
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A pH-enhanced resolution in benchtop NMR spectroscopy†
Paulina
Putko
*,
Javier A.
Romero
and
Krzysztof
Kazimierczuk
 *
*
Centre of New Technologies, University of Warsaw, Banacha 2C, 02-097 Warsaw, Poland. E-mail: k.kazimierczuk@cent.uw.edu.pl
First published on 26th February 2024
Abstract
NMR spectroscopy is one of the most potent methods in analytical chemistry. NMR titration experiments are particularly useful since they measure molecular binding affinities and other concentration-dependent effects. These experiments, however, require a long series of measurements. An alternative to these serial measurements has recently been presented, exploiting a pH (or generally – a concentration) gradient along the NMR tube. The proposed experiment, although efficient, was based on the sensitivity- and hardware-demanding chemical shift imaging (CSI) method. Thus, it is practically limited to high-resolution NMR spectrometers. This paper proposes modifying and adapting the approach to the popular and cost-efficient benchtop NMR machines. Instead of CSI, we use a device that shifts the NMR tube vertically to measure the spectra of different sample volumes, which have different pH values due to the established gradient along the tube. We demonstrate the potential of the method on the test samples of L-tyrosine and 2,6-lutidine, and two real samples from the food industry – an infant formula and an energy drink. The proposed method boosts spectral resolution and allows for the sampling of a broader range of pH values when compared to the original approach.
1. Introduction
The pH of a sample is one of the most important parameters in organic chemistry, medicinal chemistry, and food sciences. Various methods for determining pH and acid dissociation constants (pKa) are available, including UV-vis spectroscopy, potentiometry, and NMR spectroscopy.1 Variable pH experiments are frequently performed to answer various research questions since molecular structures are often highly dependent on pH. In particular, a series of NMR measurements with varying pH can give information about chemical structures, conformations and reaction mechanisms.2–4Various experimental approaches are used to determine pKa values by NMR spectroscopy. In the conventional procedure, it is necessary to prepare several samples at different pH or adjust the pH of a single sample between successive NMR measurements.5,6 Recently, Wallace et al.7,8 proposed an alternative procedure involving the use of a pH gradient along the NMR tube and ‘single shot’ NMR experiments providing spectra at different ‘slices’ of a sample (and thus at different pH values). The studied solution should contain several pH indicators to measure the actual pH in a given ‘slice’. The method has been extended to measure ligand binding affinities with saturation transfer difference NMR.9 In the current study, we further exploit the perspective of using an established pH gradient within the sample.
Varying the pH of a sample changes the resonance frequencies, with a profound effect on the nuclei that are close to protonation sites. Thus, pH variation can be used as a simple trick to resolve the crowded regions of an NMR spectrum. This procedure could be particularly useful in low-field machines, like benchtop NMR spectrometers (BT-NMR), given their intrinsically lower sensitivity and resolution. The most common applications of BT-NMR are studies of mixtures in industrial applications.10–12 The spectra of mixtures are exceptionally crowded and here variable-pH experiments could be considered a solution to the low-resolution problem. However, changing the pH of a sample can be cumbersome. In addition, many pH titration steps are needed to shift resonance peaks sufficiently slow to track them accurately.
This paper proposes using samples with a pH gradient along the NMR tube to boost the resolution of spectra acquired on a 43 MHz benchtop spectrometer. The sample preparation is similar to that proposed by Wallace et al.,7 but the measurement does not require pulse field gradients. Instead, we vertically shift the NMR tube using a mechanical device called a SWAPE or Sweeping Apparatus for Polarization Enhancement,13 which is synchronized with the acquisition pulse sequence. The low resolution of BT-NMR prohibits the use of many pH indicators, as it is suggested in the original method.7 For this reason, we propose to use only two indicators and aim at identifying and resolving pH-sensitive peaks in a mixture rather than a precise determination of pKa. First, we test the method with simple samples of L-tyrosine and 2,6-lutidine. Then, we demonstrate its usefulness and applicability to the food and health industries by detecting melamine in infant formula and taurine in energy drinks.
2. Materials and methods
2.1. Test sample of L-tyrosine in a pH gradient
L-Tyrosine (Sigma Aldrich, ≥98%) was dissolved in 2 ml of D2O to obtain a final concentration of 8.0 ± 0.2 mM. We also added pH indicators (Sigma Aldrich, ≥98%): 2 μl of 2,6-lutidine and 1.00 ± 0.05 mg of sodium formate. Then, a pH gradient was established as described below (section 2.4).2.2. Test sample of 2,6-lutidine in a pH gradient
2,6-Lutidine was dissolved in 2 mL D2O to obtain a final concentration of 10 mM. Next, the pH of the sample was adjusted to 11.00 with NaOH. Then, a pH gradient was established as described below (section 2.4). A series of thirty 10 mM samples with pH values from 2.0 to 9.0 was also prepared. We used a digital pH meter (SevenCompact S210, Mettler Toledo, Switzerland) to check the pH in each sample from the conventional titration series. The position of the D2O peak was set to 4.75 ppm in all spectra.2.3. Food products
A sample of 90 ± 0.5 mg of infant formula was dissolved in 2 mL of DMSO-d6 to obtain a final concentration of 450 ± 0.25 g L−1. The sample was shaken for 1 min on a vortex mixer and 5 min in an ultrasonic bath. Then 0.50 ± 0.05 mg of melamine (Sigma Aldrich, ≥98%) was added to the sample to obtain a final concentration of 250 ± 0.03 g L−1.2 ml sample of Red Bull energy drink (Red Bull Polska, Poland) was degassed for 10 min in an ultrasonic bath. Then, the pH of the sample was adjusted to 11.00 with NaOH. The two critical ingredients of an energy drink, taurine and citric acid, were purchased from Sigma Aldrich (Steinheim, Germany). The 10 mM samples of these compounds were prepared by dissolving them in D2O and then used for measuring reference pH titration curves.
