
Tuning the electro-catalytic activity of the Zn–Cu MOF/rGO nanocomposite as a novel enzyme-free electrochemical sensor for the detection of the oxytocin hormone†
Md Zainul
Abedeen
a,
Manish
Sharma
a,
Himmat Singh
Kushwaha
 a and
Ragini
Gupta
*ab
a and
Ragini
Gupta
*ab
aMaterials Research Centre, Malaviya National Institute of Technology Jaipur, Jaipur 302017, India. E-mail: rgupta.chy@mnit.ac.in
bDepartment of Chemistry, Malaviya National Institute of Technology Jaipur, Jaipur 302017, India
First published on 14th November 2024
Abstract
Oxytocin (OXY), a peptide hormone and neurotransmitter essential for biological processes with nine distinct amino acid residues, has received significant attention due to its illegal use in food adulteration and stimulating milk ejection in cattle. Herein, for the first time, electrochemical detection of oxytocin (OXY) is reported using a novel nanocomposite consisting of a Zn–Cu metal–organic framework (Zn–Cu MOF) decorated on reduced graphene oxide (rGO). An octahedral surface morphology with a crystalline structure of 45 nm in size, formation of a metal–oxygen bond, an enhanced pore diameter of 6.8 nm, a specific surface area of 70.8 m2 g−1, and a pore volume of 0.08 cm3 g−1 were revealed by different characterization techniques. The electro-catalytic activity of the Zn–Cu MOF/rGO nanocomposite increased substantially owing to the synergistic effect, which is evident from cyclic voltammetry (CV) when compared to those of the Zn-MOF, the Cu-MOF, Zn MOF/rGO, Cu MOF/rGO, and Zn–Cu MOF, keeping other parameters the same. Moreover, the electrochemical impedance spectroscopy (EIS) spectra reveal the excellent conductivity of the nanocomposite. The experimental parameters, viz. electrolyte pH (5), supporting electrolyte (0.1 M ABS), and volume of coating (12 μL), were optimized. The differential pulse voltammetry (DPV) technique was adopted to determine the OXY with the lowest limit of detection (LOD) of 1.1 nM (S/N = 3) with a linear range of 40–400 nM. The analytical application of the modified electrode was examined by spiking OXY in pasteurized toned milk, skimmed powder milk, animal milk, and RO water, with a good recovery range of 95–106%.
1. Introduction
Food adulteration has been practiced since ancient times; its detection is essential due to the ill effects of the contaminants that affect human beings. Oxytocin (OXY) is a neuropeptide hormone consisting of nine amino acids (cysteine (Cys), tyrosine (Tyr), isoleucine (Iso), glutamine (Glu), asparagine (Asp), cysteine (Cys), proline (Pro), leucine (Leu), and glycinamide (Gly)) primarily authorized for use in hospitals for medical emergencies.1 Additionally, OXY promotes growth when injected into plant parts such as stems and given to milk-producing animals in the form of injection to increase the milk extraction quantity.2 Uncontrolled and non-physiological intake of OXY can result in hormonal imbalance, nervous breakdown, memory loss, and sterility due to the chance that the residues of hormones can accumulate and may be passed down the food chain through the intake of milk.3 According to the Food and Drug Adulteration Prevention Act of 1960, oxytocin is prohibited from being used in vegetables. Furthermore, oxytocin has been classified as a Schedule H1 medication under the Drug and Cosmetic Act of 1940, meaning that it can only be supplied with a doctor's prescription.4The detection of OXY has been attempted using several analytical techniques such as high-performance liquid chromatography (HPLC),5 capillary electrophoresis (CE),6 and liquid chromatography-mass spectrometry (LC-MS),7 which require sophisticated instruments and expertise in handling, are time-consuming, lack on-site detection, and need large amounts of solvents and reagents. Furthermore, S. Rastogi et al. developed a colorimetric approach for detecting OXY in vegetable samples using cysteamine-functionalized gold nanoparticles (Cys-AuNPs).8 Another approach proposed by Rastogi et al. discusses the RGB colorimetric method used to assess the oxytocin in milk, fruits, and vegetables, considering the Cys-AuNP aggregation principle.9 Digital images of the color shift brought about by the addition of Cys-AuNP aggregation were captured and utilized to measure the analyte's R, G, and B intensities to examine its concentration objectively. This method is based on colors, so there is a high chance that some interfering substances may give the same colors.10 The electrochemical method is a good choice due to its high sensitivity, the possibility of on-site detection, less time consumption, and more comprehensive linearity range.11 Electrochemical sensors are advantageous in many fields, including the food industries, environmental monitoring, and the healthcare sector, but they still suffer from some problems. Fouling of the electrode surface is one of the major concerns that lead to alterations in the electrochemical properties, leading to problems in selectivity and sensitivity.12 Moreover, potentiostatic control may be impacted, and problems with signal perception and interpretation may arise due to the electromechanical instability of the electrical double layer, which is a source of noise in sensor signals.13 Despite these drawbacks, continuous research endeavors to overcome these obstacles by emphasizing the development of more resilient and long-lasting electrochemical sensors, aiming to enhance performance in real samples.14 In this regard, Liu et al. demonstrated an electrochemical assay to quantify OXY, which responds in milliseconds but suffers from problems such as a narrow detection range, the need for waveform modifications, and tedious electrode modifications.15
Asai Kai et al. proposed the detection of OXY using an electrochemical method employing boron-doped diamond (BDD) electrodes,16 in which the CV curve shows an oxidation peak at +0.7 V (vs. Ag/AgCl) of OXY in a phosphate buffer solution, attributed to the oxidation of the tyrosyl moiety's phenolic group. Additionally, the linearity of the current observed with BDD microelectrodes in flow injection analysis (FIA) spans the 0.1–10.0 μM oxytocin concentration range with a 50 nM detection limit. Nano-scale materials have attracted much attention in various scientific studies as they have been widely used in multiple applications. Moreover, carbon nanofibers (CNFs) and chitosan were combined to develop environmentally friendly disposable membranes.17 Its surface was studied and further functionalized to use the membrane as a bio-electrode to detect OXY. CNF has many reactive carbon edges, low electrical resistance, high porosity, and reinforcing ability. The developed bio-electrode demonstrated an LOD of 24.98 ± 11.37 pg mL−1 quantified using electrochemical impedance spectroscopy. Moreover, P. S. Sharma et al. developed an electrochemical sensing platform using plastic antibodies instead of traditional biological receptors for sensing OXY.18 Molecularly imprinted polymers (MIPs) coated on gold (Au) electrodes consisting of miniaturized microfluidic cells were used for the electrochemical sensing of OXY, using impedimetric sensing techniques, which specifically bound with the target analyte.
