology recommended by IUPAC.13a However, we want to emphasize on the fact that with only three replicates, we cannot determine and claim an accurate value of LOD but rather a relevant concentration range reachable with our experimental methodology (i.e., probe, concentration of probe, spectrofluorometer instrument, set of micropipettes, quartz cell, lab operator, not only the sensing molecule!).According to the tutorial shared by Evard et al.,13b we used the following equation:
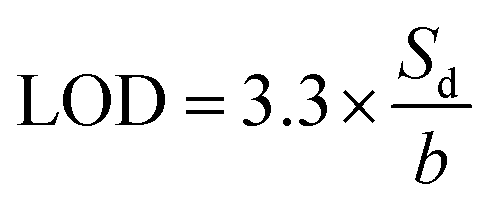
The assumptions used to apply this equation are the following:
- Normal distribution of the data,
- Homoscedasticity (homogeneity of the variance values) of the dataset (Hartley test),14 or the Fmax test using the following equation:

- Linearity of the calibration function (r2).
To determine the LOD of probes, we used data following Table S2 (entries 1–10)‡ at the F.I./F.I. control maximum (i.e., values at t = 15 min after addition of cyanide ions) which was the time that enabled us to obtain the best compromise between high fluorogenic response vs. limited spontaneous hydrolysis of probes. For each probe, we plotted the mean value of F.I. against the concentration of KCN to establish the calibration function. We then calculated the LOD using either the residual std dev. of the calibration curve or the intercept of the calibration function.
Fluorescence-based assays with PLE
All assays were performed at 25 °C (using a grant instrument LT ecocool 100 combined with a temperature controller grant instrument T ecocool 100, connected to the spectrofluorometer cell rack) and conducted with continuous mechanical stirring using small stirrer blades. Probe's concentration in a 3.0 mL fluorescence quartz cell was set to 5.0 μM in PB. The volume of the probe's solution was always 2.0 mL. The following sets of detection parameters were used:- Ex/Em 565/595 nm, slits 5 nm, PMT voltage = 417 V for the detection of released resorufin,
- Ex/Em 330/450 nm, slits 5 nm, PMT voltage = 430 V for the detection of released umbelliferone.
Fluorescence emission of released phenol was monitored at the suitable Ex/Em wavelength pair, over time with measurements every 60 s (duration of assay: 120 min). 1 U (37 μL) of PLE was added after 5 min of incubation in buffer alone. Blank experiments to assess the stability of the probes in PB were achieved in the same way but without adding PLE.
Results and discussion
Synthesis of fluorescent probes equipped with Kim's cyanide triggering unit
The preparation of all fluorescent probes considered in the present study requires the prior synthesis of the cyanide triggering unit (under its carboxylic acid form) initially devised by the Kim group for the implementation of the fluorogenic intramolecular crossed-benzoin reaction onto the resorufin scaffold.9 This compound named 2′-formyl-[1,1′-biphenyl]-2-carboxylic acid S2 was readily obtained through a Suzuki–Miyaura cross-coupling reaction between methyl 2-iodobenzoate and 2-formylphenylboronic acid, followed by ester saponification, according to protocols reported by Penhoat et al.15 (for synthetic Scheme S1 and experimental details, see the ESI‡). Next, effective conversion of S2 into active 1-hydroxy-7-azabenzotriazole (HOAt) esters was achieved by brief treatment with uronium-type coupling reagent HATU and DIEA.16 Subsequent O-acylation of