Then, for both food product samples, a pH gradient was established as described below (section 2.4).
2.4. Establishing a pH gradient
All the above samples were prepared using thin-walled NMR tubes 9′′ in length and 5 mm in outer-diameter (see Fig. 1). The first step in establishing a pH gradient was to add 20 μl of 25% w/w hydrochloric acid to the bottom of the NMR tube using a glass pipette with O-rings (for precise insertion). Then, six glass beads 2 mm in diameter (Merck, Germany) were washed in methanol to be later introduced into the NMR tube.7 In the next step, a crystal of bromothymol blue indicator and one-mole equivalent of NaOH were added to each analyte sample and shaken for 1 min on a vortex mixer. Finally, a prepared analyte sample was added along the wall of the tube. The addition of NaOH was omitted for the Red Bull energy drink sample and the test sample of 2,6-lutidine.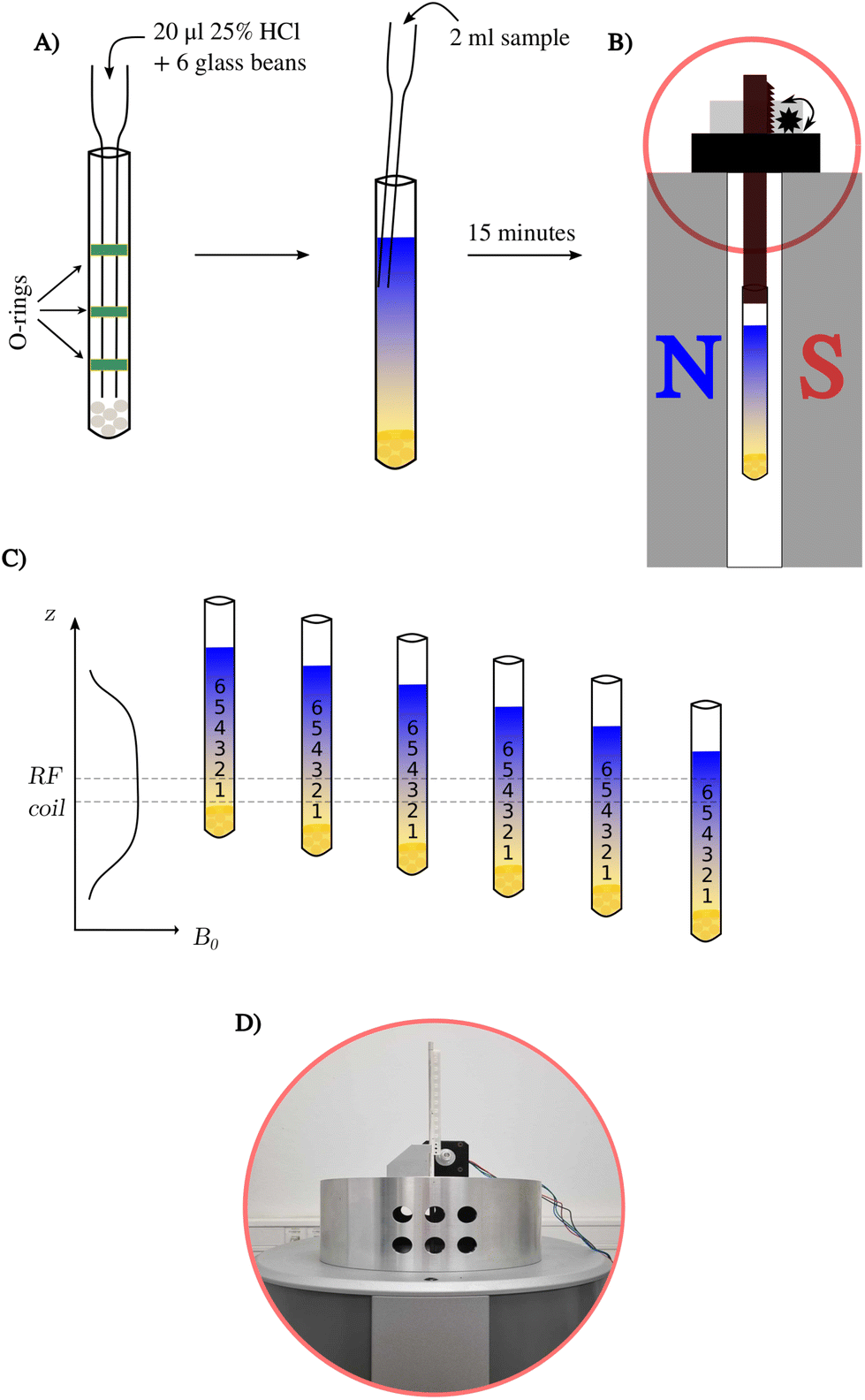 | ||
| Fig. 1 Scheme of a pH gradient experiment using 43 MHz BT-NMR. (A) General sample preparation procedure, (B) sample in the SWAPE device inside a BT-NMR spectrometer, (C) idea of measuring a series of 1H NMR spectra, and (D) a photo of the SWAPE motor marked in red on panel (B). | ||
2.5. 1H NMR measurements at 43 MHz
1H NMR experiments were performed on a Spinsolve Carbon 43 benchtop spectrometer (Magritek, Germany) at a temperature of 298 K. We used the SWAPE device13 to mechanically change the vertical position of the sample. The L-tyrosine and Red Bull samples were measured at 16 different positions starting from 2 cm above the bottom of the NMR tube, and