Graphene-based materials have garnered significant academic attention in detection research related to the creation of electrochemical sensors using effective electro-catalysts due to their easy functionalization, excellent conductivity, and faster electron transfer. Previous investigations revealed that graphene's electrochemical properties can be further improved by doping it with heteroatoms (N, P, and B).19 The charge polarization between dopants and carbon atoms increases electron transport, enhancing the conductivity of the graphene lattice. Thomas et al. reported using nitrogen and phosphorus co-doped graphene (NPG) modified electrodes for the electrochemical detection of OXY.20 Square wave stripping voltammetry (SWSV) was used to study the electro-oxidation behavior of OXY in a 0.1 M PBS electrolyte solution. There were two ranges where the oxidation peak current was linear: 0.1–10 nM and 15–95 nM, with the lowest LOD found to be 40 pM. The author used platinum as a working electrode, which is not cost-effective, and the doping of heteroatoms on graphene derived from coke, whose quality is more important.
Metal–organic frameworks (MOFs) are crystalline porous nano-scale materials with excellent attributes such as high specific surface area, tunable pore size, and various topologies that make them useful in different applications.21 Problems such as chemical stability and electron transfer hindrance arise with the monometallic MOF. Recent research shows that bimetallic MOFs can act as better electro-catalysts than monometallic MOFs due to the synergistic effect, larger active sites, and porosity of the former.22 MOFs are less conductive because combining metal ions and organic linkers leads to poor orbital overlap, making electron movement difficult.23,24 Copper offers higher conductivity than zinc, improving the sensor signal.25 In this instance, a particular composite combining MOFs with graphene has garnered much interest lately.26
Integrating two metal ions demonstrates the synergistic catalytic activity, resulting in improved selectivity, higher active sites, and better electro-activity.23 Also, the electro-catalytic responses of different electrodes were assessed, revealing that the bimetallic MOF is a better electro-catalyst than the monometallic MOF. Moreover, adding rGO enhances its conductivity, thereby increasing its sensitivity. Therefore, the Zn–Cu MOF/rGO nanocomposite was chosen due to more active sites, higher active surface area, and an enhanced electron transfer rate. To date, a bimetallic MOF/rGO nanocomposite has not been utilized for the electrochemical detection of the oxytocin hormone. Herein, the one-pot solvothermal method is being reported for the synthesis of the Zn–Cu MOF/rGO nanocomposite, which was further explored using various characterization techniques such as FE-SEM, HR-TEM, XRD, BET, Raman spectroscopy, and FT-IR. Tuning of different materials (Zn-MOF, Cu-MOF, Zn MOF/rGO, Cu-MOF/rGO, Zn–Cu MOF, and Zn–Cu MOF/rGO) has been explored towards the sensing of OXY. After decorating the MOF on an rGO sheet, its specific surface area and porosity have increased substantially, leading to excellent electro-catalytic behavior. Moreover, spiking and recovery analysis found the modified electrode suitable for real-sample applications in milk samples and water.
2. Experimental section
2.1 Synthesis of the Zn–Cu MOF, reduced graphene oxide (rGO), and the Zn–Cu MOF/rGO nanocomposite
The metal–organic framework was synthesized according to the previously reported literature using a solvothermal approach.27 In brief, zinc nitrate hexahydrate (45 mmol), copper(II) nitrate trihydrate (138 mmol), and terephthalic acid (1.57 g) were mixed in the beaker containing ethanol (30 mL) and DMF (30 mL). Stirring was done at room temperature for 3 hours using a magnetic stirrer to ensure proper mixing. Then, the mixture was transferred to a stainless steel autoclave (100 mL) and kept in a hot air oven at 85 °C for 20 hours. The mixture was washed with the DMF and ethanol using centrifugation at 7500 rpm for 5 min each, and the precipitate was kept in a vacuum oven at 80 °C for 8 h.Graphene oxide (GO) was synthesized using the Modified Hummers’ method, taking graphite as an initial precursor material.28 In brief, graphite flakes (3 g) and H2SO4 (150 mL) were added to the round-bottomed flask and stirred using a magnetic stirrer for one hour, in an ice bath with the temperature maintained at 5 °C. After that, KMNO4 (9 g) was added to the above mixture as an oxidizing agent and then sonicated (5 min), followed by stirring (25 min), which was repeated 12 times. DI water (600 mL) was added to the solution to quench the reaction and left for two hours, and then hydrogen peroxide (45 mL) was added, which was then sonicated (15 min) and stirred (45 min). The mixture was washed with 1 M HCl to remove excess metal ions using centrifugation at 7500 rpm and then washed many times with DI water until the pH of the solution became neutral and dried under vacuum (80 °C, 10 h) to obtain a sheet-like structure. Reduced graphene oxide (rGO) was synthesized by adding GO to the DI water in a beaker and then sonicated for 4 hours. To this, a suitable amount of hydrazine hydrate (750 μL) was added dropwise while continuously stirring for 4 hours and then washed using centrifugation at 7500 rpm with 1 M HCl and DI water until the pH became neutral.
The nanocomposite was synthesized using a solvothermal method, taking Cu(NO3)2·3H2O and Zn(NO3)2·6H2O at a molar ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, terephthalic acid (0.39 g) and rGO (5 wt%) dissolved in a beaker containing ethanol and DMF at the same ratio. The mixture was agitated continuously for 3 hours, then transferred to the autoclave, and kept in a hot air oven for 20 hours at 85 °C (Fig. 1). Washing was done with the ethanol and DMF using centrifugation at 7500 rpm thrice each and placed in a vacuum oven for 8 hours at 80 °C. Moreover, the materials viz. Cu-MOF/rGO, Zn-MOF, Cu-MOF, and Zn-MOF/rGO, were synthesized following the same protocol (Table S1†). In Zn–Cu MOF/rGO-A, the molar ratio of the Zn and Cu salts was 1:1, and the rGO weight percent was kept at 5%. In Zn–Cu MOF/rGO, the molar ratio of Zn and Cu salts was 1:3, and the rGO weight percent was kept at 5%. In Zn–Cu MOF/rGO-B, the molar ratio of Zn and Cu salt was 3:1, and the rGO weight percent was kept at 5%. All the reagents, materials, and instrumentation information are discussed in the ESI (Text S1 and S2†).
:1, terephthalic acid (0.39 g) and rGO (5 wt%) dissolved in a beaker containing ethanol and DMF at the same ratio. The mixture was agitated continuously for 3 hours, then transferred to the autoclave, and kept in a hot air oven for 20 hours at 85 °C (Fig. 1). Washing was done with the ethanol and DMF using centrifugation at 7500 rpm thrice each and placed in a vacuum oven for 8 hours at 80 °C. Moreover, the materials viz. Cu-MOF/rGO, Zn-MOF, Cu-MOF, and Zn-MOF/rGO, were synthesized following the same protocol (Table S1†). In Zn–Cu MOF/rGO-A, the molar ratio of the Zn and Cu salts was 1:1, and the rGO weight percent was kept at 5%. In Zn–Cu MOF/rGO, the molar ratio of Zn and Cu salts was 1:3, and the rGO weight percent was kept at 5%. In Zn–Cu MOF/rGO-B, the molar ratio of Zn and Cu salt was 3:1, and the rGO weight percent was kept at 5%. All the reagents, materials, and instrumentation information are discussed in the ESI (Text S1 and S2†).
