
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficacy of tyrosine kinase inhibitors examined by a combination of Raman micro-spectroscopy and a deep wavelet scattering-based multivariate analysis framework†
Irina
Schuler
ab,
Martin
Schuler
 ab,
Tatjana
Frick
ab,
Dairovys
Jimenez
ab,
Abdelouahid
Maghnouj
c,
Stephan
Hahn
c,
Rami
Zewail
d,
Klaus
Gerwert
ab and
Samir F.
El-Mashtoly‡
*abe
ab,
Tatjana
Frick
ab,
Dairovys
Jimenez
ab,
Abdelouahid
Maghnouj
c,
Stephan
Hahn
c,
Rami
Zewail
d,
Klaus
Gerwert
ab and
Samir F.
El-Mashtoly‡
*abe
aCenter for Protein Diagnostics, Ruhr-University Bochum, Bochum, Germany. E-mail: Samir.Elmashtoly@leibniz-ipht.de
bDepartment of Biophysics, Ruhr-University Bochum, Bochum, Germany
cDepartment of Molecular GI-Oncology, Clinical Research Center, Ruhr-University Bochum, Bochum, Germany
dDepartment of Computer Science & Engineering, Egypt-Japan University of Science and Technology, New Borg El-Arab, Egypt
eBiotechnology Program, Institute of Basic and Applied Science, Egypt-Japan University of Science and Technology, New Borg El-Arab, Egypt
First published on 19th February 2024
Abstract
HER2 is a crucial therapeutic target in breast cancer, and the survival rate of breast cancer patients has increased because of this receptor's inhibition. However, tumors have shown resistance to this therapeutic strategy due to oncogenic mutations that decrease the binding of several HER2-targeted drugs, including lapatinib, and confer resistance to this drug. Neratinib can overcome this drug resistance and effectively inhibit HER2 signaling and tumor growth. In the present study, we examined the efficacy of lapatinib and neratinib using breast cancer cells by Raman microscopy combined with a deep wavelet scattering-based multivariate analysis framework. This approach discriminated between control cells and drug-treated cells with high accuracy, compared to classical principal component analysis. Both lapatinib and neratinib induced changes in the cellular biochemical composition. Furthermore, the Raman results were compared with the results of several in vitro assays. For instance, drug-treated cells exhibited (i) inhibition of ERK and AKT phosphorylation, (ii) inhibition of cellular proliferation, (iii) cell-cycle arrest, and (iv) apoptosis as indicated by western blotting, real-time cell analysis (RTCA), cell-cycle analysis, and apoptosis assays. Thus, the observed Raman spectral changes are attributed to cell-cycle arrest and apoptosis. The results also indicated that neratinib is more potent than lapatinib. Moreover, the uptake and distribution of lapatinib in cells were visualized through its label-free marker bands in the fingerprint region using Raman spectral imaging. These results show the prospects of Raman microscopy in drug evaluation and presumably in drug discovery.
Introduction
Breast cancer (BC) is the second most common cancer and the main reason for cancer-related deaths in women worldwide. Globally, an estimated 2.26 million new cases and 680![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths were recorded in 2020.1 Although significant advances have been made over the past few decades in the prevention, diagnosis, and treatment of breast cancer, it remains a major global health problem.2 More importantly, distant (metastatic) recurrence is a significant clinical issue and is responsible for the majority of BC deaths,3 where the five-year survival of patients with localized BC is 99% but decreases to 27% when diagnosed with late-stage disease.1
000 deaths were recorded in 2020.1 Although significant advances have been made over the past few decades in the prevention, diagnosis, and treatment of breast cancer, it remains a major global health problem.2 More importantly, distant (metastatic) recurrence is a significant clinical issue and is responsible for the majority of BC deaths,3 where the five-year survival of patients with localized BC is 99% but decreases to 27% when diagnosed with late-stage disease.1
Depending on the severity and stage of the cancer, different therapies can be used to treat breast cancer. Surgery, radiation therapy, chemotherapy, and hormone therapy are the most popular forms of treatment. In recent years, targeted therapy has emerged as a potentially effective form of treatment.4 Targeted therapy aims to block or inhibit the specific molecules and pathways that promote tumour growth and progression, offering improved treatment outcomes and fewer side effects compared to traditional chemotherapy. This type of therapy has demonstrated significant promise in the treatment of breast cancer.4,5
The well-known role of the epidermal growth factor receptors (EGFR or HER family) in the growth and proliferation of cancer cells makes them one of the most important targets in cancer therapy6,7 The HER receptors have an extracellular ligand-binding domain, transmembrane domain, and an intracellular tyrosine kinase (TK) domain. When a ligand binds to the HER proteins, these receptors homo- or hetero-dimerize, which activates downstream signalling pathways that encourage cell division, growth, and inhibit apoptosis.8 HER2 does not have a known ligand, but the other HER proteins prefer it as a dimerization partner. Besides, HER2 overexpression or amplification causes abnormal dimerization and signalling. In these “HER2-positive” tumours, the effectiveness of HER2-targeted drugs is most apparent.
Trastuzumab was the first humanized monoclonal antibody (mAb) developed to target HER2-positive breast cancer. Although it showed remarkable success in the treatment of HER2-positive breast cancer, a substantial number of patients develop therapeutic resistance and disease relapse.9,10 After that, pertuzumab mAb was developed and approved by the Food and Drug Administration (FDA) in the U.S.A. for the treatment of HER2-positive breast cancer at a high risk of recurrence in combination with trastuzumab and chemotherapy.11–13
Lapatinib, a small-molecule tyrosine kinase inhibitor (TKI), is a reversible dual receptor kinase inhibitor that binds to the cytosolic ATP-binding pocket, a competitor to ATP. It binds non-covalently to the EGFR and the HER2 receptor, putting the receptor in an inactive conformation.14,15 Lapatinib is used in combination with capecitabine or letrozole, chemotherapeutic agents, for late stages of breast cancer with metastasis.16In vitro studies suggested that lapatinib overcomes trastuzumab resistance mediated by the upregulation of the insulin-like growth factor 1 receptor (IGF1R).17 Furthermore, neratinib, a second-generation TKI, targets EGFR, HER2, and HER4, inhibiting the receptor autophosphorylation.18 Therefore, it blocks the downstream signals and leads to cell cycle arrest and apoptosis.19 It is reported that neratinib inhibits cellular growth in trastuzumab-resistant cell lines and is synergistic with trastuzumab.19,20 Neratinib is approved by the FDA to be used as the extended adjuvant treatment of patients with HER2-positive breast cancer after completing trastuzumab therapy. It overcomes the trastuzumab and lapatinib resistance and acts in combination with trastuzumab.20,21 In addition, the FDA approved neratinib in combination with capecitabine for patients with advanced or metastatic HER2-positive breast cancer.
Raman microscopy has been utilized in numerous biological, therapeutic, and drug discovery applications, as well as in other fields.22–26 It has enormous potential for differentiating between various cell types and ultimately can screen for cancer cells.27–29 Besides, Raman microscopy has been applied to study cell–drug interactions. For instance, the effectiveness of numerous anti-cancer drug candidates and medications, including antibodies, small-molecule inhibitors such as TKIs, and chemotherapeutic agents, has been studied using Raman micro-spectroscopy.30–36 Moreover, the literature is well documented with label-free distribution of small molecule inhibitors or drug carriers, including functional groups such as an alkyne or nitrile, or isotopic labelling (deuterium) in cells by Raman microscopy.30,37–39 Based on the fact that these functional groups show Raman bands in the silent region of the cell spectrum (1800–2800 cm−1), they can be employed as label-free markers. However, few reports showed the potential of Raman imaging for monitoring small molecule inhibitors in cells using Raman bands in the fingerprint region (600–1800 cm−1).31,40,41
Recently, there has been growing interest in the potential of machine learning (ML) in the analysis of Raman spectroscopy for a wide range of applications.22,42,43 Principal component analysis (PCA) and clustering techniques are examples of unsupervised models that have been used in ML applications. Others have used supervised learning techniques like partial least-squares (PLS), linear discriminant analysis (LDA), support vector machine (SVM), and deep learning techniques.
In the present study, we used Raman micro-spectroscopy in combination with a deep wavelet scattering-based multivariate analysis framework for the first time to examine the potency and effectiveness of lapatinib and neratinib on breast cancer cells. This approach was successful in discriminating between control cells and drug-treated cells with high accuracy, compared to PCA. Lapatinib or neratinib induced large spectral changes in the Raman spectra upon cell treatment with different doses of the drugs. Both lapatinib and neratinib showed suppression of ERK and AKT phosphorylation, growth inhibition, cell cycle arrest, and apoptosis, as indicated by the in vitro assays. These findings imply that a cellular response to TKI such as cell cycle arrest and apoptosis is the reason behind the observed Raman spectral alteration in the cells following TKI treatment. Additionally, the Raman bands of lapatinib in the fingerprint region were used to track its label-free distribution inside cells. The current findings show Raman micro-spectroscopy's capability for screening anticancer drugs and suggest that it has potential for drug discovery.