resorufin (sodium salt) or umbelliferone was conducted in dry MeCN at room temperature, and purification by flash-column chromatography over silica gel provided probes IND-1 and 1 in the pure form and with satisfactory isolated yields (51% and 59% respectively). Conversely, N-acylation of 7-amino-4-methylcoumarin was found to be less effective and the resulting amide-based probe 2 was recovered in the pure form only with a yield of 12% (purification by semi-preparative RP-HPLC purification using a C8 column). In addition to the simple construction of these two-component chemodosimeters (triggering unit + fluorophore), we devised original synthetic routes towards more sophisticated probes 3 and 4 bearing a traceless cleavable linker (Schemes 1 and 2). Indeed, it is now well established that the incorporation of such a structural unit enables one to solve problems such as poor aqueous stability and low fluorogenic reactivity, negatively impacting the response time.12 In this context, we first considered the installation of the well-known and reliable para-hydroxybenzyl moiety using a multi-step synthetic route whose key step is the selective phenol esterification of para-hydroxybenzyl alcohol (PHBA) with an acyl-Bunte salt intermediate readily generated under the conditions reported by Liao et al.17 (Scheme 1). Thus, acid anhydride S3 was reacted with PHBA in the presence of sodium thiosulfate (Na2S2O3) and Na2CO3 in dry DMF at room temperature to give aryl ester S4 in 44% yield after purification by column chromatography over silica gel. Thereafter, bromination of benzyl alcohol was achieved with an excess of PBr3 in dry THF to yield S5 which finally reacted with umbelliferone under conventional conditions currently used for phenol alkylation (i.e., K2CO3, DMF). Several purification attempts have led to identify semi-preparative RP-HPLC over a C8 column as the most appropriate method to recover probe 3 with a high purity and a moderate yield. | ||
| Scheme 1 Synthetic route towards umbelliferone-based probe 3 bearing the PHBA-type spacer [Ar = argon, FC (SiO2) = flash-column chromatography over silica gel, MsCl = mesyl chloride, O/N = overnight, RT = room temperature, TEA = triethylamine]. | ||
 | ||
| Scheme 2 Synthetic route towards umbelliferone-based probe 4 bearing the acetal-type spacer [TBAHSO4 = tetrabutylammonium hydrogenosulfate, O/N = overnight, RT = room temperature]. | ||
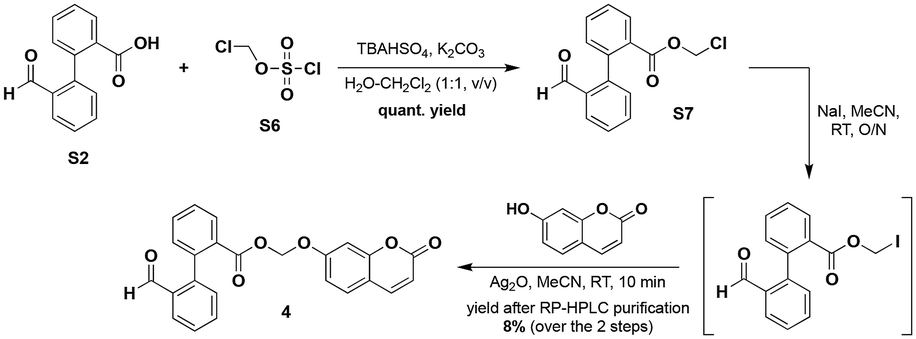
A second traceless linker belonging to the class of acetals, namely methylene aryloxy ester, was also regarded and the corresponding coumarin-based probe 4 was successfully synthesised according to a multi-step route inspired by that published by the Lavis group for the preparation of stable fluorogenic esterase substrates based on the fluorescein scaffold18 (Scheme 2).
Iodomethyl ester, prepared through phase transfer alkylation of 2′-formyl-[1,1′-biphenyl]-2-carboxylic acid S2 and subsequent Finkelstein iodination of S7, was reacted with umbelliferone in the presence of Ag2O to give 4 which was subjected to purification by semi-preparative RP-HPLC (C8 column). The structures of all probes were unambiguously confirmed by IR, NMR and mass spectrometry (see the ESI‡). Furthermore, their high purity (>98%) was confirmed by RP-HPLC analyses and found to be ideally suited for their use in fluorescence assays.