 | ||
| Fig. 1 Schematic illustration of the synthesis of the Zn–Cu MOF/rGO nanocomposite. | ||
2.2 Preparation of Zn–Cu MOF/rGO/GCE
Initially, the GCE was cleaned using a micro-cloth polishing pad with a 0.05 μm alumina slurry and DI water to get a mirror-like surface. The nanocomposite Zn–Cu MOF/rGO, isopropanol, and Nafion were mixed in a vial to create the suspension of the composite material, which was then sonicated for 15 minutes. An aliquot of this dispersed sample was dropcast onto the GCE designated as Zn–Cu MOF/rGO/GCE and used for further experimental study. Other modified electrodes were prepared using the same protocol.2.3 Real sample preparation
Analysis of OXY in the real sample was done to verify its applicability; different milk samples were bought from a nearby supermarket. The pre-treatment of the milk sample was done according to a general procedure.29 In brief, the milk (5 g) was mixed in a beaker containing trichloro-acetic acid (61 mM, 5 mL) and methanol solvent (4.9 M, 35 mL). The resultant mixture was sonicated for 15 minutes, and then the supernatant was collected using centrifugation was filtered out, and kept for use in further experiments.3. Results and discussion
3.1 Materials characterization
The synthesized Zn–Cu MOF/rGO surface morphology was observed from a field emission scanning electron microscope (FE-SEM) image in which the Zn–Cu MOF appears to be in an octahedral shape decorated on the rGO sheet. Fig. 2(a–c) shows the images of the nanocomposite recorded at the magnifications of 5000, 10000, and 20000. Fig. S2† shows FE-SEM images of the Zn–Cu MOF. Furthermore, elemental mapping was performed to verify the percentage composition of the different elements in the nanocomposite (Fig. S1(a)†). The energy dispersive spectrometer (EDS) spectrum of Zn–Cu MOF/rGO shows significant characteristic peaks corresponding to carbon (74%), copper (9%), zinc (1%), oxygen (6%), and nitrogen (10%) (Fig. S1(b)†). Based on the elemental mapping of the different materials, copper (Fig. S1(c)†), oxygen (Fig. S1(d)†), carbon (Fig. S1(e)†), zinc (Fig. S1(f)†), and nitrogen (Fig. S1(g)†), it was concluded that the elements were uniformly distributed. The insertion of the aromatic nitrogen (N2) moiety in the nanocomposite was due to the hydrazine treatment of GO (graphene oxide).30
 | ||
| Fig. 2 FE-SEM image of the Zn–Cu MOF/rGO nanocomposite at different magnifications: (a) 5000, (b) 10000, and (c) 20000. (d–f) High-resolution transmission electron microscopy (HR-TEM) images of the Zn–Cu MOF/rGO nanocomposite. (g) Corresponding SAED pattern of the Zn–Cu MOF/rGO nanocomposite. | ||
Fig. 2(d–f) shows an HR-TEM image of the Zn–Cu MOF/rGO nanocomposite in which an octahedral-shaped MOF decorated on the rGO sheet was observed. The image clearly depicts the successful formation of the Zn–Cu MOF/rGO nanocomposite recorded at different magnifications. Fig. 2(g) shows the selected area electron diffraction (SAED) pattern employing different lattice planes such as (330), (321), (222), and (211), verifying the polycrystalline nature of the material, and its interlayer planes is illustrated in Fig. S3† with a lattice fringe width of 0.21 nm. Fig. 3(a) shows Raman spectra of rGO, the Zn–Cu MOF, and Zn–Cu MOF/rGO. In the spectra of the Zn–Cu MOF, the peaks at 1610 cm−1, 1430 cm−1, 1105 cm−1, 865 cm−1, and 490 cm−1 are attributed to the stretching modes of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C of the aromatic rings of ligands, the symmetric vibration of the terephthalic acid carboxylates, stretching of C–C of the aromatic rings, the C–H bond of the MOF, and metal oxide peak, respectively.31 The Raman spectra of rGO exhibit three characteristic peaks at 1350 cm−1 ascribed to the D band, 1592 cm−1 attributed to the G band, and 2760 cm−1 corresponding to the 2D band.32 The D band denotes sp3 hybridized carbon and degree of randomness (defects); the G band ascribes to the vibration of sp2-hybridized carbon. The appearance of an extra peak (2D band) in the nanocomposite spectrum indicates successful MOF decoration on the rGO sheet.
C of the aromatic rings of ligands, the symmetric vibration of the terephthalic acid carboxylates, stretching of C–C of the aromatic rings, the C–H bond of the MOF, and metal oxide peak, respectively.31 The Raman spectra of rGO exhibit three characteristic peaks at 1350 cm−1 ascribed to the D band, 1592 cm−1 attributed to the G band, and 2760 cm−1 corresponding to the 2D band.32 The D band denotes sp3 hybridized carbon and degree of randomness (defects); the G band ascribes to the vibration of sp2-hybridized carbon. The appearance of an extra peak (2D band) in the nanocomposite spectrum indicates successful MOF decoration on the rGO sheet.
 | ||
| Fig. 3 (a) Raman spectra of rGO, the Zn–Cu MOF, and Zn–Cu MOF/rGO, (b) FT-IR spectra, (c) XRD graph of the Zn–Cu MOF, and Zn–Cu MOF/rGO, N2 adsorption–desorption isotherm, pore size distribution (inset), (d) the Zn–Cu MOF, and (e) Zn–Cu MOF/rGO obtained from the Brunauer–Emmett–Teller (BET) technique. | ||
Furthermore, Fig. 3(b) gives information on the functional groups in the synthesized material evaluated using FTIR spectroscopy. The prominent peak at 1630 cm−1 and 1393 cm−1 is attributed to the asymmetric and symmetric stretching of the carboxylate groups, indicating a ligand in the framework.33 The other significant peaks available at 1500 cm−1 are attributed to the C–C stretching of the aromatic ring, 1085 cm−1 corresponds to the C–O stretching, and 745 cm−1 is related to the C–H bending vibration of benzene.34,35 Usually, the peak at 940 cm−1 is observed due to the O–H bending vibration of the ligand in the raw material of the terephthalic acid, which has disappeared, indicating the attachment of the ligand to the metal ions.36 The peak visibility around 560 cm−1 indicates the metal–oxide formation in the nanocomposite material.