Experimental section
Cell culture
SK-BR-3 breast cancer cell line was purchased from the American Type Culture Collection (ATCC). The passaging of the SK-BR-3 cells was performed in Dulbecco's Modified Eagle Medium (DMEM). Cells were washed with phosphate-buffered saline (PBS) to remove dead cells; after this 3 ml of trypsin–EDTA were added for 3 to 5 minutes to the cells. After the incubation time, the detachment of the cells was checked under the microscope and 7 ml of DMEM was added. The suspension was transferred into a Falcon tube and centrifuged for 3 minutes at room temperature and 1500g. The supernatant was sucked from the tube and the pellet was resuspended in 10 ml fresh DMEM. Finally, around 1 ml of cells was diluted to the final volume of 10 ml DMEM in a new Petri dish. Cells were incubated until it reached a confluence of around 80% at 37 °C under a 5% CO2 atmosphere in an incubator.For the Raman measurements, 5 ml of DMEM was added to the slides in six-well plates and a variable number of cells (ca. 70 μl) were added during a splitting on the slides. Cells were then incubated at 37 °C under a 5% CO2 atmosphere until they reached a confluence of around 60%. At this juncture, the media in the wells was discarded and fresh media with different concentrations of drugs or without treatment (control) were added. The drugs used in the experiments are lapatinib and neratinib, each in three different concentrations of 0.1, 0.5, and 1 μM. The drugs were incubated for 16 h in the incubator. After that, the media was sucked away, and the untreated and treated cells were fixed with 4% paraformaldehyde (PFA) for 15 min at room temperature. Then, PFA was discarded, and the slides were washed three times with PBS and finally stored in PBS at 4 °C until the measurements.
Confocal Raman micro-spectroscopy
For the Raman micro-spectroscopic imaging, the WITec alpha 300AR confocal Raman microscope was used as described in previous studies.30,44–46 Two excitation sources, a frequency-doubled Nd-YAG laser of 532 nm (Crystan Laser, Reno, USA) and a single-frequency diode laser of 785 nm (Toptica Photonics AG, Munich, Germany), were used. The lasers and the Zeiss microscope were connected to each other through the wavelength-specific single-mode fiber. An achromatic lens collected the laser beam which passed through the holographic band-pass filter. The Nikon NIR APO (60×/1.00NA) water immersion objective was used to focus the beam on the sample. The microscope had a piezoelectrically driven microscope scanning stage, which has an x–y-resolution of 3 nm and a z-resolution of 0.3 nm. The sample was placed on the microscope scanning stage and covered with PBS.Raman back-scattered radiation was collected through the water immersion objective and passed into the holographic edge filter into the multimode fiber (50 μm diameter). Then, the beam was passed into the 300 mm focal length monochromator which incorporated a 600 mm−1 grating blazed at 500 nm. The spectra were detected with a back-illuminated deep-depletion charge-coupled device (CCD) camera operating at −60 °C. Before the sample measurement, the spectrograph and the laser intensity were calibrated to get a high measurement precision and a high signal-to-noise ratio. Raman imaging measurements were conducted by raster scanning the laser beam over the cells and getting a full Raman spectrum at each pixel with an integration time of 0.5 seconds per pixel and a pixel resolution of 500 nm.
Around 50 SK-BR-3 cells per treated/untreated cohort and concentration were measured for the evaluation of lapatinib and neratinib using 532 nm excitation wavelength.
Deep wavelet scattering-based multivariate analysis framework
In this work, we investigated the potential of deep wavelet scattering networks in uncovering discriminatory features from Raman spectra. A deep wavelet scattering-based multivariate analysis framework is presented for the analysis of Raman spectra. Fig. 1 shows the building blocks of the proposed framework. At the core of the proposed framework is a deep wavelet scattering network. A wavelet scattering network is a deep architecture that can extract deep low-variance features.47–50 Wavelet scattering networks share several appealing properties with convolutional neural networks (CNNs) such as multi-scale representation, non-linearity, and sparsity. Furthermore, wavelet scattering networks use predefined wavelet and scattering filters. This makes them appealing in resource-aware applications that are constrained in data or computational resources. Next, a task-aware low-dimension representation was obtained from the wavelet scattering space using the minimum redundancy maximum relevance (MRMR) algorithm.51 Principal component analysis (PCA) and independent component analysis (ICA) were then applied to the low-dimensional scattering-based representation. | ||
| Fig. 1 Scattering-based multivariate analysis framework for Raman spectra. | ||
Deep wavelet scattering network (DWSN)
A DWSN was constructed by iteratively repeating three basic operations. These are namely convolution with a wavelet filter, applying a non-linearity operator, and averaging using the scaling function. Complex Morlet wavelets were used to construct the wavelet scattering network. Let x be an input 1D Raman spectrum signal. Let ψ and φ be the mother wavelet and scaling functions used to build the network's filter banks. φJ is a low-pass filter used to introduce invariance at scale J. {ψjk}jk∈Λk is the filterbank constructed by dilating of the mother wavelet. Λk is the family of wavelet filters with octave resolution Qk. In the proposed framework, the wavelet scattering network was constructed using two layers with 8 wavelets per octave in each layer. The mth order scattering coefficients are then defined as in eqn (1). | (1) |
The final scattering matrix is then calculated by aggregating the scattering coefficients from all the orders as given in eqn (2) where l is the maximal decomposition order.
| Sx = {Smx}0<m<l | (2) |
A scattering-based embedding of the Raman spectra is then obtained by aggregating the scattering coefficients into a one-dimensional vector.
Task-aware low dimensional scattering-based representation
The scattering representation is redundant and over-complete.48,49 Hence, next, a smaller subspace of the scattering space is learnt using the MRMR algorithm.51 The MRMR algorithm aims at finding the optimal subset of feature space that has maximum relevance with respect to the response variable y and minimum redundancies. The algorithm ranks the features using the mutual information quotient (MIQ) defined in eqn (3), where Vx is a measure of the relevance of the feature y to the response variable c. Wy is a measure for the redundancy of the feature y and I(y, c) is the mutual information between the feature y and the response variable c. | (3) |
| Vy = I(y, c) | (4) |
 | (5) |
Scattering-based multivariate analysis
Having discovered the low dimensional discriminative sub-space in the scattering domain, the final step is to uncover hidden modes of variations in the wavelet scattering domain. This is done by applying PCA in the scattering sub-space.Scattering principal component analysis (scattering PCA)
PCA is a multivariate analysis method that is commonly used in the analysis of Raman spectra. Despite its ability to produce compact representations, using standard PCA on Raman spectra inherently suffers several limitations. For instance, in the case of a limited training set, the sample covariance matrix estimated by PCA becomes a poor estimator of the true covariance matrix. This inherently increases the model's sensitivity to outliers. Moreover, the degree of freedom of a PCA-based representation is limited by the number of training samples. Hence, rather than direct application of PCA on raw Raman spectra, we applied the PCA on the low-dimensional scattering representation obtained in section 3.2. This helps alleviate the limitations of traditional PCA related to becoming poor estimators in the case of limited training data, inherent higher model scale sensitivity, and inherent global nature of the discovered PCA variation modes.Applying PCA in the scattering domain can be viewed as a Gaussianization process where linear transformation is applied in the scattering domain to the axes of maximal variance. Only the first K − 1 principal components are relevant where K − 1 is less than the original dimensionality of the scattering embedding space and less than the training dataset.