Aqueous stability, fluorogenic response of two-component probes for cyanide ions and revised sensing mechanism
We first examined the fluorescence response of IND-1 and 1 to cyanide analyte through in vitro time-course assays, conducted in PBS (10 mM phosphate + 2.7 mM KCl + 137 mM NaCl, pH 7.5) at 25 °C, and under experimental conditions closely modelled on those used by the Kim group,9 to allow fair and accurate comparisons of our respective results. Briefly, 10 μM solutions of probes IND-1 and 1 were incubated in PBS alone or in PBS with different amounts of KCN (1–20 equiv.). Firstly, for IND-1, we observed a spontaneous hydrolysis of this probe upon its incubation in PBS alone as supported by the gradual increase of fluorescence emission at 595 nm (assigned to the emissive phenolate form of the released resorufin), and a quite similar fluorescence kinetics curve was obtained after addition of 10 equiv. of KCN (Fig. 2A). These disappointing results about the sensing performances of IND-1 differ completely from those reported by the Kim group. Furthermore, at 10 μM, we noted partial and gradual precipitation of IND-1 in this buffer (see Fig. S13 in the ESI‡) thus distorting quantitative analysis of its fluorogenic activation. We also evidenced a self-quenching of the released resorufin at concentrations higher than 5 μM, as well supported by the concentration-dependent fluorescence study displayed in Fig. S11 (see the ESI‡). These further observations enable us to affirm that the reported LOD value of 4 nM does not reflect the real sensing performances of this fluorescent chemodosimeter in PBS. In order to minimize these adverse effects related to a too large working concentration, we repeated fluorescence assays at 1.0 μM. However, under these more diluted conditions, the aqueous instability was also largely favoured (see Fig. S12 in the ESI‡). Even more surprisingly, for the coumarin-based probe 1, we also noted a non-negligible hydrolysis upon its incubation in PBS alone, thus suggesting that the ester linkage of cyanide-responsive trigger is fragile whatever the phenol-based fluorophore used (Fig. 2B). However, its fluorogenic response at 450 nm towards cyanide ions (10 equiv. of KCN) is a bit better than that obtained with IND-1. According to normalized data (Fig. 2C), the two probes showed a modest maximum of fluorescence intensity (F.I.) ratio. | ||
| Fig. 2 (A-B) Time-dependent changes in the fluorescence intensity of fluorogenic probes IND-1 and 1 in the presence of KCN, in PBS (pH 7.5) at 25 °C. (C) Normalised mean values of F.I. to control condition (PBS alone). (D) Bar chart of the normalised mean values of fluorescence intensity to control condition after 15 min of incubation with KCN for IND-1 and 1. All experiments were independently repeated thrice. Error bars represent standard deviation. See Table S3‡ for parameters used for fluorescence kinetics. | ||
To solve inherent problems associated with the poor aqueous stability of IND-1 and 1, we next studied the spectral behavior of aniline-based probe 2 which has a more stable amide linkage between triggering and reporter units, under the same conditions described above. Unfortunately, this chemodosimeter was found to be unreactive towards cyanide ions as supported by the lack of a fluorogenic response (see Fig. S8 in the ESI‡). Due to the stabilization of the electrophilic carbonyl carbon by nitrogen lone-pair delocalisation,19 the last step of the crossed-benzoin reaction involving intramolecular nucleophilic attack of the cyanohydrin carbanion did not occur (Fig. 3).
 | ||
| Fig. 3 Revised detection mechanism of cyanide ions based on the fluorogenic intramolecular crossed-benzoin reaction leading to the release of a fluorescent phenol (resorufin for probe IND-1 and umbelliferone for probe 1). Please note: for probes 3 and 4, an elimination step (release of PHBA or formaldehyde respectively) completes this domino process. | ||