Fig. 3(c) shows the XRD spectra of the bimetallic MOF (Zn–Cu MOF) and the Zn–Cu MOF/rGO nanocomposite, and the existence of the sharp diffraction peaks indicates the crystalline nature of the as-synthesized nanocomposite. The significant diffraction peaks at 24.5°, 34.9°, 37.8°, and 43.1° correspond to the lattice planes (211), (222), (321), and (330), denoting that both the metals Zn and Cu are present, and exhibit a polycrystalline structure37 (JCPDS card number: 01-071-0397). Moreover, a broad diffraction peak observed at 26.5° indicates the presence of hexagonal graphitic layers with the lattice plane of (002).38 Also, it can be observed that the rGO has not disrupted the crystalline nature of the Zn–Cu MOF, and a small shift in the diffraction peak angle and decrease in the peak intensity illustrates that the Zn–Cu MOF has been successfully decorated on the rGO sheet. Some unidentified peaks of smaller intensity may be due to the guest molecules such as solvents trapped within the MOF surface.39 The average crystalline sizes were calculated to be 25 nm and 45 nm for the Zn–Cu MOF and the Zn–Cu MOF/rGO nanocomposite, respectively, using the Debye–Scherrer equation. It can be seen that the spatial arrangement of the bimetallic MOFs consisting of the two different metals existing in the same secondary-building units (SBU) shows stronger synergistic interactions. Moreover, the two metals with similar coordination spheres and ionic radius will likely exist in the same SBU. Given that Cu and Zn ions have comparable ionic sizes, both Cu and Zn ions may contribute to forming the octahedral structure.27 The even distribution of Cu and Zn elements may confirm the EDS mapping shown in Fig. S1.† According to the earlier study by T. Qi et al., oxygen atoms in the MOF framework octahedrally coordinate the Zn and Cu atoms.40 Furthermore, the pseudo octahedral consists of many edge/corner centers connected along the crystallographic plane direction, forming three-dimensional layers separated by organic linkers.41 The MOF network facilitates ion accessibility, which improves mass transport along the bimetallic MOF material and, in turn, reveals exposed active sites, good stability, and electro-catalytic activity.25 Also, the edge/corner-rich structure increases the effective catalytic sites. Generally, the addition of highly conductive reduced graphene oxide has no remarkable effects on bimetallic MOF crystallization.42
BET was conducted to know about the porosity, pore volume, and specific surface area of the nanocomposite. Fig. 3(d and e) shows the N2 adsorption–desorption isotherm following the type IV isotherm and pore size distribution diagram as shown in the inset of the Zn–Cu MOF and Zn–Cu MOF/rGO.43 The mean pore diameter, volume, and specific surface area have increased substantially to 6.41 nm, 0.08 cm3 g−1, and 70 m2 g−1 of Zn–Cu MOF/rGO compared to those of the Zn–Cu MOF (Table 1). These results show that on successful incorporation of the MOF on the rGO sheets, the porosity has increased and possesses a mesoporous structure.44
| Pore diameter (nm) | Volume (cm3 g−1) | Specific surface area (m2 g−1) | |
|---|---|---|---|
| Zn–Cu MOF | 3.4 | 0.03 | 35 |
| Zn–Cu MOF/rGO | 6.8 | 0.08 | 70.8 |
3.2 Electrochemical behavior of the Zn–Cu MOF/rGO nanocomposite
The electrochemical efficacies of the electrodes, including the bare GCE, Zn-MOF/GCE, Cu-MOF/GCE, Zn-MOF/rGO/GCE, Cu-MOF/rGO/GCE, Zn–Cu MOF/GCE, and Zn–Cu MOF/rGO/GCE, were evaluated by recording the CV at the scan rate of 50 mV s−1 employing a redox pair [Fe(CN)6]3−/4− as shown in Fig. 4(a). The anodic peak current values of the Zn–Cu MOF/rGO/GCE and bare GCE are 0.206 mA and 0.096 mA, respectively, while the cathodic peak current values are 0.204 mA and 0.094 mA. Moreover, the difference between the anodic and cathodic peak potential values are 178 mV and 577 mV for the Zn–Cu MOF/rGO/GCE and bare GCE, respectively. The increase in the current and decrease in the difference in the peak potential (anodic and cathodic peak potential) indicate that the faster electron transfer kinetics, enhanced behavior of oxidation and reduction leading to improvement in the electro-catalytic activity for the modified GCE.45 Moreover, the intensity of the peak current in the case of other modified electrodes (the Zn-MOF, the Cu-MOF, Zn-MOF/rGo, Cu-MOF/rGO, and the Zn–Cu MOF) was observed to be less than that of Zn–Cu MOF/rGO (Table S2†). This effect might be related to the synergistic effect between the two metal ions leading to the beneficial surface modification and faster electron transfer between the redox pair of [Fe(CN)6]3−/4− ions and the surface of modified electrodes.46 | ||
| Fig. 4 (a) CV profile of the different materials coated on the GCE, (b) electrochemical impedance spectroscopy (EIS) spectra of the different modified electrodes in an electrolyte solution containing 5 mM [Fe (CN)6]3−/4−/0.1 M KCl, (Resistance of the supporting electrolyte (R1), double layer capacitance (Cdl), Warburg impedance (Zw), (c) CV curves of the different modified electrodes without the electro-active species in the electrolyte solution (0.1 M ABS) (pH 5), and (d) CV curves of the rGO modified GCE with and without the electro-active species, OXY (0.1 M ABS) (pH 5). | ||
The electro-active surface area is an essential physio-chemical parameter influencing the accumulation efficiency of analytes onto the electrode surface. The chance of the electro-catalytic reaction occurring is greatly enhanced when the exposed surface area at the electrode-electrolyte interface is higher.47 The Randles–Sevcik equation (eqn (i)) calculates the electro-active surface area of the different electrodes.
| Ipa = 2.69 × 105n3/2AD1/2v1/2C | (i) |
Here, the anodic peak current is represented as Ipa, the scan rate is taken as ν(V s−1), the diffusion constant is denoted as D (D = 7.20 × 10−6 cm2 s−1), the electron transfer number transferred is taken as n, and concentration is represented as C (mol cm−3). Using the above equation, the active surface areas of the different modified electrodes were found to be 0.060 cm2, 0.002 cm2, 0.0035 cm2, 0.032 cm2, 0.0160 cm2, 0.035 cm2, and 0.127 cm2 for the bare GCE, Zn-MOF/GCE, Zn-MOF/rGo/GCE, Cu-MOF/GCE, Cu-MOF/rGO/GCE, Zn–Cu MOF/GCE, and Zn–Cu MOF/rGO/GCE, respectively (Table S2†).
Additionally, the cyclic voltammetry curve was recorded for Zn–Cu MOF/rGO-A and Zn–Cu MOF/rGO-B at the scan rate of 50 mV s−1 under 10 mM K3[Fe(CN)6]/K4[Fe(CN)6] electrolyte conditions (Fig. S4†). From the CV analysis, it was seen that the maximum active surface area was obtained for Zn–Cu MOF/rGO compared to other synthesized materials (Table S2†). Moreover, the oxidation and reduction peak current were greatly enhanced, and the difference between the oxidation and reduction peak potential was reduced on modifying the GCE with Zn–Cu MOF/rGO, illustrating the faster electron transfer rate and higher number of available active sites. Therefore, this material was taken for further study.