each successive value of the sample represents a step of 0.5 cm, while the active region of the RF coil is 1 cm. The infant formula sample was measured using only eight volumes (steps of 1.0 cm). Averaging 32 scans per measurement extended the acquisition time to 32 minutes to record all 16 different volumes.3. Results and discussion
We tested the applicability of a pH gradient to BT-NMR on four different samples. The previously described experimental setup involved the use of high-field NMR with pulsed field gradients (PFGs) and chemical shift imaging (CSI).7 However, BT-NMR machines (like ours) usually lack a PFG. Even those equipped with a PFG, usually produce gradients in a transversal direction to the NMR tube. Thus, using CSI to collect spectra from different slices with different pH values is impossible. Instead, we propose to mechanically shift the sample along the vertical dimension to gain access to different pH values. Compared to CSI, the disadvantage of this approach is the line-broadening caused by signal averaging within a single volume. This effect is shown in Fig. 2. The advantage is better sensitivity and a broader ‘pH-window’ than for CSI experiments on a static sample. The sensitivity aspect is crucial for low-field NMR, while access to a broad range of pH values allows for measurement of several pKa values over a sample volume much larger than the active detection region of the RF coil.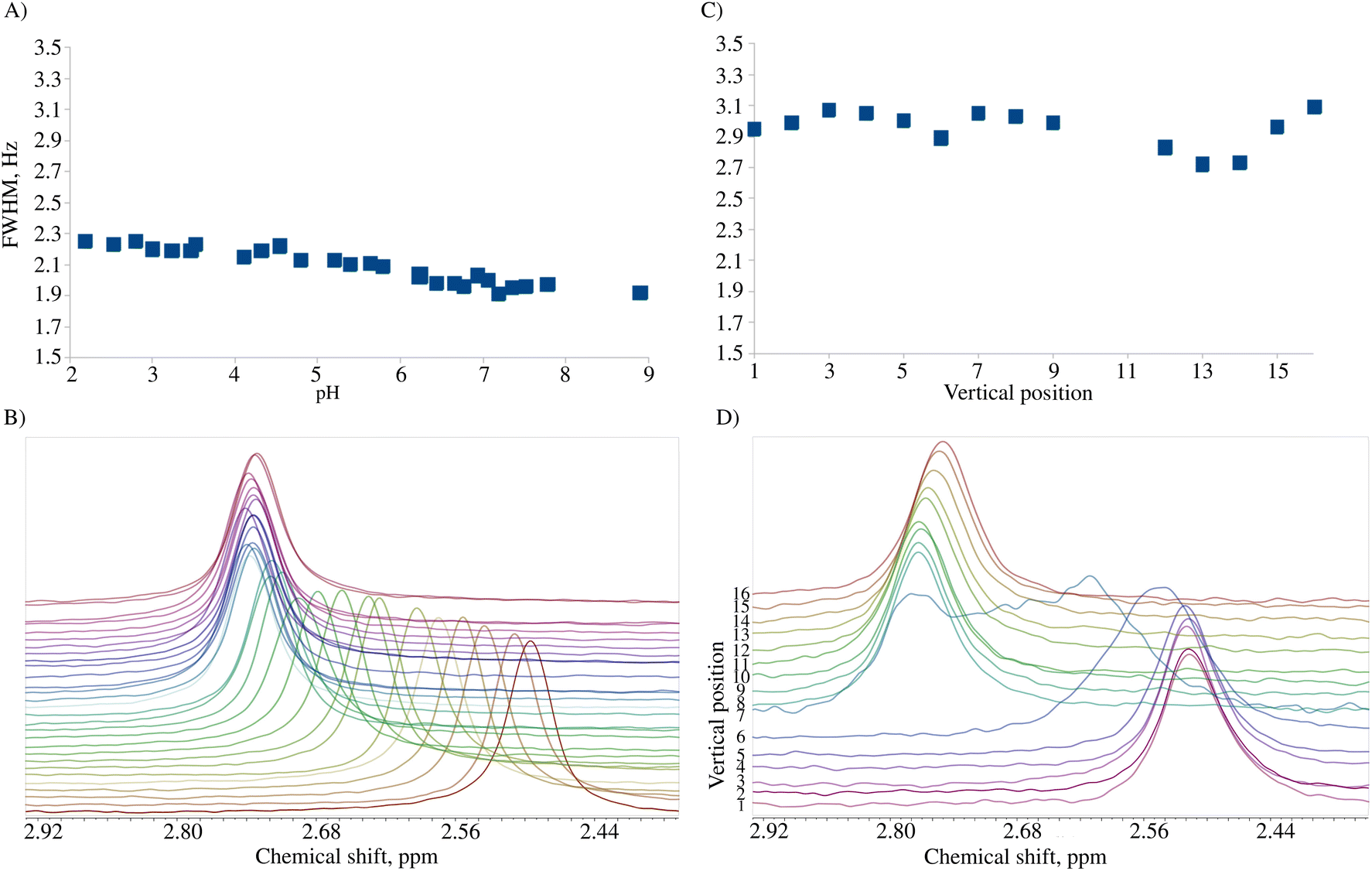 | ||