Data preparation and pre-processing
The original data constitute a 3D tensor of the Raman spectra at each pixel of the specimen collected. Prior to evaluating the proposed multivariate analysis framework, several pre-processing steps have been conducted. Cells were extracted from each specimen and labelled as being either control drug-free, lapatinib-treated, or neratinib-treated. Fig. S1 (ESI†) shows examples of the cell cropping process. The average Raman spectrum over the pixels for each cell is then calculated and given the appropriate label. The Raman spectra were subjected to several standard pre-processing steps as illustrated in Fig. S2 (ESI†). This includes cosmic-spike removal, automated autofluorescence background subtraction using the Vancouver algorithm,52 normalization, baseline correction, and selection of the region of interest between 500 to 1800 cm−1 and 2800 to 3100 cm−1.Scattering subspace selection
A low-dimensional sub-space was extracted from the Raman embedding space using the method described above. Fig. S3 (ESI†) shows the output of applying the MRMR algorithm in the scattering embedding space. Fig. S3† suggests that the dimension of the inherent sub-space of interest is less than 200. Fig. S4 and Table S1 in the ESI† provide further insights into the impact of the dimensionality of the scattering embedding sub-space on the subsequent multivariate analysis and classification stages. The reported results in Fig. S4 and Table S1† are for the case of control vs. neratinib-treated cells with doses of 1.0 μM concentration. Fig. S4† shows the extracted scattering-PCA components from sub-spaces with dimensions of 8, 16, 150, and 400. Table S1† compares the average classification accuracy obtained for scattering sub-space of dimensions 8, 16, 150, and 500. It is evident that the discriminability of the framework starts to decrease once the sub-space dimension goes beyond the inherent relevant sub-space discovered in Fig. S3.† Based on the abovementioned insights, we have selected the dimensionality of the scattering embedding space to be 16 × 1, resulting in a considerable reduction in the memory and computational requirements of subsequent stages.Results and discussion
Breast cancer cells were first well characterized by several in vitro assays. Then the efficacy of TKIs was investigated using a combination of Raman micro-spectroscopy and ML.Cellular response to lapatinib and neratinib by RTCA
To determine whether SK-BR-3 breast cancer cells respond to lapatinib and neratinib or not, a label-free xCELLigence RTCA technology was used. This technology is a real-time, non-invasive method, and impedance-based detection of cell viability.53 RTCA monitors cellular proliferation, migration, and invasion by observing the growth, shape, and damage of the cell over an extended period of time. In this approach, gold electrodes are used to measure the changes in the impedance induced by adherent cells. The impedance signal expressed in the cell index value increases over time as cells divide reflecting cell proliferation, while it decreases when cells die and detach from the well surface. The kinetics of SK-BR-3 cellular proliferation and response to different concentrations (0.1, 0.5, and 1.0 μM) of lapatinib (panel A) and neratinib (panel B), as well as cells without drug treatment (control, black traces) were assessed by the xCELLigence platform as displayed in Fig. 2 (see also the ESI†). The used drug concentrations in the present study are slightly smaller and higher than detected lapatinib (1.47–11.5 μg mL−1) and neratinib (5.8–143.3 ng mL−1) concentrations, respectively, in the plasma of patients diagnosed with HER2 positive metastatic breast cancer or solid tumours, after treatment with a single oral dose of either lapatinib (1250–1500 mg d−1) or neratinib (200–400 mg d−1).54–58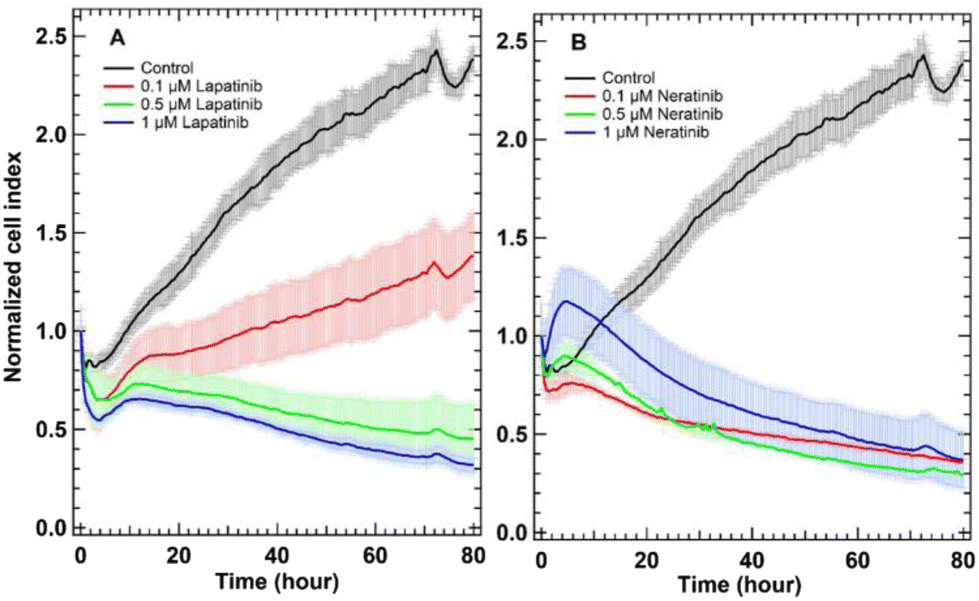 | ||
| Fig. 2 RTCA of untreated SK-BR-3 cells (control) and cells treated with lapatinib (A) and neratinib (B). The panels show the control cells in black, cells treated with 1 μM drug in blue, cells treated with 0.5 μM drug in green, and cells treated with 0.1 μM drug in red. | ||
In Fig. 2, the cell index value of the control SK-BR-3 cells (black traces) increased over time implying that cells still proliferate. The cell index value for cells treated with lapatinib (panel A, red trace) indicated that cells were still growing but the cell index values were lower than those of the control and decreased to around 1.5 after 80 hours of cell incubation with lapatinib. This means that lapatinib reduces the proliferation of cells. In contrast, cells treated with the same concentration of neratinib (panel B, red trace) steadily detached from the plates, and the cell index values remained near 0.5.
Furthermore, the cell index values of lapatinib-treated cells (panel A, 0.5 and 1.0 μM) and neratinib-treated cells (panel B, 0.1–1.0 μM) slightly decreased and then almost remained without a significant change near 0.5 but not increased significantly as in the control nor decreased to near zero as in the case of cells treated with higher drug concentrations.31 The RTCA is used to investigate the cytotoxic/cytostatic effect of different drugs on cells. Cytotoxic compounds induce cellular necrosis, apoptosis, or cytostasis.59 Cytostatic compounds interfere with cell division or growth. By treating SK-BR-3 cells with different concentrations of lapatinib and neratinib, the concentration-dependent cell response is observed. Overall, neratinib and lapatinib show in most of the concentrations a cytostatic effect by interfering with the cell-cycle machinery leading to growth arrest.60 Therefore, the results indicate that SK-BR-3 cells responded to lapatinib and neratinib and thus, we used in the Raman and other in vitro assays cells treated with 0.1–1.0 μM of these drugs for less than 30 hours of incubation (16–24 hours). This is because the respective cell indices demonstrate values between 0.6 and 1.0, implying that cells are viable under these conditions.
Furthermore, the cell viability assay, MTT assay, was performed as shown in Fig. S5 in the ESI,† and the results indicated that neratinib seems to be more cytotoxic at lower concentrations. The MTT assay also validates the rather cytostatic effect of 0.1 μM lapatinib and an increased cytotoxic effect with higher lapatinib concentrations. In addition, the MTT assay in combination with the RTCA demonstrates that the viability of cells treated with a low concentration of 0.1–1 μM of both drugs is very high between 80 and 100% after 16 h. Therefore, concentrations of 0.1–1 μM of both drugs were used in the experiments of the present study.
Moreover, the cell cycle analysis (Table S2 in the ESI†) was performed and the treatment of SK-BR-3 human breast cancer cells with different concentrations of lapatinib and neratinib (0.1–1.0 μM) resulted in a significant reduction in the G2/M phases and an increase in the G1 phase. These results demonstrate clearly that both drugs induce a cell cycle arrest in the G1 phase. The RTCA and the cell cycle results show that the drugs have a cytostatic effect on the cancer cells in the first 24 hours.
Apoptosis assay
In the apoptosis assay, two types of cells are monitored: the apoptotic cells and the necrotic cells. During the apoptosis, phosphatidylserine is translocated from the inner to the outer plasma membrane of cells, exposing it to the extracellular environment.61 Annexin V coupled to fluorescein is used to detect the phosphatidylserine in this assay.62 Necrotic cells are detected by red fluorescent PI, which binds to free nucleic acids in cells, therefore, marking dead cells.63 SK-BR-3 cells were stained not only with PI but also with Annexin V. Table 1 shows that in the untreated cells around 8% of cells were apoptotic and less than 1% were necrotic cells. On the one hand, treatment of SK-BR-3 cells with either lapatinib or neratinib almost did not lead to significant necrosis since the assay shows that there are always less than 1% necrotic cells, in comparison with untreated cells. On the other hand, cells underwent apoptosis when treated with either 0.1 μM lapatinib or neratinib, leading to around 17%. At a higher concentration (1.0 μM), around 25% and 18% apoptotic cells were detected in the case of neratinib- and lapatinib-treated cells, respectively.| Concentrations of drugs in μM | Apoptotic cells in % | Necrotic cells in % | Dead cells in % | |
|---|---|---|---|---|
| Control | — | 7.8 | 0.6 | 1.4 |
| Neratinib | 0.1 | 17.1 | 0.3 | 0.8 |
| 1.0 | 25.4 | 0.9 | 2.0 | |
| Lapatinib | 0.1 | 17.3 | 0.3 | 0.7 |
| 1.0 | 18.3 | 0.3 | 0.7 |
The apoptosis assay shows that the apoptosis rate of cells is more than doubled after the treatment with either of the drugs. These results correlate with the RTCA results especially in the first 16 h (Fig. 2) because the cell indices in the RTCA decreased by around a maximum of 20% of the initial indices before treatment. Taken together the results of all in vitro assays suggest that lapatinib and neratinib induce cell cycle arrest and cell apoptosis after cell treatment with the drug.