The EIS study evaluated the interface characteristics of the modified electrodes. Fig. 4(b) shows the Nyquist plot for Zn–Cu MOF/rGO, Zn–Cu MOF, and bare GCE in the frequency range of 0.1–100 kHz, and its Randles equivalent circuit is shown in the inset. The semi-circular part is attributed to high frequency, followed by the linear region related to low frequency.48 The value of the charge transfer resistance (Rct) equal to the diameter of the semi-circular region, which was found to be 93 Ω for Zn–Cu MOF/rGO/GCE, 200 Ω for Zn–Cu MOF/GCE, and 245 Ω for the bare GCE. The Zn–Cu MOF/rGO/GCE exhibits a lower Rct value than the bare GCE, indicating a more facile route for the electron transfer rate and enhanced electro-catalytic activity. After decorating the MOF on the rGO sheet, electron conductance between the active site and the electrolyte has increased significantly, which might be attributed to the synergistic effect leading to the excellent electrochemical behavior of the Zn–Cu MOR/rGO/GCE.49
Cyclic voltammetry (CV) was recorded for the different materials modified on the GCE in an electrolyte consisting of 0.1 M ABS (pH 5), keeping the volume of coating on the GCE the same without the analyte in the solution (Fig. 4c). The peak current intensity was maximum for Zn–Cu MOF/rGO compared to the other different electrodes modified (Table S3†). Zn-MOF/GCE has a slightly higher redox peak current intensity than the bare GCE, and on addition of the rGO to the Zn-MOF, the intensity of the redox peak current has increased significantly. On modifying the GCE with the Cu-MOF, the oxidation peak current was 0.208 mA, which increased to 0.246 mA with the addition of rGO to the Cu-MOF. During the modification of the GCE with the bimetallic Zn–Cu MOF, the oxidation peak current increased to 0.258 mA; this increment may be due to the synergistic interaction between the two metal ions, leading to faster electron movement.39 The conductivity was increased further with the addition of rGO to the bimetallic MOF, as revealed from the enhancement of the peak current intensity. Also, the difference between the oxidation and reduction peak potentials reduced for Zn–Cu MOF/rGO, signifying higher electro-catalytic ability and an indication of the more active site. It can also be observed from the DPV curve (Fig. S5(a)†) that there is a slight change in the oxidation peak potential of the Cu-MOF and Zn–Cu MOF, so it can be inferred that Cu(II) ions may be responsible for the binding with the OXY moieties. Here, Zn(II) ions provide the synergistic effect, while the rGO was used to enhance the conductivity of the MOF. Furthermore, to check the influence of rGO on the OXY sensing, the CV (Fig. 4d) and DPV were recorded at different concentrations of the OXY (Fig. S5(b)†). It is assumed that there was no change in the behavior, demonstrating no effect on the sensing properties but rather only an increase in the conductivity.
 | ||
| Fig. 5 (a) Cyclic voltammetry (CV) profile of Zn–Cu MOF/rGO/GCE recorded at different scan speeds in 0.1 M ABS (pH 5) containing 100 nM OXY, (b) corresponding plot of peak current vs. scan speed, (c) corresponding plot of the logarithm of anodic peak current vs. logarithm of scan speed, and (d) corresponding linear plot between anodic peak potential (Ep) and the logarithm of scan speed. | ||
As illustrated in Fig. 5(b), the oxidation peak current shows a linear relation with the scan speed, whose equation is expressed as I (mA) = 0.0005v (mV s−1) + 0.216 with a correlation coefficient of 0.99. This linearity demonstrates that the process is a surface-controlled electro-catalytic reaction at the surface electrode. Fig. 5(c) shows the linear plot between the logarithmic scan speed and the logarithm of the anodic peak current whose slope is less than 0.5, indicating diffusion-controlled process on the electrode surface.51 Furthermore, a slight shift in the anodic peak potential was observed with the increase in the scan speed (25–225 mV s−1), as shown in Fig. 5(d), demonstrating the irreversibility of the electrode process.52 The linear equation is given as Ep = 0.052logv + 0.22 with a correlation coefficient of 0.98. The number of electrons transferring in the process can be evaluated using Laviron's equation, which is expressed as follows:53
 | (ii) |
Here, the Faraday constant (F) is taken as 96500, temperature (T) is 298 K, and the R is taken as the universal gas constant. Since the process is reversible, the electron transfer coefficient (α) is assumed to be 0.5.54 On equating the slope of the above equation with the slope of the linear relationship between the anodic potential and logarithm of scan speed, the number of electrons transferring in the process was found to be 2.2 (≈2). The electron transfer rate (Ko) can be found using Laviron's equation (eqn (iii)) using the scan speed of 0.05 V s−1.53
 | (iii) |
The electron transfer rate was calculated to be 1.94 s−1 by substituting the value in the above equation. The Brown–Anson model can determine the average surface concentration (Γ) of ionic species on the electrode surface.55
 | (iv) |
The linear expression between the scan speed and peak current intensity is expressed as Ip = 0.0005v + 0.216. On equating the slope of the above linear relationship line with the Brown–Anson model, the average surface concentration is 1.05 × 10−9 mol cm−3 using the active surface area as A = 0.127 cm2.
 | ||
| Fig. 6 (a) Cyclic voltammetry profile of the Zn–Cu MOF/rGO/GCE in the presence of the different supporting electrolytes, (b) effect of volume of coating on the GCE in terms of peak current obtained from the DPV curve, (c) CV of modified electrodes for different pH values of supporting electrolyte, (d) corresponding change in the intensity of current and potential with a change in pH value (electrolyte: 0.1 M ABS, scan speed: 50 mV s−1), (e) variation of the CV profile for the different concentrations ranging from 40 to 400 nM, and (f) corresponding linear calibration plot. | ||
Fig. 6(b) shows the variation of peak current intensity obtained from the DPV curve by coating the GCE with the different amounts of the Zn–Cu MOF/rGO nanocomposite. Electrode modification was done by drop-casting different volumes of the suspension and used for further analysis in 0.1 M ABS electrolyte after drying the coating. The maximum peak current was obtained when the coating on the GCE was 12 μL in 0.1 M ABS electrolyte solution (pH 5) consisting of 100 nM OXY. The lower peak current was obtained when the coating volume on the GCE was too low due to the thin layer, as it did not cover the electrode surface adequately. Furthermore, an increase in the coating on the GCE increases the thickness, which may hinder the transfer of electrons. Therefore, the optimum coating (12 μL) was considered for further study.