| Fig. 2 Line-width in conventional samples (A and B) and pH gradient samples (C and D). (A) Full width at half maximum (FWHM) of the 2,6-lutidine methyl group signal for a conventional titration series with pH from 2.0 to 9.0, (B) the corresponding spectra (pH 2.0 at the bottom), (C) FWHM in the pH gradient experiment 24 h after sample preparation, and (D) the corresponding spectra (lowest pH at the bottom). | ||
3.1. A pH gradient test sample
To test the concept of vertically shifting the pH gradient sample, we prepared a solution containing three compounds with pH-sensitive chemical shifts: L-tyrosine, 2,6-lutidine, and sodium formate. The pH values at different volumes of the sample can be calculated using the modified Henderson–Hasselbach equation (eqn (1))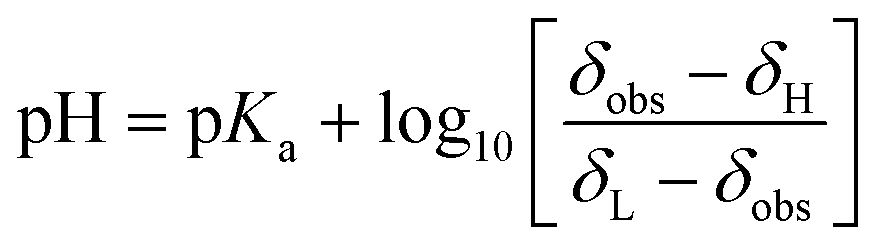 | (1) |
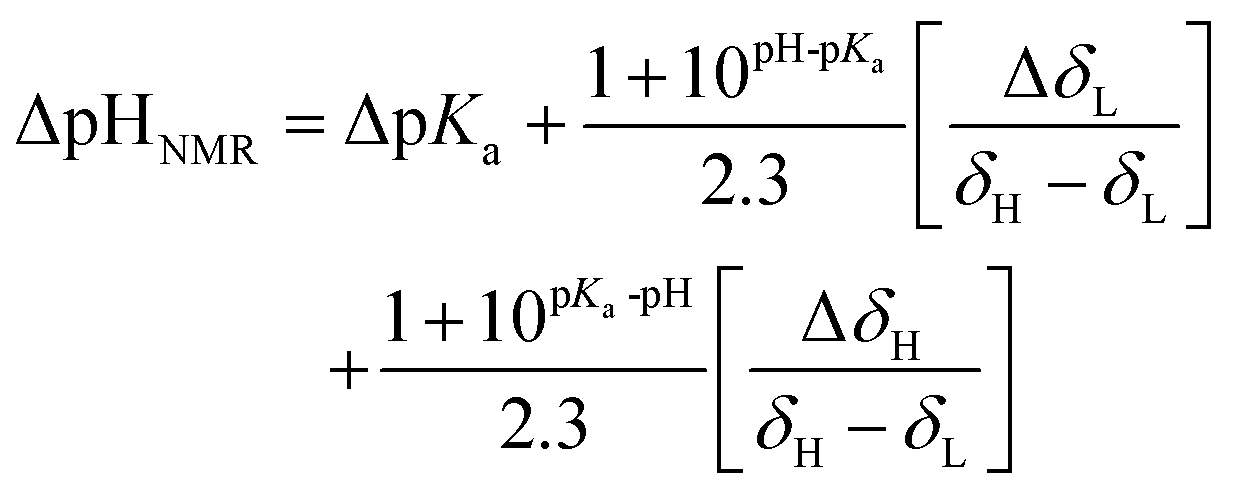 | (2) |
To use the above equations and relate the chemical shift of an indicator with a pH in a given volume of a sample, one has to know pKa values. To confirm the literature values, we performed a series of conventional 1H NMR experiments (see the ESI†). The results allowed us to estimate the pH in the volumes of pH gradient samples containing pH indicators.
Fig. 3A shows the chemical shift changes as a function of sample position. Importantly, we have observed both chemical shift transitions (corresponding to pKa1 and pKa2) for L-tyrosine. This is impossible with the original approach by Wallace et al.,7 where a much smaller pH window is sampled. While mechanically shifting the NMR tube using SWAPE provides access to as much as 80 mm of the sample, employing CSI on a static sample allows for measurement of the spectra in a range of 10–15 mm depending on the probe.