Internalization of EGFR and HER2 receptors
The above-discussed assays mainly monitor whole-cell responses that do not allow a pathway-specific conclusion of the stated cellular changes. To address the effect of the two TKIs, lapatinib and neratinib, more specifically, fluorescence staining and western blotting of the targeted receptors were conducted. As TKIs, lapatinib and neratinib bind to the intracellular ATP binding site of the EGFR and HER2 receptors and block their downstream signalling.14–16,18,64 To monitor the two receptors, which are the targets of the drugs, fluorescence staining of these receptors was performed. Cells were labelled with specific antibodies for EGFR and HER2, whereas the DRAQ-5 dye targeted the nucleus. The fluorescence imaging of the untreated cells (control), the EGF-treated cells (positive control), and lapatinib- and neratinib-treated cells are displayed in Fig. 3. In these images, EGFR, HER2, and the nucleus are visualized. In the control cells, EGFR and HER2 are distributed evenly and in high amounts mostly in the outer cell membrane but also in smaller amounts in the cytoplasm (panels A and B). After EGF treatment, the EGFR and HER2 receptors were internalized and localized in clusters in the cytoplasm (panels E and F). This is expected since EGF is a specific ligand of the EGFR; therefore, the binding leads to an activation of the receptor and is followed by internalization, where the receptor gets either recycled or degraded.65,66 | ||
| Fig. 3 SK-BR-3 cells with fluorescence stained EGFR (A, E, I and M), HER2 (B, F, J and N), and nucleus (C, G, K and O). The figure displays control cells (A–D) and cells incubated with EGF (E–H) as a positive control, lapatinib (I–L), and neratinib (M–P). Panels D, H, L, and P display the overlays of all staining. | ||
The fluorescence imaging of cells treated with different concentrations of either lapatinib or neratinib shows a distribution of the receptors in the membrane but also a large internalization of the receptors in the cytoplasm (Fig. 3(I, J, M and N) and see also Fig. S6 and S7 in the ESI†), in comparison with the control. Lapatinib and neratinib bind to both receptors and this leads, without further activation of a ligand, to an internalization of receptors. Hence, the downstream signals, which are activated by the receptor phosphorylation, are presumably blocked leading to an inhibition of proliferation and cell cycle arrest.
The treatment with lapatinib and neratinib leads to the internalization of EGFR and HER2 independent of the drug concentration. As mentioned before, internalization is in part due to ligand binding, but if compared to control levels of internalization, drug-treated cells display elevated levels of internalization. In conclusion, the fluorescence imaging shows the receptor-specific drug effect as an increased number of receptor–drug complexes seems to get internalized demonstrated by receptor clusters in the cells.
Furthermore, western blotting was applied to evaluate the potency and efficacy of drug candidates by monitoring individual proteins marked by antibodies and single signal transduction pathways. The results (Fig. S8 in the ESI†) showed that the drugs inhibited the phosphorylation of downstream signalling pathways of EGFR. Therefore, the growth and proliferation of SK-BR-3 cells were inhibited by the used drug concentrations as shown by both RTCA (Fig. 2) and western blot results (Fig. S8†).
Cellular response to lapatinib and neratinib by Raman micro-spectroscopy
Lapatinib and neratinib were developed to treat HER2-positive breast cancer in metastatic/advanced forms.67 Neratinib inhibits the receptor autophosphorylation by binding irreversibly to Cys-773 and Cys-805 in EGFR and HER2 in the ATP binding pocket, respectively.18 Lapatinib binds reversibly to the cytoplasmic ATP-binding site of the kinase, preventing receptor phosphorylation.14 SK-BR-3 cells were treated separately with two TKIs, lapatinib and neratinib, and cells were well characterized by several in vitro assays. The results indicated the inhibition of the receptors, cell-cycle arrest, and cell apoptosis. Raman measurement of the untreated and lapatinib- and neratinib-treated cells was performed under similar experimental conditions. Around 50 cells of each group were measured. After the measurement, the data were clustered with HCA to obtain the average spectra of each cell, which were later merged to get the mean spectra of each group and the results are displayed in Fig. 4. The mean spectra of cells (a–d) depict the same characteristic Raman peaks at 2800–3050 cm−1 with two prominent peaks at 2935 cm−1 and 2879 cm−1 (C–H-symmetric stretching vibrations for lipids and proteins), around 1657 cm−1 (amide I), 1448 cm−1 (CH–CH2 bending modes), around 1340–1250 cm−1 (amide III (C–N stretching, N–H bending, proteins) and/or PO2 asymmetric stretching), and 1311 cm−1 (CH3 and CH2 twisting or bending of lipids and collagen).30,33,68,69 Moreover, two smaller peaks are displayed in the spectra: the peak around 1093 cm−1, which is typical of the symmetric O–P–O-stretching vibrations of the DNA backbone or the C–C stretching of phospholipids and the phenylalanine ring breathing mode around 1006 cm−1.30,68 The biochemical changes upon drug treatment are explored by calculating the Raman difference spectra for different drug concentrations. For this purpose, the mean Raman spectra of drug-treated cells (b–d) were subtracted from the mean Raman spectrum of the untreated cells (a), and the results are displayed in Fig. 5.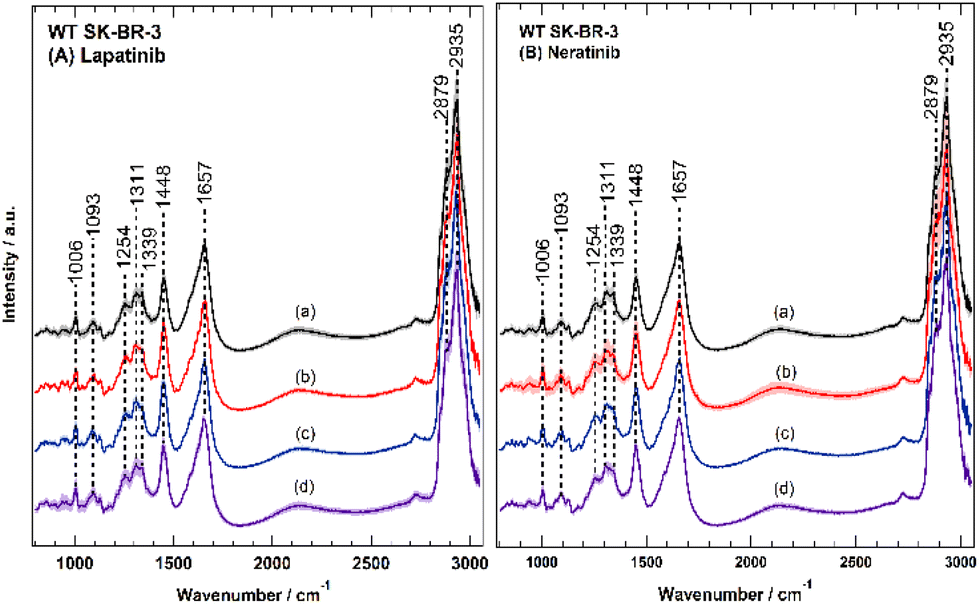 | ||
| Fig. 4 Raman mean-spectra of SK-BR-3 cells untreated or treated with either lapatinib (A) or neratinib (B) in different concentrations. Spectra a represent the control cells without drug treatment. In panel (A), the spectra b, c, and d represent the spectra of cells treated with lapatinib concentrations of 0.1 μM (b), 0.5 μM (c), 1 μM (d), respectively. In panel (B), the spectra b, c, and d represent the spectra of cells treated with neratinib concentrations of 0.1 μM (b), 0.5 μM (c), 1 μM (d), respectively. | ||
 | ||
| Fig. 5 Raman difference spectra of SK-BR-3 treated with lapatinib (A) and neratinib (B). Spectra (a–c) are obtained by subtracting the drug-treated cells from the untreated cells (control), in which cells were treated with different drug concentrations of 1 μM (a), 0.5 μM (b), and 0.1 μM (c). | ||
Fig. 5 clearly demonstrates that both drugs with different concentrations significantly influence the SK-BR-3 cells. The largest spectral changes can be seen in C–H stretching vibrations of lipids and proteins (2850–3050 cm−1), amide I region (around 1650 cm−1), and CH–CH2 bending modes (1448–1450 cm−1, representing lipids, collagen, and phospholipids).70 A smaller change can be also observed at 1590 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching vibration) in panel A and at 1612 cm−1 (CC stretching vibrations of aromatic rings) in panel B.71–73 On the one hand, the difference spectra of lapatinib strikingly show an inverse drug concentration effect, meaning that the observed biochemical changes increase with decreasing the concentration of lapatinib. On the other hand, the cellular response to different concentrations of neratinib is almost similar. These results suggest differences in the cellular response to lapatinib and neratinib at least on a cellular level.
N stretching vibration) in panel A and at 1612 cm−1 (CC stretching vibrations of aromatic rings) in panel B.71–73 On the one hand, the difference spectra of lapatinib strikingly show an inverse drug concentration effect, meaning that the observed biochemical changes increase with decreasing the concentration of lapatinib. On the other hand, the cellular response to different concentrations of neratinib is almost similar. These results suggest differences in the cellular response to lapatinib and neratinib at least on a cellular level.