The effect of the different pH values of the 0.1 M ABS consisting of 100 nM OXY in Zn–Cu MOF/rGO/GCE was investigated by recording the CV curve (Fig. 6(c)). The change in the oxidation peak current was observed when the pH of the electrolyte was varied, and the maximum current intensity was observed when the pH of the electrolyte was 5, revealing the lowest detection limit and highest sensitivity at this pH value, which is attributed to the charge interaction on the electrode surface. Furthermore, the current intensity started decreasing at a higher pH due to the decrease in the adsorption of the OXY. Also, the Zn–Cu MOF/rGO nanocomposite contains functional groups that may deprotonate at a higher pH, leading to a negatively charged surface resulting in the electrostatic repulsion and a decrease in the possibility of adsorption of analytes. Furthermore, when the pH of the electrolyte was increased, the oxidation peak potential of OXY was changed and had a linear relationship with the pH variation demonstrated by the linear equation Ep = −0.09 pH + 0.805, as shown in Fig. 6(d). The slope of the equation compared with that of the Nernst equation E = −0.059 pH + c is almost equal, revealing the equal number of protons and electrons transferred during the reaction.56
The electrochemical technique is often a recognition element between a novel nano-scale material and an analyte. The sensory layer undergoes significant alteration upon binding, leading to changes in the charge and surface density.57Fig. 6(e) illustrates the CV profile of the electrode of the different concentrations of OXY (40–400 nM) at a sweep rate of 50 mV s−1 and its linear calibration curve is demonstrated in Fig. 6(f). With the increase in the concentration of OXY, the anodic peak current decreases without a change in the shape of the CV curve, which is attributed to the synergistic effect of the bimetallic MOF having a high adsorption capacity.58
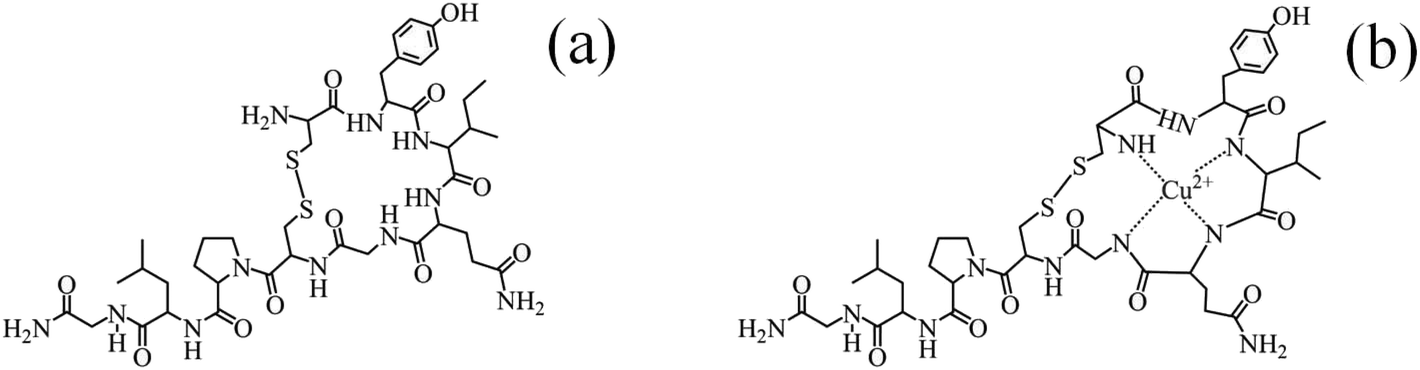 | ||
| Fig. 7 (a) The chemical structure of OXY and (b) plausible mechanism of the formation of the oxytocin-copper complex.59,65 | ||
 | ||
| Fig. 8 (a) Differential pulse voltammetry response curves for the different concentrations of OXY ranging from 40 to 400 nM in 0.1 M ABS (pH 5), (b) corresponding linear calibration curve, (c) selectivity: change in peak current response in the presence of 100 nM OXY and other interfering ions of the same concentration as 0.1 M ABS (pH 5) obtained from the DPV curve, and (d) intensity of the peak current of five different electrodes. | ||
The limit of detection (LOD) was found to be 1.1 nM using the formula (LOD = 3 m/σ), where m is the slope of the calibration plot, and σ is the standard deviation, which was obtained from the response signal of five blank measurements.66 Furthermore, the analytical parameters of the proposed electrochemical sensor based on Zn–Cu MOF/rGO/GCE for OXY sensing are compared with previously reported OXY sensors in terms of the detection method, LOD, and linear range (Table 2). Overall, the demonstrated non-enzymatic electrochemical sensor displayed superior analytical performance regarding linear range and LOD compared to the conventional approach. However, few recently reported works show better analytical performance than the presented approach due to enzyme-based sensing.
| Techniques | Linearity range (ng mL−1) | LOD (ng mL−1) | Ref. |
|---|---|---|---|
| Enzyme immunoassay (EIA) | 0.010–0.250 | 0.03 | 2 |
| UV-HPLC | 23.75–12500 |
4.1 | 5 |
| Capillary electrophoresis | 150–4000 | 50 | 6 |
| LC-MS | 15–115 | 2 | 7 |
| Cysteamine functionalized gold nanoparticles (Cys-Au NPs)/colorimetric | 1660–8330 | 1079 | 8 |
| Cysteamine functionalized gold nanoparticles/RGB colorimetric method | 1320–7960 | 1220 | 9 |
| Boron-doped diamond electrode/electrochemical | 100.7–10.7 | 50.35 | 16 |
| Chitosan-carbon nanofiber (CNF)/electrochemical | 10–105 | 0.024 | 17 |
| MIP/Au electrode/electrochemical | 60420–106 |
60420 |
18 |
| N, P co-doped graphene electrode/electrochemical | 0.1–10 15–95 | 0.04 | 20 |
| Surface plasmon resonance sensor | 0.01–1 | 0.003 | 68 |
| Zn–Cu MOF/rGO/GCE | 40–400 (40–400 nM) | 1.1 (1.1 nM) | This work |
Moreover, the five different electrodes were separately modified with the Zn–Cu MOF/rGO nanocomposite, keeping the same optimized parameter to determine 100 nM OXY in the supporting electrolyte (Fig. S8†). The current intensity of all electrodes was almost constant, as is evident from Fig. 8(d), whose relative standard deviation (RSD) was 2.5%, revealing satisfactory reproducibility. Furthermore, CVs were recorded in the same electrolyte solution to investigate the stability of the proposed non-enzymatic electrochemical sensor. This study shows that peak current intensity stays almost constant after 25 cycles with a scan rate of 50 mV s−1 (Fig. S9(a) & (b)†). Also, to assess the long-term stability, the modified electrode was kept in the desiccator for fourteen days. The result showed that peak current intensity declined to 98% after the first week and approximately 92% after the second week, indicating adequate sensor stability.
:3. Then, different amounts of OXY were added according to the standard addition method. First, the reading was taken and recorded without the addition of OXY. After that, a known concentration was added to the electrolyte. Each of the pre-processed samples was spiked with a different amount of OXY, and we can estimate the recovered concentration of the OXY using a linear calibration plot obtained from the DPV response curve. The Zn–Cu MOF/rGO/GCE demonstrates a considerable recovery range of 95–106% (Table 3), suggesting that the applicability of the electrochemical sensor for the determination of OXY in real samples without any influence from the interfering ions, and its good recovery value demonstrates the significance of the sensor.