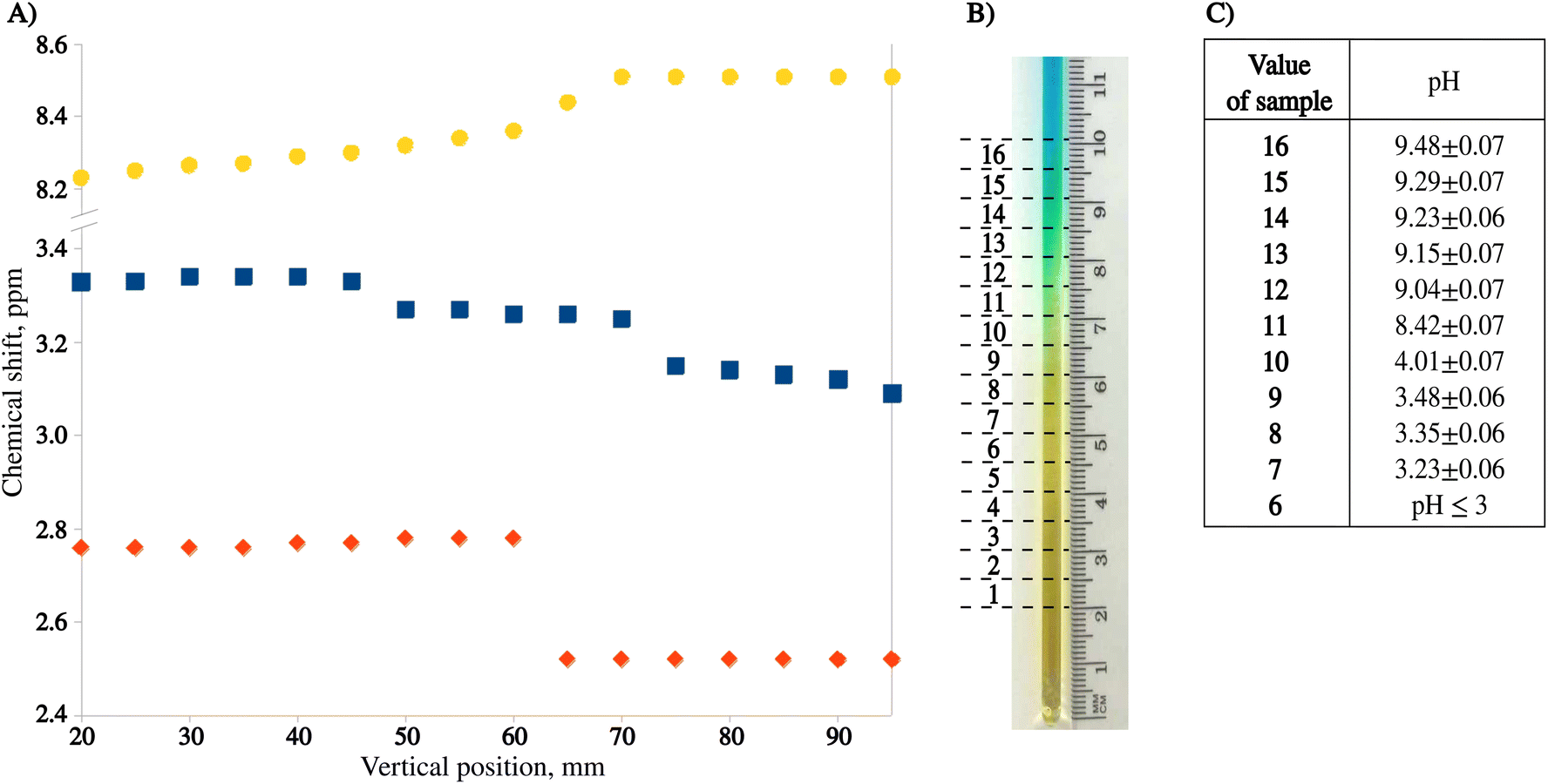 | ||
| Fig. 3 Method testing results. (A) Chemical shift profiles of sodium formate (yellow), L-tyrosine (blue), and 2,6-lutidine (orange) 24 h after sample preparation. (B) Photo of the pH gradient 24 h after sample preparation with 16 measured volumes marked. (C) Relations between pH and volume number (6–16) using chemical shifts of NMR indicators. These relations were determined using eqn (1) and chemical shifts of 2.6-lutidine (volumes 12–16, pKa = 6.877), and formate (volumes 7–11, pKa = 3.637). | ||
The sample can be measured as early as 20 minutes after preparation, but the optimal window for measurements is 24 hours after preparation when the color transition of bromomethyl blue is approximately in the middle of the sample (see Fig. 3B). Fig. 3C presents pH values calculated from eqn (1) for 11 volumes of the pH gradient sample. As can be seen, the “pH gradient” is heavily non-linear, which means that the actual pH values can only be calculated for the volumes with pH close to the pKa values of the pH indicators used. In BT-NMR, it is not possible to use too many indicators due to common peak overlap. Thus, the actual applicability of the pH gradient approach for the determination of pKa values is somewhat limited. Still, however, the pH gradient can be helpful to detect and resolve “pH-sensitive” peaks.