SK-BR-3 cells were well characterized by several in vitro assays, and the results indicated cell-cycle arrest, cell apoptosis, and inhibition of the receptors (Tables 1 and S2 and Fig. S8†). Therefore, it is reasonable to conclude that the observed Raman spectral changes upon drug treatment (Fig. 6) reflect the cellular response to the drug, including cell-cycle arrest, apoptosis, and receptor inhibition.
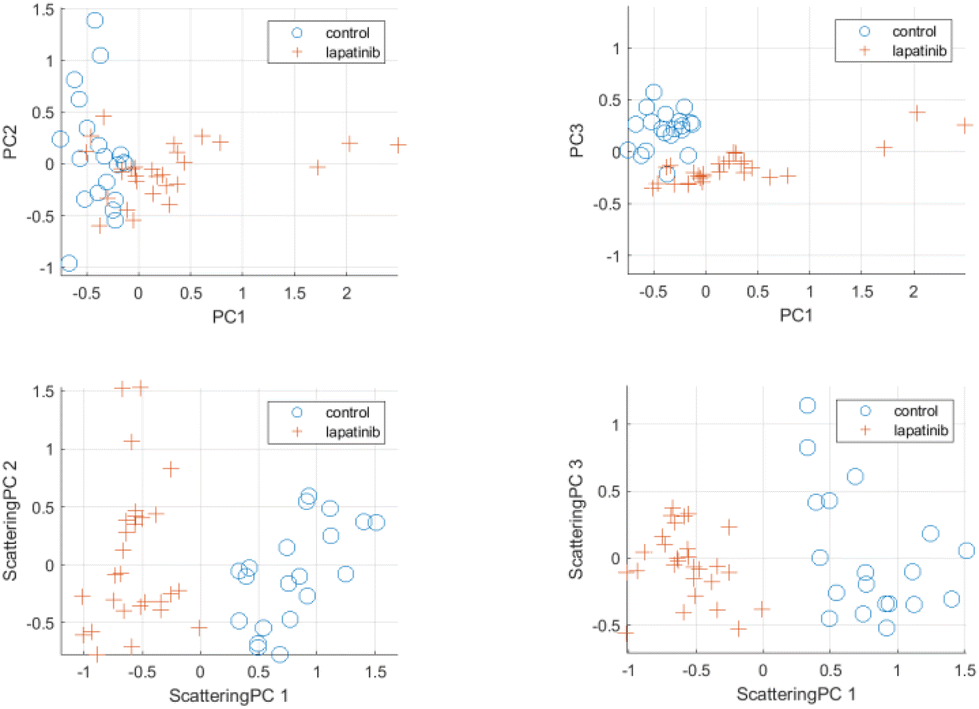 | ||
| Fig. 6 Scattering PCA multivariate analysis results for control cells and cells treated with lapatinib at 0.5 μM concentration. Top row: Principal components (PCs) using standard PCA-based analysis. Bottom row: Scattering principal components using proposed analysis framework. | ||
Scattering-based multivariate analysis
Scattering-based multivariate analysis is conducted by applying PCA on the low-dimensional scattering subspace for the Raman spectra of cells in the dataset. PCA aims at extracting directions of maximum variance within the scattering embedding space. The extracted principal components in the scattering domain are referred to as scattering principal components (scattering PC). For the sake of comparison, standard principal components (PCs) were extracted using the commonly used PCA-based multivariate analysis of raw Raman spectra. Fig. 6 and 7 (see also Fig. S9 and S10 in ESI†) provide visualization for the discriminating power of the proposed scattering-based multivariate analysis compared to the standard PCA-based analysis. Fig. 6 shows the scatter plots for the first three scattering-principal components (ScatPC1, Scat-PC2, Scat-PC3) compared to the PCs. It is evident that the scattering-based principal components tend to better differentiate between the drug-free cells and lapatinib-treated cells. The same observation can be seen in Fig. 7 for the case of neratinib-treated cells.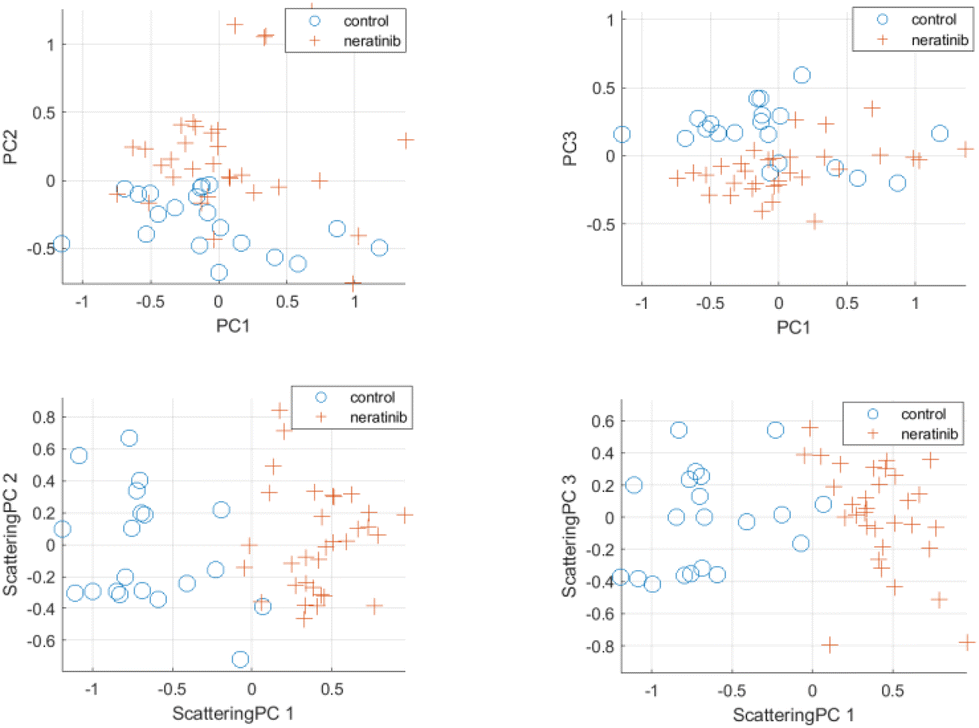 | ||
| Fig. 7 Scattering-based PCA multivariate analysis results for control cells and cells treated with neratinib at 0.5 μM concentration. Top row: Principal components (PCs) using standard PCA-based analysis. Bottom row: Scattering principal components using proposed analysis framework. | ||
Next, we have evaluated the ability of the scattering-based multivariate analysis framework to discriminate between control cells and drug-treated cells. The Support Vector Machine (SVM) was used as the classifier in all experiments. Given the limited size of the training dataset, leave-one-out cross-validation was used to evaluate the accuracies in all the experiments. Results were compared against commonly used PCA and independent component analysis (ICA-based) analysis frameworks. Classification results are summarized in Tables S3 and S4 in the ESI† for the case of lapatinib- and neratinib-treated cells, respectively. In the classification experiment, detecting a control cell is referred to as a negative case, and detecting a drug-treated cell is referred to as a positive case. Corresponding confusion matrices are presented in Fig. S11 and S12† for the cases of doses with 0.5 μM and 1 μM concentrations, respectively.
Overall, the scattering-based multivariate framework has demonstrated superior performance in differentiated control vs. treated cells at different drug doses. In the case of lapatinib-treated cells, compared to the PCA-based framework, the scattering-based multivariate framework has achieved an improvement in accuracy of 10.42% and 10.71% for the 0.5 μM and 1 μM concentrations, respectively. Compared to the ICA-based framework, an average accuracy improvement of 2.1% was achieved. In the case of neratinib-treated cells, the scattering-based multivariate framework has outperformed both PCA-based and ICA-based frameworks with an average accuracy improvement of 5.7% and 3.75% compared to the PCA-based and ICA-based frameworks, respectively.
F1-score is also used to assess the overall ability of the framework to identify drug-treated cases while minimizing false positives and false negative cases. Compared to the PCA-based framework, the proposed framework has achieved an average improvement of 15.63% and 4.87% for the case of lapatinib-treated and neratinib-treated cells, respectively. Confusion matrices for the leave-one-out cross-validation experiments are presented in Fig. S11 and S12 in the ESI.† In terms of false positive rates (FPR), the proposed scattering PCA-based framework has achieved an average False Positive Rate (FPR) of 2.5%, compared to 15% in the case of the PCA-based framework. In terms of false negative rates (FNR), the proposed framework has achieved an average false negative rate (FNR) of 0.775% compared to an FNR of 10.625% in the case of the PCA-based framework. In conclusion, a scattering-based multivariate framework was applied here successfully for the first time on Raman data, and this approach could differentiate between drug-free cells and drug-treated cells with high accuracy compared to the classical PCA.