| Real sample | Spiked concentration | Recovered concentration | Recovery (%) | Standard deviation (%) |
|---|---|---|---|---|
| Pasteurized toned milk | 50 | 53 | 106 | 3.6 |
| 150 | 155 | 104 | 3.9 | |
| Skimmed powder milk | 40 | 38 | 95 | 4 |
| 150 | 160 | 106 | 2.5 | |
| Animal milk | 45 | 43 | 95.5 | 4.5 |
| 110 | 115 | 104 | 3.5 | |
| RO water | 45 | 48 | 106 | 4 |
| 160 | 155 | 97 | 3.8 |
4. Conclusion
This work proposes a non-enzymatic electrochemical sensor for detecting oxytocin using the Zn–Cu MOF/rGO nanocomposite synthesized via a one-pot solvothermal approach. The analytical results reveal that Zn–Cu MOF/rGO shows better electro-catalytic activity towards oxytocin detection compared to the bare GCE, Zn-MOF/GCE, Cu-MOF/GCE, Zn-MOF/rGO, Cu-MOF/rGO, and Zn–Cu MOF/GCE, which might be due to the synergistic effect, excellent redox characteristics and higher electron transfer rate. Voltammetric techniques reveal that the proposed sensor has a more comprehensive linear range, excellent selectivity, enhanced conductivity, lower LOD, and highly stable and satisfactory reproducibility for OXY analysis. Moreover, the applicability of the sensor was analyzed by spiking OXY in milk and water, giving a satisfactory recovery range. Such a novel material opens a new avenue for use in electrochemical sensors, and can be integrated into developed devices for on-site application, is easy to use, and hence can be preferred over other conventional materials.Author contributions
Md Zainul Abedeen: conceptualization, methodology, data curation, investigation, visualization, and writing – original draft. Manish Sharma: visualization and formal analysis. Himmat Singh Kushwaha: validation, supervision, and visualization. Ragini Gupta: conceptualization, methodology, supervision, visualization, and writing – review & editing.Data availability
All the data will be made available on reasonable request to the corresponding author.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
The authors would like to acknowledge and thank MNITJ for the institute fellowship. The authors also acknowledge the Department of Chemistry and Materials Research Centre (MRC) of the MNITJ for providing experimental and characterization facilities. The authors also acknowledge the Department of Science and Technology (DST), India (DST/TMD-EWO/WTI/2K19/EWFH/2019/222(C)).References
- M. H. Walter, H. Abele and C. F. Plappert, Front. Endocrinol., 2021, 12, 1409 CrossRef PubMed.
- B. Prakash, V. Paul, H. Kliem, U. Kulozik and H. H. Meyer, Anal. Chim. Acta, 2009, 636, 111–115 CrossRef CAS PubMed.
- M. Matsuzaki, H. Matsushita, K. Tomizawa and H. Matsui, J. Physiol. Sci., 2012, 62, 441–444 CrossRef CAS PubMed.
- M. Mishra, S. Ali and M. Das, Toxicol. Mech. Methods, 2014, 24, 342–346 CrossRef CAS PubMed.
- M. Mishra, S. Ali and M. Das, Food Anal. Methods, 2013, 6, 1308–1319 CrossRef.
- A. R. Solangi, S. Q. Memon, A. Mallah, M. Khuhawar and M. Bhanger, Biomed. Chromatogr., 2009, 23, 1007–1013 CrossRef CAS PubMed.
- C. M. Karbiwnyk, K. C. Faul, S. B. Turnipseed, W. C. Andersen and K. E. Miller, J. Pharm. Biomed. Anal., 2008, 48, 672–677 CrossRef CAS PubMed.
- S. Rastogi, V. Kumari, V. Sharma and F. J. Ahmad, Food Anal. Methods, 2022, 15, 2972–2983 CrossRef.
- S. Rastogi, V. Kumari, V. Sharma and F. J. Ahmad, Anal. Biochem., 2022, 656, 114886 CrossRef CAS PubMed.
- L. Sankhla and H. S. Kushwaha, J. Electron. Mater., 2024, 53, 1896–1902 CrossRef CAS.
- M. Z. Abedeen, M. Sharma, H. S. Kushwaha and R. Gupta, TrAC, Trends Anal. Chem., 2024, 176, 117729 CrossRef CAS.
- S. Menon, M. R. Mathew, S. Sam, K. Keerthi and K. G. Kumar, J. Electroanal. Chem., 2020, 878, 114596 CrossRef CAS PubMed.
- C. Ferrag and K. Kerman, Front. Sens., 2020, 1, 583822 CrossRef.
- L. Sankhla, M. Z. Abedeen, R. Gupta and H. S. Kushwaha, J. Electrochem. Soc., 2024, 171, 066505 CrossRef CAS.
- F. A. Liu, N. Ardabili, I. Brown, H. Rafi, C. Cook, R. Nikopoulou, A. Lopez, S. Zou, M. R. Hartings and A. G. Zestos, J. Electrochem. Soc., 2022, 169, 017512 CrossRef CAS PubMed.
- K. Asai, T. A. Ivandini and Y. Einaga, Sci. Rep., 2016, 6, 32429 CrossRef CAS PubMed.
- S. Mehrotra, P. Rai, K. Gautam, A. Saxena, R. Verma, V. Lahane, S. Singh, A. K. Yadav, S. Patnaik and S. Anbumani, Food Chem., 2023, 418, 135965 CrossRef CAS PubMed.
- P. S. Sharma, Z. Iskierko, K. Noworyta, M. Cieplak, P. Borowicz, W. Lisowski, F. D'Souza and W. Kutner, Biosens. Bioelectron., 2018, 100, 251–258 CrossRef CAS PubMed.
- H. Laddha, P. Yadav, P. Sharma, M. Agarwal and R. Gupta, J. Ind. Eng. Chem., 2024, 131, 257–264 CrossRef CAS.
- R. Thomas and M. Balachandran, Food Chem., 2023, 400, 134106 CrossRef CAS PubMed.
- M. Z. Abedeen, H. Laddha, M. Sharma, R. Gupta and H. S. Kushwaha, J. Electroanal. Chem., 2023, 948, 117810 CrossRef CAS.
- K. Liu, Y. Chen, X. Dong, Y. Hu and H. Huang, Electrochim. Acta, 2023, 456, 142441 CrossRef CAS.
- M. Narayanan, N. P. S. Chauhan and P. Perumal, RSC Adv., 2023, 13, 5565–5575 RSC.
- N. G. Balasubramaniyan and P. Perumal, Anal. Methods, 2024, 16, 624–638 RSC.
- L. Chen, H.-F. Wang, C. Li and Q. Xu, Chem. Sci., 2020, 11, 5369–5403 RSC.
- M. Cao, Y. Zou, Y. Zhang, T. Zeng, Q. Wan, G. Lai and N. Yang, Electrochim. Acta, 2022, 409, 139967 CrossRef CAS.
- X. Wang, S. Li, L. Xie, X. Li, D. Lin and Z. Zhu, Ceram. Int., 2020, 46, 15858–15866 CrossRef CAS.
- N. Sharma, V. Sharma, Y. Jain, M. Kumari, R. Gupta, S. Sharma and K. Sachdev, Synthesis and characterization of graphene oxide (GO) and reduced graphene oxide (rGO) for gas sensing application, Macromolecular symposia, Wiley Online Library, 2017, p. 1700006.
- Q. Cao, H. Zhao, L. Zeng, J. Wang, R. Wang, X. Qiu and Y. He, Talanta, 2009, 80, 484–488 CrossRef CAS PubMed.
- S. Park, Y. Hu, J. O. Hwang, E.-S. Lee, L. B. Casabianca, W. Cai, J. R. Potts, H.-W. Ha, S. Chen and J. Oh, Nat. Commun., 2012, 3, 638 CrossRef PubMed.
- M. Sharma, K. Chaudhary, M. Kumari, P. Yadav, K. Sachdev, V. C. Janu and R. Gupta, Mater. Today Chem., 2020, 18, 100379 CrossRef CAS.
- Y. Jain, M. Kumari, H. Laddha and R. Gupta, ChemistrySelect, 2019, 4, 7015–7026 CrossRef CAS.