A second feature of the Kim's reaction-based cyanide sensing strategy related to the catalytic mechanism assumed for the activation of IND-1 raised issues. Indeed, in view of the concomitant release of fluorescent resorufin, 9,10-phenanthrenequinone and the cyanide anion itself (Fig. 3), the recycling of the latter analyte should be effective to produce formal molecular target amplification through its further reaction with multiple reporter molecules IND-1. Curiously, this valuable asset was not mentioned and studied whereas it would be particularly useful for achieving trace-level detection,20 and in the present case, detection of substoichiometric amounts of CN− ions. Furthermore, for practical sensing applications of such a probe, a dramatic different methodology from the one used by the Kim group for LOD determination, should be implemented to perform quantification of analytes, through prior generation of a calibration curve using known concentrations of cyanide ions and for a given set time point. In order to gain insights into the activation mechanism of probes IND-1 and 1 and to evaluate the conversion rate, reaction mixtures corresponding to their incubation in PBS alone or in the presence of 10 equiv. of KCN were subjected to RP-HPLC-ESI-MS analyses (both full scan and single ion monitoring (SIM) modes; the latter one being considered to enhance the detection sensitivity of poorly ionizable compounds such as 9,10-phenanthrenequinone) (see Fig. 4 for illustrative examples of UV and SIM elution profiles obtained for the reaction of IND-1, and S16–S18 in the ESI‡). After 4 h of incubation whatever the probe studied and the conditions used (i.e., the absence or presence of CN− analyte), we observed in all elution profiles a peak assigned to the phenol-based fluorophore (resorufin, tR = 2.9 min, m/z = 214.1 [M + H]+, calcd for C12H8NO3+ 214.0; umbelliferone, tR = 2.5 min, m/z = 163.1 [M + H]+, calcd for C9H7O3+ 163.0). We also noticed the formation of 2′-formyl[1,1′-biphenyl]-2-carboxylic acid (S2, tR = 3.5 min, m/z = 225.1 [M − H]−, calcd for C14H9O3− 225.2), thus proving that undesirable hydrolysis of the ester linkage of IND-1 and 1 readily occurs in PBS. For the assays conducted with cyanide analyte (10 equiv.), two further peaks were observed at tR = 3.7 min and tR = 4.0 min, identified as 9,10-phenanthrenequinone (MS(ESI+): m/z = 209.1 [M + H]+, calcd for C14H9O2+ 209.1) and cyanohydrin intermediate 5 (MS(ESI+): m/z = 236.2 [M + H]+, calcd for C15H10NO2+ 236.1) respectively. As clearly illustrated in Fig. 4, the independent and unambiguous detection of released phenol (resorufin), compound S2, 9,10-phenanthrenequinone and cyanohydrin 5 was most conveniently achieved using ESI-MS SIM mode tuned to the mass expected for the molecular ion of species to identify. In chromatograms with SIM(+) adjusted to 209.0 and 236.0 ± 0.5 u.ma., three and two peaks were observed respectively (Fig. 4E and F). It is easy to explain why without compromising our mechanistic hypothesis: (1) starting probe IND-1 and 2′-formyl[1,1′-biphenyl]-2-carboxylic acid S2 were detected on SIM(+) channel 209.0 ± 0.5, due to their propensity to form 9,10-phenanthrenequinone during the ESI+ ionization process; (2) on SIM(+) channel 236.0 ± 0.5, it was possible to detect released resorufin because the sodium adduct of its molecular ion has the same mass (calcd mass for C12H7NNaO3+ 236.0). Lastly, the predominance of starting probe IND-1 (tR = 4.4 min) even after 4 h of reaction with KCN (Fig. 4B), confirmed the negative impact of its poor solubility in PBS on its reactivity. A possible hypothesis is that partial precipitation and formation of aggregates may complicate the accessibility of the triggering moiety to cyanide ions. This poor reactivity was not observed with umbelliferone-based probe 1 (Fig. S16 in the ESI‡). The formation of 5 as a stable product in PBS was not considered by the Kim group, and two distinct reaction pathways may be suggested to explain it: (1) nucleophilic addition of CN− onto activated carbonyl of newly formed 9,10-phenanthrenequinone, and (2) premature ending of the cyanide-mediated intramolecular crossed-benzoin reaction. We ruled out the first hypothesis by conducting further assay involving incubation of commercial 9,10-phenanthrenequinone with 10 equiv. of cyanide ions in PBS (containing 1% DMSO) and the non-formation of 5 was unambiguously confirmed by RP-HPLC-ESI-MS analyses (see Fig. S19 in the ESI‡). Consequently, we assumed that the last elimination step leading to the release of CN− and formation of 9,10-phenanthrenequinone is not really favoured in PBS, and the activation mechanism of the probe can be revised as shown in Fig. 3, with 5 as the major end product. Our study clearly demonstrates the non-catalytic feature of this intramolecular crossed-benzoin reaction explaining among others why the magnitude of the fluorogenic response grows when probes are incubated with increasing amounts of analyte.