Label-free distribution of lapatinib in cells by Raman spectral imaging
Label-free Raman micro-spectroscopy has been used tremendously in the last two decades to monitor the uptake and distribution of drugs or drug carriers in cells. However, it is very challenging because drugs or their carriers often accumulate at lower concentrations within cells. It is well documented that the Raman sensitivity of a drug of interest can be improved when it contains an alkyne or nitrile functional group, or isotopic labelling (deuterium or 13C) because these groups produce Raman bands in the silent region of the cell spectrum (1800–2800 cm−1).22,74 Recently, it has been shown that small-molecule drugs or drug candidates can be visualized in cells using a Raman marker band in the fingerprint region (700–1800 cm−1). This is because the small-molecule drugs have sharp and strong Raman bands in the fingerprint region, while those of cells are broad bands. For instance, the distribution of tyrosine kinase inhibitors such as sorafenib and sunitinib was monitored in cells using Raman bands located at 1028 cm−1 and 1320/1570 cm−1, respectively.31,75 In SRS imaging study, Fu et al.40 used also the Raman bands of nilotinib, chloroquine, and GNF and GNF at 1300 cm, 1370, and 1600 cm−1 to display their distribution in the cells. Besides, the distribution of doxorubicin and squalene-doxorubicin in cancer cells was detected through several Raman bands in the fingerprint region.76We have previously reported the neratinib distribution in cells using label-free Raman micro-spectroscopy.37 In the present study, the uptake and distribution of lapatinib in breast cancer cells were investigated. The Raman spectrum (a) of pure lapatinib displays bands around 794, 1002, 1031, 1064, 1135, 1209, 1269, 1295, 1363, 1390, 1443, 1488, 1525, 1578, 1611, and 1625 cm−1 as depicted in Fig. 8A. The strong Raman bands of lapatinib at 794, 1209, 1363, 1390, 1525, 1611, and 1625 cm−1 are possible candidates for visualizing the localization of lapatinib in cells. However, the Raman spectrum of SK-BR-3 cells (c) reveals Raman bands near 1002 cm−1 (ring-breathing mode of phenylalanine), 1250–1340 cm−1 (amide III), and ∼1659 cm−1 (amide I) that overlap with lapatinib Raman bands near 1002, 1611, and 1625 cm−1. Therefore, it is reasonable to conclude that lapatinib Raman bands at 794, 1209, 1363, and 1525 cm−1 (spectrum a), which have no corresponding bands in the cell spectrum (c), can be used as label-free marker candidates to visualize the localization within cells. Xu et al. used the Raman band near 1363 cm−1 to monitor the distribution of lapatinib in A549 cells by SRS imaging.41
 | ||
| Fig. 8 (A) The Raman spectra of free lapatinib (a), the average Raman spectrum of the lapatinib-containing cluster in SK-BR-3 cells shown in panel (E) (b), the average Raman spectrum of the lapatinib-containing cluster in SK-BR-3 cells shown in panel (I), and the average Raman spectrum of the control cells (d). (B–I) Raman imaging of SK-BR-3 cells treated with 10 μM lapatinib for 4 h. Raman images reconstructed from the band's intensities at 1420–1470 cm−1 (B) and 1515–1535 cm−1 (C). (D) Overlay of panels B and C. (F–H) Cross-section Raman images along the x–z axis of the same cells. Scanning positions are indicated by the white line in panel B. (E and I) represent the distribution of lapatinib (red structure) in cells. (E and I) Lapatinib-containing clusters obtained by K-means based on the Raman data shown in panels B and F. | ||
Fig. 8(B–I) shows the Raman micro-spectroscopic imaging results for SK-BR-3 cells incubated for four hours with approximately 10 μM lapatinib. At this concentration, most of the cells are viable, as confirmed by RTCA (Fig. 2) and the cytotoxicity assay (Fig. S5†). Fig. 9B depicts an integrated Raman intensity image of the Raman band in the range of 1425–1470 cm−1 of SK-BR-3 cells treated with lapatinib, reflecting the different cellular components. Since lapatinib does not have any Raman bands in this region, panel (B) does not show any contribution from lapatinib. To monitor the lapatinib distribution within cells, a Raman intensity image was constructed around the 1515–1535 cm−1 band (panel C). The subcellular spatial distribution of lapatinib in SK-BR-3 cells is obtained in panel (D) by overlaying images (B) and (C). To confirm that lapatinib is localized within SK-BR-3 cells but not precipitated on the cell surface, cross-section Raman imaging of the same cell along the indicated x–z axis was also acquired as shown in panels (F–H), demonstrating that lapatinib was internalized within SK-BR-3 cells. The reproducibility of these results was confirmed as shown in Fig. S13–S15 in the ESI.† Therefore, Raman micro-spectroscopy shows the lapatinib uptake and distribution in cells employing Raman marker bands in the fingerprint region.
 | ||
| Fig. 9 (A) Lapatinib-containing cluster of K-means based on Raman data shown in red. (B) Fluorescence image of the same cells that shows the EGFR (green) and the nucleus (blue). (C) Fluorescence image of the same cells that shows the HER2 (red) and the nucleus (blue). (D) Overlay of (B) and (C). (E) Overlay of the lapatinib-containing cluster (red) (A) and EGFR (green) (B) and the overlaid regions are shown in yellow. (F) Overlay of the lapatinib-containing cluster (red) (A) and HER2 (green) (C) and the overlaid regions are shown in yellow and white. | ||
The Raman spectral imaging results were analysed utilizing K-means and the drug cluster and the average Raman spectrum of the lapatinib-containing cluster within cells are shown in Fig. 8(E and I) and 8A (b and c), respectively. The K-means image shows more lapatinib-containing clusters than the corresponding integrated Raman intensity image of the lapatinib band (1515–1535 cm−1) (panel B). This is because the K-means clustering assigns each pixel exclusively to a cluster generating a better-quality image than the univariate Raman intensity image. It is worth mentioning that K-means of the Raman spectra were used in this section but not in the scattering-based analysis.
The average Raman spectrum of the lapatinib-containing cluster within cells (Fig. 8A (spectrum b and c)) reveals several Raman bands, similar to those observed for pure lapatinib (spectrum a), especially those observed at 1002, 1209, 1363, 1525, 1611, and 1625 cm−1. Cellular contribution to the spectra of the lapatinib-containing cluster (b and c) is detected as indicated by the Raman bands located at 1002, 1210–1380, 1443, and 1659 cm−1.
Interestingly, the spectra of the lapatinib-containing cluster (b and c) are different from that of pure lapatinib (a), where the Raman bands at 794 and 1488 cm−1 of pure lapatinib (spectrum a) disappeared in the spectra of the lapatinib-containing cluster (spectrum b and c). These results suggest that lapatinib is metabolized within cells. In contrast, in other measurements, a small broad band at 794 cm−1 appeared (Fig. S13 in the ESI†) instead of complete disappearance as shown in Fig. 8A.
Lapatinib is a dual inhibitor and binds to the intracellular ATP binding site of the EGFR and HER2 receptors and blocks their downstream signalling.14–16,18,64 The distribution of lapatinib, EGFR, and HER2 in the same cells was achieved by performing fluorescence imaging of the same cells after Raman micro-spectroscopic imaging and the results are depicted in Fig. 9. The K-means of the Raman results showing the lapatinib-containing cluster (shown also in Fig. 8E) and fluorescence imaging of EGFR and HER2 are shown in panels A, B, and C, respectively. The lapatinib-containing cluster (red) from (A) is overlaid with the EGFR from (B) and with the HER2 from (C) and the results are displayed in (E) and (F), respectively. Parts of lapatinib are colocalized with the target receptors, EGFR and HER2, as indicated by the overlaid regions shown in yellow colour in panels (E) and (F). These results imply that some of the lapatinib binds to EGFR and HER2 receptors, blocking their phosphorylation and then inhibiting their downstream signalling and subsequently cellular proliferation.14–16,18,64
Conclusions
A deep wavelet scattering-based multivariate analysis framework is presented for the analysis of Raman spectra of control versus drug-treated breast cancer cells. The Raman results detected large lapatinib- and neratinib-induced differences in breast cancer cells. The potent effect of lapatinib and neratinib on breast cancer cells is thus demonstrated by the Raman micro-spectroscopic results. In addition, the results showed the discriminating power of the proposed scattering-based multivariate analysis compared to the standard PCA-based analysis. The lapatinib- and neratinib-treated SK-BR-3 cells exhibit reduced cellular proliferation and inhibition of the phosphorylation of ERK and AKT, as demonstrated by the in vitro RTCA and western blot assays, respectively. Furthermore, lapatinib and neratinib have a cytostatic effect on the cancer cells and induced apoptosis, as demonstrated by the results of the cell cycle, apoptosis assays, as well as RTCA. Therefore, the observed Raman spectral changes upon drug treatment are produced as a result of cell-cycle arrest, apoptosis, and receptor inhibition. These results also concur with the clinical studies as well. Additionally, the results of the Raman spectral imaging have demonstrated the uptake and label-free distribution in cells and colocalization of lapatinib with EGFR and HER2 in cancer cells. These findings reveal that Raman micro-spectroscopy is capable of detecting drug uptake in the fingerprint region, as demonstrated in this case with lapatinib. When the efficacy data for lapatinib and neratinib are combined with their distribution in cells, it expands the ability of Raman micro-spectroscopy to track drug pharmacodynamics and pharmacokinetics. This will help with the next stage of pre-clinical evaluation and drug discovery, which is the evaluation of drugs in an in vivo setting like mouse models.Author contributions
S. F. E.-M., K. G., A. M. and S. H. designed research; I. S., T. F., D. J., and M. S. performed research; I. S., M. S., T. F., A. M., and S. F. E.-M. analysed data; R. Z. developed and performed the multivariate analysis; S. F. E.-M. and R. Z. wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Protein Research Unit Ruhr within Europe (PURE), funded by the Ministry of Innovation, Science and Research (MIWF) of North-Rhine Westphalia, Germany (Grant Number 233-1.08.03.03-031-68079), and the Center for Protein Diagnostics (PRODI), funded by the Ministry of Culture and Science (MKW) of the State of North Rhine-Westphalia, Germany (Grant Number 111.08.03.05-133974).References
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, CA-Cancer J. Clin., 2021, 71, 209–249 CrossRef PubMed.