- M. Sharma, P. Sharma, L. Yadav, V. C. Janu and R. Gupta, Sep. Purif. Technol., 2023, 124264 CrossRef CAS.
- M. Sharma, P. Dhiware, P. Sharma, V. C. Janu and R. Gupta, ACS Appl. Eng. Mater., 2023, 1, 2649–2662 CrossRef CAS.
- L. Yadav, P. Yadav, H. Laddha, M. Sharma, P. Sharma, M. Agarwal and R. Gupta, Inorg. Chem. Commun., 2024, 161, 112083 CrossRef CAS.
- Y. Wang, K. Kretschmer, J. Zhang, A. K. Mondal, X. Guo and G. Wang, RSC Adv., 2016, 6, 57098–57102 RSC.
- S. Gopi, A. M. Al-Mohaimeed, W. A. Al-onazi, M. S. Elshikh and K. Yun, J. King Saud Univ., Sci., 2021, 33, 101379 CrossRef.
- S. Rani, S. Kapoor, B. Sharma, S. Kumar, R. Malhotra and N. Dilbaghi, J. Alloys Compd., 2020, 816, 152509 CrossRef CAS.
- N. K. Ravikumar and P. Perumal, J. Mater. Res., 2024, 1–13 Search PubMed.
- T. Qi, Y. Zhao, S. Chen, W. Li, X. Guo, Y. Zhang and C. Song, Mol. Catal., 2021, 514, 111870 CrossRef CAS.
- M. F. Sanad, A. R. Puente Santiago, S. A. Tolba, M. A. Ahsan, O. Fernandez-Delgado, M. Shawky Adly, E. M. Hashem, M. Mahrous Abodouh, M. S. El-Shall, S. T. Sreenivasan and N. K. Allam, J. Am. Chem. Soc., 2021, 143(10), 4064–4073 CrossRef CAS PubMed.
- J. Meng, Y. Zhou, H. Chi, K. Li, J. Wan and Z. Hu, ChemistrySelect, 2019, 4, 8661–8670 CrossRef CAS.
- M. Sharma, H. Laddha, P. Yadav, Y. Jain, K. Sachdev, V. C. Janu and R. Gupta, Mater. Today Commun., 2022, 32, 104020 CrossRef CAS.
- P. Sharma, H. Laddha, M. Agarwal and R. Gupta, Microporous Mesoporous Mater., 2022, 338, 111982 CrossRef CAS.
- N. Gao, C. He, M. Ma, Z. Cai, Y. Zhou, G. Chang, X. Wang and Y. He, Anal. Chim. Acta, 2019, 1072, 25–34 CrossRef CAS PubMed.
- M. Guler, V. Turkoglu, A. Bulut and M. Zahmakiran, Electrochim. Acta, 2018, 263, 118–126 CrossRef CAS.
- E. Narayanamoorthi, P. Arul, N. Gowthaman and S. A. John, Electrochim. Acta, 2022, 409, 139994 CrossRef CAS.
- M. Khairy, H. A. Ayoub and C. E. Banks, Food Chem., 2018, 255, 104–111 CrossRef CAS PubMed.
- M. Singh, N. Jaiswal, I. Tiwari, C. W. Foster and C. E. Banks, J. Electroanal. Chem., 2018, 829, 230–240 CrossRef CAS.
- M. A. Kachouei, F. Hekmat, H. Wang, G. A. Amaratunga, H. E. Unalan and S. Shahrokhian, Electrochim. Acta, 2022, 428, 140952 CrossRef CAS.
- Y.-T. Xue, Z. Chen, X. Chen, G.-C. Han, X.-Z. Feng and H.-B. Kraatz, Electrochim. Acta, 2024, 144009 CrossRef CAS.
- H. Naeim, F. Kheiri, M. Sirousazar and A. Afghan, Electrochim. Acta, 2018, 282, 137–146 CrossRef CAS.
- E. Laviron, J. Electroanal. Chem. Interfacial Electrochem., 1979, 101, 19–28 CrossRef CAS.
- A. J. Bard, L. R. Faulkner and H. S. White, Electrochemical methods: fundamentals and applications, John Wiley & Sons, 2022 Search PubMed.
- M. W. Ahmad, B. Dey, G. Sarkhel, D.-J. Yang and A. Choudhury, J. Electroanal. Chem., 2022, 920, 116646 CrossRef CAS.
- S. J. Malode, K. Prabhu, S. S. Kalanur, N. Meghani and N. P. Shetti, Chemosphere, 2023, 312, 137302 CrossRef CAS PubMed.
- J. Attia, S. Nir, E. Mervinetsky, D. Balogh, A. Gitlin-Domagalska, I. Alshanski, M. Reches, M. Hurevich and S. Yitzchaik, Sci. Rep., 2021, 11, 7051 CrossRef PubMed.
- B. Dey, M. W. Ahmad, B. H. Kim, T. Kamal, D.-J. Yang, C. N. Patra, S. S. Hossain and A. Choudhury, Anal. Bioanal. Chem., 2023, 415, 3487–3501 CrossRef CAS PubMed.
- W. Bal, H. Kozlowski, B. Lammek, K. Rolka and L. D. Pettit, J. Inorg. Biochem., 1992, 45, 193–202 CrossRef CAS PubMed.
- L. Joly, R. Antoine, F. Albrieux, R. Ballivian, M. Broyer, F. Chirot, J. Lemoine, P. Dugourd, C. Greco and R. Mitric, J. Phys. Chem. B, 2009, 113, 11293–11300 CrossRef CAS PubMed.
- T. Wyttenbach, D. Liu and M. T. Bowers, J. Am. Chem. Soc., 2008, 130, 5993–6000 CrossRef CAS PubMed.
- J. J. O'Sullivan, K. S. Uyeda, M. J. Stevenson and M. C. Heffern, RSC Chem. Biol., 2023, 4, 165–172 RSC.
- K. K. Tadi, I. Alshanski, E. Mervinetsky, G. Marx, P. Petrou, K. M. Dimitrios, C. Gilon, M. Hurevich and S. Yitzchaik, ACS Omega, 2017, 2, 8770–8778 CrossRef CAS PubMed.
- K. K. Tadi, I. Alshanski, M. Hurevich and S. Yitzchaik, Surfaces, 2018, 1, 90–95 CrossRef.
- I. Alshanski, D. E. Shalev, S. Yitzchaik and M. Hurevich, JBIC, J. Biol. Inorg. Chem., 2021, 26, 809–815 CrossRef CAS PubMed.
- H. Laddha, P. Yadav, Y. Jain, M. Sharma, M. Reza, M. Agarwal and R. Gupta, J. Mol. Liq., 2022, 346, 117088 CrossRef CAS.
- N. Elgrishi, K. J. Rountree, B. D. McCarthy, E. S. Rountree, T. T. Eisenhart and J. L. Dempsey, J. Chem. Educ., 2018, 95, 197–206 CrossRef CAS.
- M. L. Yola, N. Atar and A. Erdem, Sens. Actuators, B, 2015, 221, 842–848 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4an01157k |
| This journal is © The Royal Society of Chemistry 2025 |