3.2. The energy drink sample
The pH of the unprocessed energy drink (ED) is 3.4. Under these conditions, the spectral peaks from taurine are entirely covered by stronger resonances from the sugar region.15 We exploited the pH gradient to resolve the peak signal of taurine and facilitate its identification, which allowed for accurate determination of its chemical shift and dependence on pH. To verify the latter, we measured a 10 mM standard taurine solution at different pH values. Fig. 4A illustrates the shift of the taurine signal. We repeated the procedure for the citric acid peak (Fig. 4B). The identification of this compound in the sample is also improved by observing the chemical shift vs. pH dependence. Fig. 4C shows a series of spectra recorded 24 hours after sample preparation. As it can be seen, the method provides better resolution and peak characterization than the constant pH experiment. Moreover, in contrast to CSI, there is no sensitivity loss as long as a large volume of the sample (ca. 2 ml) can be measured.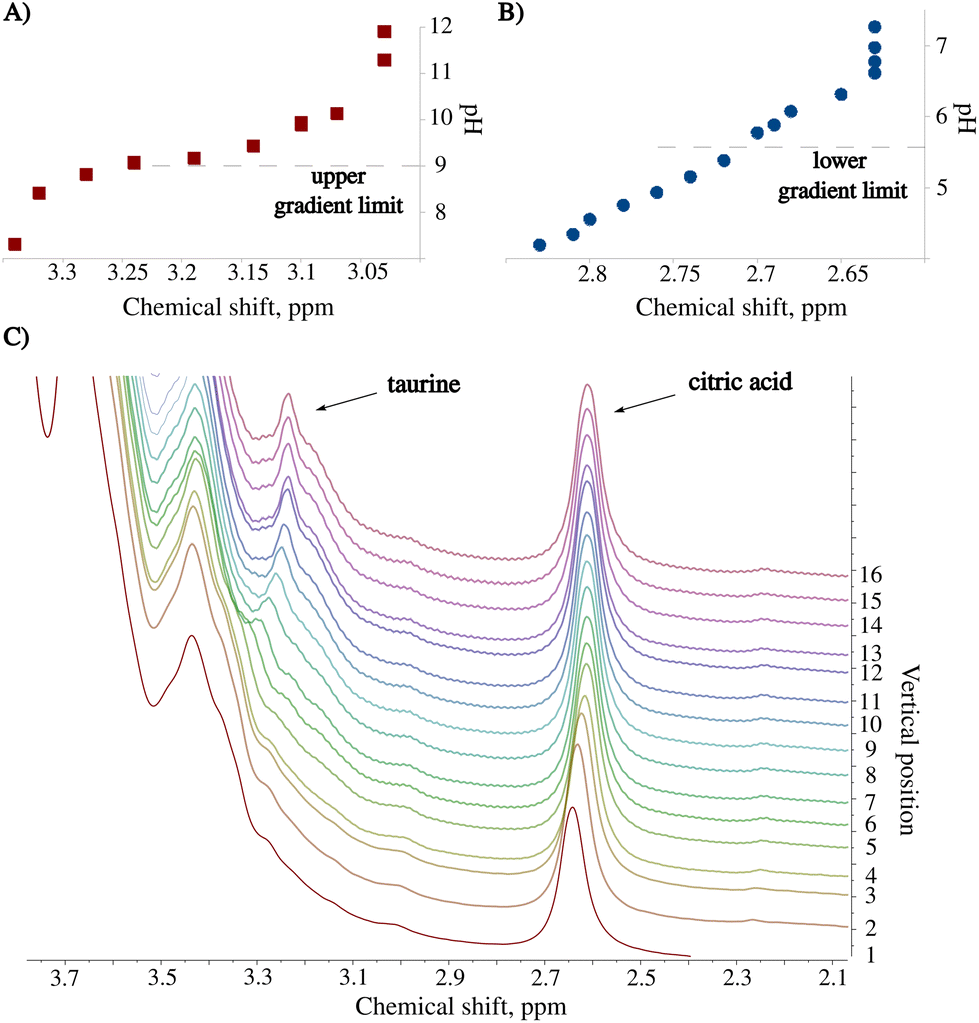 | ||
| Fig. 4 Results from the analysis of the Red Bull energy drink sample measured using BT-NMR with the SWAPE apparatus. Titration curves of a standard solution containing 10 mM of (A) taurine and (B) citric acid. The dashed lines indicate the pH range resulting from the pH gradient. (C) A region from a series of 1H NMR spectra showing how signals from the nuclei close to protonation sites shift with varying pH. | ||
3.3. Detecting melamine in infant formula
A recent crisis with melamine contamination of infant formula in China has pointed out a severe deficiency in current food control systems.16 In many of the recorded cases, the contamination led to the development of kidney stones and in some cases, it even led to kidney failure and consequently, death.171H NMR with an added pH dimension can identify melamine-contaminated infant formula based on pH. Fig. 5 shows the stack of spectra of melamine-contaminated infant formula acquired using a pH gradient. The spectra were collected 24 hours after sample preparation.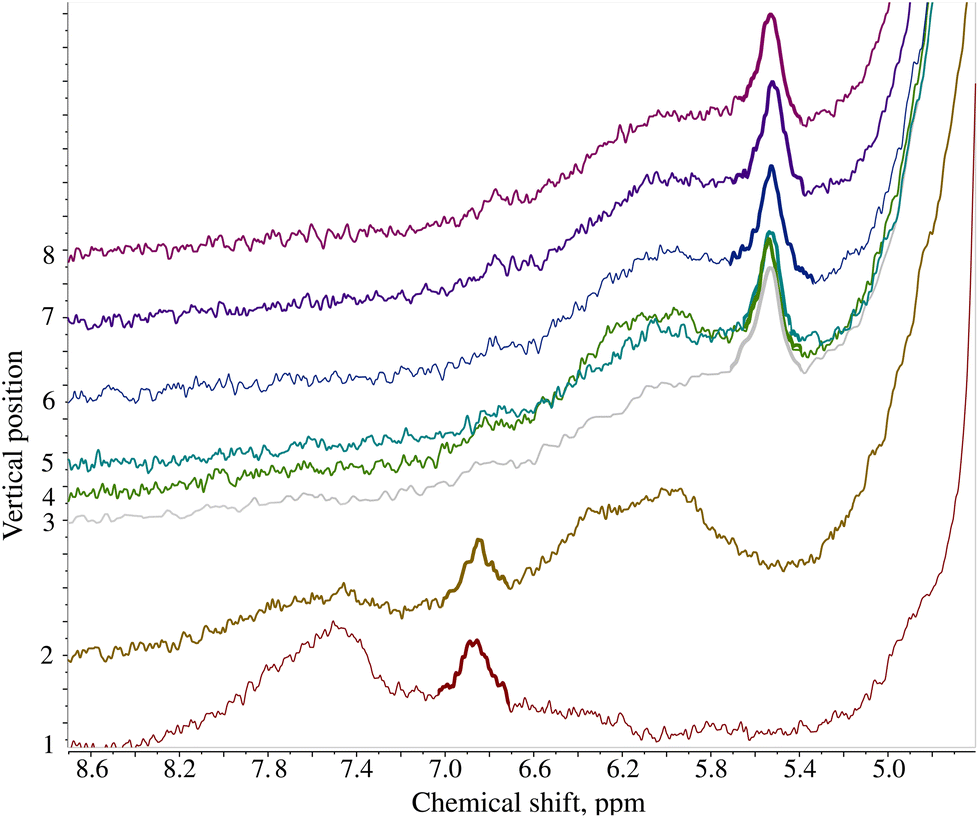 | ||