- A. E. Giuliano, J. L. Connolly, S. B. Edge, E. A. Mittendorf, H. S. Rugo, L. J. Solin, D. L. Weaver, D. J. Winchester and G. N. Hortobagyi, CA-Cancer J. Clin., 2017, 67, 290–303 CrossRef PubMed.
- H. Dillekås, M. S. Rogers and O. Straume, Cancer Med., 2019, 8, 5574–5576 CrossRef PubMed.
- S. M. Swain, M. Shastry and E. Hamilton, Nat. Rev. Drug Discovery, 2023, 22, 101–126 CrossRef CAS PubMed.
- R. V. J. Chari, Acc. Chem. Res., 2008, 41, 98–107 CrossRef CAS PubMed.
- H. Masuda, D. Zhang, C. Bartholomeusz, H. Doihara, G. N. Hortobagyi and N. T. Ueno, Breast Cancer Res. Treat., 2012, 136, 331–345 CrossRef CAS PubMed.
- R. Roskoski, Pharmacol. Res., 2014, 79, 34–74 CrossRef CAS PubMed.
- S. J. Fuller, K. Sivarajah and P. H. Sugden, J. Mol. Cell. Cardiol., 2008, 44, 831–854 CrossRef CAS PubMed.
- J. Baselga, Ann. Oncol., 2010, 21, vii36–vii40 CrossRef PubMed.
- R. Nahta and F. J. Esteva, Cancer Lett., 2006, 232, 123–138 CrossRef CAS PubMed.
- G. Von Minckwitz, M. Procter, E. De Azambuja, D. Zardavas, M. Benyunes, G. Viale, T. Suter, A. Arahmani, N. Rouchet, E. Clark, A. Knott, I. Lang, C. Levy, D. A. Yardley, J. Bines, R. D. Gelber, M. Piccart and J. Baselga, N. Engl. J. Med., 2017, 377, 122–131 CrossRef CAS PubMed.
- S. M. Swain, J. Baselga, S.-B. Kim, J. Ro, V. Semiglazov, M. Campone, E. Ciruelos, J.-M. Ferrero, A. Schneeweiss, S. Heeson, E. Clark, G. Ross, M. C. Benyunes and J. Cortés, N. Engl. J. Med., 2015, 372, 724–734 CrossRef CAS PubMed.
- A. Schneeweiss, S. Chia, T. Hickish, V. Harvey, A. Eniu, R. Hegg, C. Tausch, J. H. Seo, Y.-F. Tsai, J. Ratnayake, V. McNally, G. Ross and J. Cortés, Ann. Oncol., 2013, 24, 2278–2284 CrossRef CAS PubMed.
- E. R. Wood, A. T. Truesdale, O. B. McDonald, D. Yuan, A. Hassell, S. H. Dickerson, B. Ellis, C. Pennisi, E. Horne, K. Lackey, K. J. Alligood, D. W. Rusnak, T. M. Gilmer and L. Shewchuk, Cancer Res., 2004, 64, 6652–6659 CrossRef CAS PubMed.
- Q. Wang, J. A. Zorn and J. Kuriyan, in Methods in Enzymology, ed. K. M. Shokat, Academic Press, 2014, vol. 548, pp. 23–67 Search PubMed.
- M. Hasmann, in Introduction to Biological and Small Molecule Drug Research and Development, ed. R. Ganellin, S. Roberts and R. Jefferis, Elsevier, Oxford, 2013, pp. 283–305 Search PubMed.
- M. Scaltriti, F. Rojo, A. Ocana, J. Anido, M. Guzman, J. Cortes, S. Di Cosimo, X. Matias-Guiu, S. Ramon Y Cajal, J. Arribas and J. Baselga, JNCI, J. Natl. Cancer Inst., 2007, 99, 628–638 CrossRef CAS PubMed.
- K. Feldinger and A. Kong, Breast Cancer, 2015, 7, 147–162 Search PubMed.
- S. K. Rabindran, Cancer Res., 2004, 64, 3958–3965 CrossRef CAS PubMed.
- A. Canonici, M. Gijsen, M. Mullooly, R. Bennett, N. Bouguern, K. Pedersen, N. A. O'Brien, I. Roxanis, J.-L. Li, E. Bridge, R. Finn, D. Slamon, P. McGowan, M. J. Duffy, N. O'Donovan, J. Crown and A. Kong, Oncotarget, 2013, 4, 1592–1605 CrossRef PubMed.
- D. M. Collins, N. T. Conlon, S. Kannan, C. S. Verma, L. D. Eli, A. S. Lalani and J. Crown, Cancers, 2019, 11, 737 CrossRef CAS PubMed.
- S. F. El-Mashtoly and K. Gerwert, Anal. Chem., 2022, 94, 120–142 CrossRef CAS PubMed.
- C. Krafft, M. Schmitt, I. W. Schie, D. Cialla-May, C. Matthäus, T. Bocklitz and J. Popp, Angew. Chem., Int. Ed., 2017, 56, 4392–4430 CrossRef CAS PubMed.
- J. Zeng, W. Zhao and S. Yue, Front. Pharmacol., 2021, 12, 630167 CrossRef CAS PubMed.
- K. Hanna, E. Krzoska, A. M. Shaaban, D. Muirhead, R. Abu-Eid and V. Speirs, Br. J. Cancer, 2022, 126, 1125–1139 CrossRef PubMed.
- Y. Qi, Y. Liu and J. Luo, PhotoniX, 2023, 4, 22 CrossRef.
- H. K. Yosef, S. D. Krauß, T. Lechtonen, H. Jütte, A. Tannapfel, H. U. Käfferlein, T. Brüning, F. Roghmann, J. Noldus, A. Mosig, S. F. El-Mashtoly and K. Gerwert, Anal. Chem., 2017, 89, 6893–6899 CrossRef CAS PubMed.
- K. Aljakouch, Z. Hilal, I. Daho, M. Schuler, S. D. Krauß, H. K. Yosef, J. Dierks, A. Mosig, K. Gerwert and S. F. El-Mashtoly, Anal. Chem., 2019, 91, 13900–13906 CrossRef CAS PubMed.
- I. W. Schie, J. Rüger, A. S. Mondol, A. Ramoji, U. Neugebauer, C. Krafft and J. Popp, Anal. Chem., 2018, 90, 2023–2030 CrossRef CAS PubMed.
- S. F. El-Mashtoly, D. Petersen, H. K. Yosef, A. Mosig, A. Reinacher-Schick, C. Kötting and K. Gerwert, Analyst, 2014, 139, 1155–1161 RSC.
- H. K. Yosef, T. Frick, M. K. Hammoud, A. Maghnouj, S. Hahn, K. Gerwert and S. F. El-Mashtoly, Analyst, 2018, 143, 6069–6078 RSC.
- K. Meister, J. Niesel, U. Schatzschneider, N. Metzler-Nolte, D. A. Schmidt and M. Havenith, Angew. Chem., Int. Ed., 2010, 49, 3310–3312 CrossRef CAS PubMed.
- H. K. Yosef, L. Mavarani, A. Maghnouj, S. Hahn, S. F. El-Mashtoly and K. Gerwert, Anal. Bioanal. Chem., 2015, 407, 8321–8331 CrossRef CAS PubMed.
- H. Nawaz, F. Bonnier, A. D. Meade, F. M. Lyng and H. J. Byrne, Analyst, 2011, 136, 2450–2463 RSC.
- H. Nawaz, A. Garcia, A. D. Meade, F. M. Lyng and H. J. Byrne, Analyst, 2013, 138, 6177–6184 RSC.
- Z. Farhane, F. Bonnier and H. J. Byrne, Anal. Bioanal. Chem., 2017, 409, 1333–1346 CrossRef CAS PubMed.