 | ||
| Fig. 4 RP-HPLC elution profiles (system A′′) of cyanide-mediated activation of fluorogenic probe IND-1 (concentration: 10 μM, in PBS (pH 7.5) + 1% DMSO (v/v)) with 10 equiv. of analyte for 4 h at 20 °C. (A) UV detection at 260 nm of control (IND-1 incubated in PBS alone for 4 h), (B) UV detection at 260 nm of the crude reaction mixture and (C–F) ESI-MS SIM channels for optimal detection of released resorufin, 2′-formyl[1,1′-biphenyl]-2-carboxylic acid S2, 9,10-phenanthrenequinone and cyanohydrin 5 respectively. | ||
Aqueous stability and cyanide sensing performances of probes bearing a traceless cleavable spacer
In order to overcome the limitations clearly identified with probes IND-1 and 1, we next assessed the fluorogenic behaviour of the two fluorescent chemodosimeters based on a cyanide-sensitive traceless cleavable spacer. Concerning the fluorogenic activation of 3, the stabilization effect caused by its PHBA-based linker is too marked and a very weak increase of fluorescence emission at 450 nm, corresponding to the release of umbelliferone, was observed over a prolonged period of time (Fig. 5A and Fig. S9 in the ESI‡). Therefore, no relevant conclusion can be drawn on a possible selective activation of this probe by cyanide against its non-specific hydrolysis. Consequently, it was not possible to determine the LOD for CN− ions. Conversely, the presence of an acetal-based linker within the structure of 4 slowed the undesired hydrolysis process without compromising cyanide-triggered umbelliferone release. This is clearly reflected by the enhancement of the normalized F.I. (Fig. 5B) with regard to the results obtained for the linker-free probe 1. The positive impact of the ester bond stabilization on the fluorogenic behaviour was also noted at lower cyanide doses. Indeed, after 15 min of incubation with an equimolar amount of KCN (Fig. 5D), the normalized F.I. response of 4 is multiplied by a factor of 1.3 which is close to the value 1.4 obtained for activation of IND-1 with 10 equiv. of KCN. This higher resistance to hydrolysis was also supported by a further fluorescence-based enzyme assay conducted with pig liver esterase (PLE) (Fig. S20 in the ESI‡). Indeed, 7-acetoxycoumarin chosen as the positive control probe was completely hydrolysed after 15 min of incubation with PLE in phosphate buffer (0.1 M, pH 7.4); fluorescence emission at 450 nm quickly reached a plateau corresponding to the quantitative release of umbelliferone. Conversely, probes 3 and 4 exhibited only 2.5% and 10% of activation respectively, and after the same incubation time. This positive result opens the way to a possible implementation of such fluorescent chemodosimeters in biological media/samples contaminated by CN− ions. | ||
| Fig. 5 (A and B) Time-dependent changes in the fluorescence intensity of fluorogenic probes 3 and 4 in the presence of KCN, in PBS (pH 7.5) at 25 °C. (C) Normalised mean values of F.I. to control condition (PBS alone). (D) Bar chart of the normalised mean values of fluorescence intensity to control condition after 15 min of incubation with KCN for IND-1, 1, 3 and 4. All experiments were independently repeated thrice. Error bars represent standard deviation values. See Table S3‡ for parameters used for fluorescence kinetics. | ||
Furthermore, as supported by statistics and data analysis (i.e., standard deviation calculation), assays conducted with 4 led to a significant improvement in the accuracy of results produced whatever the conditions tested. Finally, as already observed for IND-1 and 1 (Fig. 2C), the maximum of F.I. ratio was obtained after 15 min of cyanide incubation (Fig. 5C). Interestingly, the plotting of normalized F.I. (at this time) as a function of KCN concentration pointed out a good correlation (r2 > 0.995) not observed with IND-1 and 1 (see Fig. S13–S15 in the ESI‡). A detectable concentration range of 4–8.5 μM was obtained whereas poor results were calculated for linker-free probes (LOD range = 23–49 μM, r2 = 0.956 for IND-1, and 12–25 μM, r2 = 0.988 for 1); these results illustrate a reliable indication of the LOD reachable with Kim's chemodosimeter approach.