| Fig. 5 1H NMR spectra of the infant formula contaminated with melamine acquired using the pH gradient. Stacked 1H NMR spectra correspond to different volumes of the NMR tube and thus different pH values. The melamine peak is marked with a bold line. The first 2 volumes correspond to pH below 5.0 causing a change in the melamine chemical shift and facilitating its identification. | ||
The solvent used for the infant formula sample (DMSO) is better suited than water to creating a pH gradient: the gradient is more stable over time due to higher viscosity. While the gradient in the D2O sample was optimal after ca. 24 h and degraded within 72 h, the DMSO sample could be measured up to 18 days after preparation. Fig. 6 shows the long-lasting stability of the pH gradient in the infant formula sample. Notably, adding both HCl and NaOH solutions introduced some water into the sample. Thus, we had two peak-shifting factors involved: the major one (pH-gradient) and a small solvent-effect (varying D2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMSO ratio). The evolution of the pH gradient profile over time reveals another advantage of using a device such as SWAPE to vertically shift the sample when compared to the CSI approach. Namely, spectra can be collected using any sample volume ranging from the bottom of the NMR tube to its very top. It was possible to observe the jump in the position of the melamine peak signal even 18 days after sample preparation. While the jump observed in volume 3 elapsed after 24 h, it shifted volume 7 after 18 days. Thanks to the SWAPE device, both volumes can be placed within the active region of the RF coil.
:DMSO ratio). The evolution of the pH gradient profile over time reveals another advantage of using a device such as SWAPE to vertically shift the sample when compared to the CSI approach. Namely, spectra can be collected using any sample volume ranging from the bottom of the NMR tube to its very top. It was possible to observe the jump in the position of the melamine peak signal even 18 days after sample preparation. While the jump observed in volume 3 elapsed after 24 h, it shifted volume 7 after 18 days. Thanks to the SWAPE device, both volumes can be placed within the active region of the RF coil.