- K. Aljakouch, T. Lechtonen, H. K. Yosef, M. K. Hammoud, W. Alsaidi, C. Kötting, C. Mügge, R. Kourist, S. F. El-Mashtoly and K. Gerwert, Angew. Chem., Int. Ed., 2018, 57, 7250–7254 CrossRef CAS PubMed.
- W. J. Tipping, M. Lee, A. Serrels, V. G. Brunton and A. N. Hulme, Chem. Soc. Rev., 2016, 45, 2075–2089 RSC.
- S. Vanden-Hehir, W. Tipping, M. Lee, V. Brunton, A. Williams and A. Hulme, Nanomaterials, 2019, 9, 341 CrossRef CAS PubMed.
- D. Fu, J. Zhou, W. S. Zhu, P. W. Manley, Y. K. Wang, T. Hood, A. Wylie and X. S. Xie, Nat. Chem., 2014, 6, 614–622 CrossRef CAS PubMed.
- F. X. Xu, E. G. Rathbone and D. Fu, J. Phys. Chem. B, 2023, 127, 2187–2197 CrossRef CAS PubMed.
- P. Pradhan, S. Guo, O. Ryabchykov, J. Popp and T. W. Bocklitz, J. Biophotonics, 2020, 13, e201960186 CrossRef PubMed.
- H. He, S. Yan, D. Lyu, M. Xu, R. Ye, P. Zheng, X. Lu, L. Wang and B. Ren, Anal. Chem., 2021, 93, 3653–3665 CrossRef CAS PubMed.
- K. Aljakouch, Z. Hilal, I. Daho, M. Schuler, S. D. Krauß, H. K. Yosef, J. Dierks, A. Mosig, K. Gerwert and S. F. El-Mashtoly, Anal. Chem., 2019, 91, 13900–13906 CrossRef CAS PubMed.
- M. K. Hammoud, H. K. Yosef, T. Lechtonen, K. Aljakouch, M. Schuler, W. Alsaidi, I. Daho, A. Maghnouj, S. Hahn, S. F. El-Mashtoly and K. Gerwert, Sci. Rep., 2018, 8, 1–11 CAS.
- S. F. El-Mashtoly, H. K. Yosef, D. Petersen, L. Mavarani, A. Maghnouj, S. Hahn, C. Kötting and K. Gerwert, Anal. Chem., 2015, 87, 7297–7304 CrossRef CAS PubMed.
- J. Bruna and S. Mallat, IEEE Trans. Pattern Anal. Mach. Intell., 2013, 35, 1872–1886 Search PubMed.
- S. Mallat, Commun. Pure Appl. Math., 2012, 65, 1331–1398 CrossRef.
- S. Mallat, Philos. Trans. R. Soc., A, 2016, 374, 20150203 CrossRef PubMed.
- R. Leonarduzzi, H. Liu and Y. Wang, Signal Process., 2018, 150, 11–19 CrossRef.
- C. Ding and H. Peng, in Computational Systems Bioinformatics. CSB2003. Proceedings of the 2003 IEEE Bioinformatics Conference. CSB2003, IEEE Comput. Soc, Stanford, CA, USA, 2003, pp. 523–528.
- J. Zhao, H. Lui, D. I. McLean and H. Zeng, Appl. Spectrosc., 2007, 61, 1225–1232 CrossRef CAS PubMed.
- N. Ke, X. Wang, X. Xu and Y. A. Abassi, in Mammalian Cell Viability, ed. M. J. Stoddart, Humana Press, Totowa, NJ, 2011, vol. 740, pp. 33–43 Search PubMed.
- K.-K. Wong, P. M. Fracasso, R. M. Bukowski, T. J. Lynch, P. N. Munster, G. I. Shapiro, P. A. Janne, J. P. Eder, M. J. Naughton, M. J. Ellis, S. F. Jones, T. Mekhail, C. Zacharchuk, J. Vermette, R. Abbas, S. Quinn, C. Powell and H. A. Burris, Clin. Cancer Res., 2009, 15, 2552–2558 CrossRef CAS PubMed.
- Y. Ito, M. Suenaga, K. Hatake, S. Takahashi, M. Yokoyama, Y. Onozawa, K. Yamazaki, S. Hironaka, K. Hashigami, H. Hasegawa, N. Takenaka and N. Boku, Jpn. J. Clin. Oncol., 2012, 42, 278–286 CrossRef PubMed.
- R. Abbas, B. A. Hug, C. Leister, J. Burns and D. Sonnichsen, Br. J. Clin. Pharmacol., 2011, 71, 522–527 CrossRef CAS PubMed.
- Q. S. C. Chu, M. E. Cianfrocca, L. J. Goldstein, M. Gale, N. Murray, J. Loftiss, N. Arya, K. M. Koch, L. Pandite, R. A. Fleming, E. Paul and E. K. Rowinsky, Clin. Cancer Res., 2008, 14, 4484–4490 CrossRef CAS PubMed.
- K. Inoue, K. Kuroi, S. Shimizu, Y. Rai, K. Aogi, N. Masuda, T. Nakayama, H. Iwata, Y. Nishimura, A. Armour and Y. Sasaki, Int. J. Clin. Oncol., 2015, 20, 1102–1109 CrossRef CAS PubMed.
- Cytotoxicity Overview, https://www.aceabio.com/applications/cytotoxicity/, (accessed November 9, 2019).
- S. Kustermann, F. Boess, A. Buness, M. Schmitz, M. Watzele, T. Weiser, T. Singer, L. Suter and A. Roth, Toxicol. in Vitro, 2013, 27, 1589–1595 CrossRef CAS PubMed.
- V. E. Kagan, J. P. Fabisiak, A. A. Shvedova, Y. Y. Tyurina, V. A. Tyurin, N. F. Schor and K. Kawai, FEBS Lett., 2000, 477, 1–7 CrossRef CAS PubMed.
- H. H. Boersma, B. L. J. H. Kietselaer, L. M. L. Stolk, A. Bennaghmouch, L. Hofstra, J. Narula, G. A. K. Heidendal and C. P. M. Reutelingsperger, J. Nucl. Med., 2005, 46, 2035–2050 CAS.
- L. C. Crowley, B. J. Marfell, A. P. Scott and N. J. Waterhouse, Cold Spring Harb Protoc., 2016, 2016 DOI:10.1101/pdb.prot087288.
- V. D'Amato, L. Raimondo, L. Formisano, M. Giuliano, S. De Placido, R. Rosa and R. Bianco, Cancer Treat. Rev., 2015, 41, 877–883 CrossRef PubMed.
- R. S. Herbst, Int. J. Radiat. Oncol., Biol., Phys., 2004, 59, S21–S26 CrossRef PubMed.
- G. Carpenter and S. Cohen, Annu. Rev. Biochem., 1979, 48, 193–216 CrossRef CAS PubMed.
- M. Martin, J. Bonneterre, C. E. Geyer, Y. Ito, J. Ro, I. Lang, S.-B. Kim, C. Germa, J. Vermette, K. Wang, K. Wang and A. Awada, Eur. J. Cancer, 2013, 49, 3763–3772 CrossRef CAS PubMed.
- R. Smith, K. L. Wright and L. Ashton, Analyst, 2016, 141, 3590–3600 RSC.
- Vibrational spectroscopy for medical diagnosis, ed. M. Diem, J. M. Chalmers and P. R. Griffiths, John Wiley & Sons, Chichester, England; Hoboken, NJ, 2008 Search PubMed.
- I. ur Rehman, Z. Movasaghi and S. Rehman, Vibrational spectroscopy for tissue analysis, CRC Press, Boca Raton, 2013 Search PubMed.
- A. Rygula, K. Majzner, K. M. Marzec, A. Kaczor, M. Pilarczyk and M. Baranska, J. Raman Spectrosc., 2013, 44, 1061–1076 CrossRef CAS.
- C. Matthäus, B. Bird, M. Miljković, T. Chernenko, M. Romeo and M. Diem, Methods Cell Biol., 2008, 89, 275–308 Search PubMed.
- N. K. Howell, G. Arteaga, S. Nakai and E. C. Y. Li-Chan, J. Agric. Food Chem., 1999, 47, 924–933 CrossRef CAS PubMed.
- S. F. El-Mashtoly, J. Med. Chem., 2020, 63, 3472–3474 CrossRef CAS PubMed.
- E. Tolstik, M. B. Gongalsky, J. Dierks, T. Brand, M. Pernecker, N. V. Pervushin, D. E. Maksutova, K. A. Gonchar, J. V. Samsonova, G. Kopeina, V. Sivakov, L. A. Osminkina and K. Lorenz, Front. Pharmacol., 2022, 13, 962763 CrossRef CAS PubMed.
- H. Rammal, A. Al Assaad, F. Dosio, B. Stella, A. Maksimenko, S. Mura, L. Van Gulick, M. Callewaert, D. Desmaële, P. Couvreur, H. Morjani and A. Beljebbar, Nanomedicine, 2021, 35, 102404 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3an02235h |
| ‡ Present address: Leibniz Institute of Photonic Technology, Jena, Germany. |
| This journal is © The Royal Society of Chemistry 2024 |