Conclusions
In summary, we have synthesised and re-assessed the fluorogenic properties of cyanide-responsive reaction-based probe IND-1. Two major weaknesses considerably impacting its sensing performances in aqueous matrices, namely poor hydrolytic stability and formation of cyanohydrin 5, have been highlighted. In order to address the issue of non-specific hydrolysis, Kim's triggering unit was attached to the phenol moiety of umbelliferone directly or through a traceless cleavable spacer, leading to three novel cyanide-responsive chemodosimeters. The very comprehensive analytical methodology used to validate them associated with a rigorous statistical analysis of fluorescence data enables us to identify acetal-based fluorescent probe 4 as the best candidate for potential practical applications, even if its fluorogenic “OFF–ON” response towards cyanide ions still remains modest (LOD: 4–8.5 μM). Furthermore, the systematic RP-HPLC-MS analysis of samples from fluorescence assays was essential to decipher precisely the activation mechanism of such cyanide-responsive probes. We hope that this clear demonstration will convince researchers working in the burgeoning field of reaction-based fluorescent probes to adopt this analytical methodology in a routine manner. Undoubtedly, this will facilitate the production of more reliable results on detection/quantification of environmental pollutants, using fluorescent chemodosimeters. To overcome the limitation associated with the modest fluorogenic “OFF–ON” response disclosed herein, the implementation of Kim's triggering moiety to zero-background “covalent-assembly” fluorescent probes whose activation leads to in situ formation of bright pyronin fluorophores21,22 is currently under investigation by our team and will be reported in due course.23 The implementation of this unusual activity-based sensing methodology could help to address the important challenge of detection of organophosphorus compounds bearing a cyanide as the leaving group (e.g., the G-type nerve agent tabun or its mimic diethyl cyanophosphate (DCNP)).24,25Author contributions
Vincent Gaumerd: investigation, formal analysis and writing – original draft. Yoan Capello: investigation. Quentin Bonnin: investigation, formal analysis and writing – review & editing. Pierre-Yves Renard: funding acquisition, supervision and writing – review & editing. Anthony Romieu: conceptualization, funding acquisition, supervision and writing – review & editing. All authors have read and agreed to the published version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.‡ All raw analytical and spectroscopic data have been deposited in dat@UBFC repositery (https://doi.org/10.25666/dataubfc-2024-07-17).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is dedicated to Prof. Jacques Coste (ENSCM, co-inventor of PyBOP® peptide coupling reagent) who passed away in 2018, first mentor of A. Romieu. Financial support from ADEME (French Agency for Ecological Transition) and AID (French Defence Innovation Agency) for the Ph.D. grant of V. Gaumerd (grant no. 2022006, 2022-2025, 36 months) and Agence Nationale de la Recherche (ANR, AAPG 2018, PRC, DetectOP_BChE, ANR-18-CE39-0014) especially for the post-doc fellowship of Dr Y. Capello is greatly acknowledged. Labex SynOrg (ANR-11-LABX-0029), Carnot Institute I2C, and the graduate school for research XL-Chem (ANR-18-EUR-0020-XLCHEM) are also acknowledged for partial financial support. The authors thank the “Plateforme d'Analyse Chimique et de Synthèse Moléculaire de l'Université de Bourgogne” (PACSMUB, https://www.wpcm.fr/) for access to analytical and molecular spectroscopy instruments. The authors also warmly thank Mrs Marie-José Penouilh (University of Burgundy, PACSMUB) for HRMS measurements, Mr Cédric Balan (University of Burgundy, ICMUB, PACSMUB) for Karl Fischer titrations, Dr Kévin Renault (CNRS, CMBC lab, Institut Curie, Orsay) for helpful suggestions about fluorescent probes bearing a traceless cleavable spacer, Drs Angélique Pipier (UBFC, ICMUB), Ibai Valverde (CNRS, ICMUB) and David Monchaud (CNRS, ICMUB) for valuable discussions about statistical analyses of fluorescence data and good practice relating to sample preparation, and Mrs Léna Bouillard (University of Burgundy) for her assistance in using dat@UBFC service (https://search-data.ubfc.fr/) to deposit our raw data.References
- D. A. Dzombak, R. S. Ghosh and G. M. Wong-Chong, Cyanide in Water and Soil - Chemistry, Risk, and Management, CRC Press, 1st edn, 2005 Search PubMed.