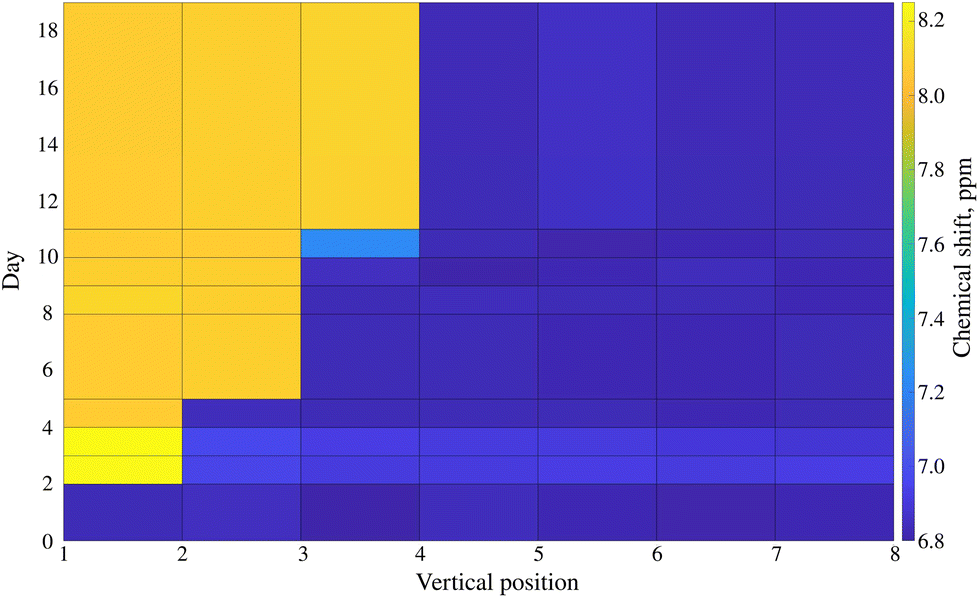 | ||
| Fig. 6 The stability of the pH gradient in the infant formula sample contaminated with melamine dissolved in DMSO. The plot presents the chemical shift of the melamine peak (marked bold in Fig. 5) as a function of sample volume position and time elapsed from sample preparation. | ||
4. Conclusions
The concept of creating a pH gradient in an NMR tube proposed by Wallace et al.7 paved the way for many new NMR experiments. Besides the one-shot measurements of pKa values, one can exploit pH variations to boost resolution, i.e., resolve crowded areas of the spectrum. Moreover, detecting ‘pH-sensitive’ peaks facilitates the resonance assignment, even without the precise determination of pKa. It is clear that peaks from compounds with protonation sites (e.g. amine oramide groups) will shift, while others will retain their positions.Unfortunately, the original pH gradient method based on chemical shift imaging cannot be implemented with most benchtop NMR machines. Many do not contain pulse field gradients necessary for the CSI experiment. Even in those that do have gradient coils, the typical direction of the field gradient is perpendicular to the NMR tube and, thus, to the pH gradient. This problem is solved by an alternative approach proposed in this paper. Replacing CSI with mechanical sample shifting enables the applicability of the method to BT-NMR and provides access to a larger sample volume. Given that the profile of pH gradient changes over time, sample shifting allowed us to obtain the specific chemical transitions more effectively than CSI.
Conflicts of interest
The authors declare no conflicting interests.Acknowledgements
The Polish National Science Centre provided financial support with a grant no. 2019/35/B/ST4/01506. Dr Dariusz Gołowicz is thanked for fruitful discussions about the use of the SWAPE device.References
- J. Reijenga, A. van Hoof, A. van Loon and B. Teunissen, Anal. Chem. Insights, 2013, 8, 53–71 CrossRef CAS PubMed.
- S. Mori, S. M. Eleff, U. Pilatus, N. Mori and P. C. V. Zijl, Magn. Reson. Med., 1998, 40, 36–42 CrossRef CAS PubMed.
- C. Gerardin, M. In, L. Allouche, M. Haouas and F. Taulelle, Chem. Mater., 1999, 11, 1285–1292 CrossRef CAS.
- F. Taulelle, M. Haouas, C. Gerardin, C. Estournes, T. Loiseau and G. Ferey, Colloids Surf., A, 1999, 158, 299–311 CrossRef CAS.
- J. Bezençon, M. B. Wittwer, B. Cutting, M. Smieško, B. Wagner, M. Kansy and B. Ernst, J. Pharm. Biomed. Anal., 2014, 93, 147–155 CrossRef PubMed.
- M. Hohmann, C. Felbinger, N. Christoph, H. Wachter, J. Wiest and U. Holzgrabe, J. Pharm. Biomed. Anal., 2014, 93, 156–160 CrossRef CAS PubMed.
- M. Wallace, D. J. Adams and J. A. Iggo, Anal. Chem., 2018, 90, 4160–4166 CrossRef CAS PubMed.
- G. Schenck, K. Baj, J. A. Iggo and M. Wallace, Anal. Chem., 2022, 94, 8115–8119 CrossRef CAS PubMed.
- S. Monaco, J. Angulo and M. Wallace, J. Am. Chem. Soc., 2023, 145, 16391–16397 CrossRef CAS PubMed.
- B. Gouilleux, J. Marchand, B. Charrier, G. S. Remaud and P. Giraudeau, Food Chem., 2018, 244, 153–158 CrossRef CAS PubMed.
- H. Metz and K. Mäder, Int. J. Pharm., 2008, 364, 170–175 CrossRef CAS PubMed.
- M. Rubens, J. V. Herck and T. Junkers, ACS Macro Lett., 2019, 8, 1437–1441 CrossRef CAS PubMed.
- J. A. Romero, K. Kazimierczuk and D. Gołowicz, Analyst, 2020, 145, 7406–7411 RSC.
- J. J. Ackerman, G. E. Soto, W. M. Spees, Z. Zhu and J. L. Evelhoch, Magn. Reson. Med., 1996, 36, 674–683 CrossRef CAS PubMed.
- K. Wegert, Y. B. Monakhova, T. Kuballa, H. Reusch, G. Winkler and D. W. Lachenmeier, Lebensmittelchemie, 2012, 66, 143–145 CrossRef.
- D. W. Lachenmeier, H. Eberhard, F. Fang, S. Birk, D. Peter, S. Constanze and S. Manfred, J. Agric. Food Chem., 2009, 57, 7194–7199 CrossRef CAS PubMed.
- C. M. E. Gossner, J. Schlundt, P. B. Embarek, S. Hird, D. Lo-Fo-Wong, J. J. O. Beltran, K. N. Teoh and A. Tritscher, Environ. Health Perspect., 2009, 117, 1803–1808 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3an02000b |
| This journal is © The Royal Society of Chemistry 2024 |