- M. Flahault, Le cyanure dans l'histoire et intoxications actuelles, Université Bordeaux 2, 2015 Search PubMed.
- For selected reviews, see: (a) J. Ma and P. K. Dasgupta, Anal. Chim. Acta, 2010, 673, 117–125 CrossRef CAS PubMed; (b) P. Yadav and M. P. Goutam, Asian J. Pharm. Pharmacol., 2020, 6, 150–163 CrossRef CAS.
- Guidelines for Drinking-water Quality, World Health Organization, Geneva, 3rd edn, 2008 Search PubMed.
- R. Jackson and B. A. Logue, Anal. Chim. Acta, 2017, 960, 18–39 CrossRef CAS PubMed.
- Z. Xu, X. Chen, H. N. Kim and J. Yoon, Chem. Soc. Rev., 2010, 39, 127–137 RSC.
- S. Singha, Y. W. Jun, S. Sarkar and K. H. Ahn, Acc. Chem. Res., 2019, 52, 2571–2581 CrossRef CAS PubMed.
- For selected reviews, see: (a) F. Wang, L. Wang, X. Chen and J. Yoon, Chem. Soc. Rev., 2014, 43, 4312–4324 RSC; (b) A. Bencini and V. Lippolis, Environ. Sci. Pollut. Res., 2016, 23, 24451–24475 CrossRef CAS PubMed; (c) P. B. Pati, Sens. Actuators, B, 2016, 222, 374–390 CrossRef CAS; (d) D. Udhayakumari, Sens. Actuators, B, 2018, 259, 1022–1057 CrossRef CAS; (e) I. Yahaya and Z. Seferoglu, Fluorescence Dyes for Determination of Cyanide, in Photochemistry and Photophysics - Fundamentals to Applications, IntechOpen, 2018, pp. 179–196 Search PubMed; (f) S. Chakraborty, S. Paul, P. Roy and S. Rayalu, Inorg. Chem. Commun., 2021, 128, 108562 CrossRef CAS; (g) E. Keleş, B. Aydıner and Z. Seferoğlu, Curr. Org. Synth., 2023, 20, 61–76 CrossRef PubMed; (h) C. I. David and H.-i. Lee, Microchem. J., 2024, 200, 110359 CrossRef; (i) A. Kumar, E. Jeong, Y. Noh and P. S. Chae, Methods, 2024, 222, 57–80 CrossRef CAS PubMed.
- J. H. Lee, J. H. Jang, N. Velusamy, H. S. Jung, S. Bhuniya and J. S. Kim, Chem. Commun., 2015, 51, 7709–7712 RSC.
- For a comprehensive review about phenol-based fluorophores, see: J. J. M. Hurley, Q. J. Meisner, C. Huang and L. Zhu, ACS Omega, 2021, 6, 3447–3462 CrossRef CAS PubMed.
- P. Lefrançois, Développement d'un microréacteur biomimétique pour l'analyse in situ d'activités enzymatiques par couplage de l’électrochimie et de la microscopie de fluorescence, Université de Bordeaux, 2017 Search PubMed.
- J. Yan, S. Lee, A. Zhang and J. Yoon, Chem. Soc. Rev., 2018, 47, 6900–6916 RSC.
- (a) G. L. Long and J. D. Winefordner, Anal. Chem., 1983, 55, 712A–724A CrossRef CAS; (b) H. Evard, A. Kruve and I. Leito, Anal. Chim. Acta, 2016, 942, 23–39 CrossRef CAS PubMed.
- G. K. Kanji, 100 Statistical Tests, SAGE Publications Ltd, 1993 Search PubMed.
- M. Penhoat, S. Leleu, G. Dupas, C. Papamicaël, F. Marsais and V. Levacher, Tetrahedron Lett., 2005, 46, 8385–8389 CrossRef CAS.
- F. Albericio, J. M. Bofill, A. El-Faham and S. A. Kates, J. Org. Chem., 1998, 63, 9678–9683 CrossRef CAS.
- W. Liao Jr., S.-Y. Lin, Y.-S. Kuo and C.-F. Liang, Org. Lett., 2022, 24, 4207–4211 CrossRef PubMed.
- L. Tian, Y. Yang, L. M. Wysocki, A. C. Arnold, A. Hu, B. Ravichandran, S. M. Sternson, L. L. Looger and L. D. Lavis, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4756–4761 CrossRef CAS PubMed.
- The Amide Linkage: Structural Significance in Chemistry, Biochemistry and Materials Science, ed. A. Greenberg, C. M. Breneman, J. F. Liebman, Wiley, 2000 Search PubMed.
- S. Goggins and C. G. Frost, Analyst, 2016, 141, 3157–3218 RSC.
- For reviews on “covalent-assembly” fluorescent probes, see: (a) X. Luo, L. Gu, X. H. Qian and Y. C. Yang, Chem. Commun., 2020, 56, 9067–9078 RSC; (b) X. Chen, Z. Huang, L. Huang, Q. Shen, N.-D. Yang, C. Pu, J. Shao, L. Li, C. Yu and W. Huang, RSC Adv., 2022, 12, 1393–1415 RSC.
- (a) Z. Lei and Y. Yang, J. Am. Chem. Soc., 2014, 136, 6594–6597 CrossRef CAS PubMed; (b) L. Song, Z. Lei, B. Zhang, Z. Xu, Z. Li and Y. Yang, Anal. Methods, 2014, 6, 7597–7600 RSC; (c) S. Debieu and A. Romieu, Org. Biomol. Chem., 2017, 15, 2575–2584 RSC; (d) S. Debieu and A. Romieu, Tetrahedron Lett., 2018, 59, 1940–1944 CrossRef CAS; (e) K. Renault, S. Debieu, J.-A. Richard and A. Romieu, Org. Biomol. Chem., 2019, 17, 8918–8932 RSC; (f) K. Renault, Y. Capello, S. Yao, S. Halila and A. Romieu, Chem. – Asian J., 2023, 18, e202300258 CrossRef CAS PubMed; (g) Z. Yang, Z. Wang, Y. Peng, H. Yang, Q. Wang, X. Jia and X. Liu, Org. Biomol. Chem., 2024, 22, 8024–8031 RSC.
- For the sole examples of cyanide-responsive “covalent-assembly” fluorescent probes respectively validated in aqueous MeCN and in a micellar medium (water with CTAB), see: (a) D. Kim, S.-Y. Na and H.-J. Kim, Sens. Actuators, B, 2016, 226, 227–231 CrossRef CAS; (b) C. Liang and S. Jiang, Analyst, 2017, 142, 4825–4833 RSC.
- For selected reviews, see: (a) L. Chen, D. Wu and J. Yoon, ACS Sens., 2018, 3, 27–43 CrossRef CAS PubMed; (b) Q. Chen, Y. Sun, S. Liu, J. Zhang, C. Zhang, H. Jiang, X. Han, L. He, S. Wang and K. Zhang, Sens. Actuators, B, 2021, 344, 130278 CrossRef CAS; (c) V. Kumar, H. Kim, B. Pandey, T. D. James, J. Yoon and E. V. Anslyn, Chem. Soc. Rev., 2023, 52, 663–704 RSC.
- (a) N. Dey, S. Jha and S. Bhattacharya, Analyst, 2018, 143, 528–535 RSC; (b) S. Mondal, B. Krishna, S. Roy and N. Dey, Analyst, 2024, 149, 3097–3107 RSC.
Footnotes |
| † A preprint was previously posted on ChemRxiv, see: https://doi.org/10.26434/chemrxiv-2024-0b4vd. |
| ‡ Electronic supplementary information (ESI) available: Experimental details related to photophysical characterisation, fluorescence-based in vitro assays and HPLC-MS analyses, synthetic procedures of compounds not disclosed in the article and analytical data of all synthesised compounds. See DOI: https://doi.org/10.1039/d4an01368a |
| This journal is © The Royal Society of Chemistry 2025 |