
Carbazole-substituted benzobisoxazoles: near-UV fluorescent emitters and ambipolar hosts for organic light-emitting diodes†
David L.
Wheeler
a,
Lloyd
Fisher
Jr
f,
Pascal
Friederich
 cde,
Christopher
Cunningham
a,
Angelar K.
Muthike
f,
Alán
Aspuru-Guzik
cd,
Theodore
Goodson
III
f and
Malika
Jeffries-EL
*ab
cde,
Christopher
Cunningham
a,
Angelar K.
Muthike
f,
Alán
Aspuru-Guzik
cd,
Theodore
Goodson
III
f and
Malika
Jeffries-EL
*ab
aDepartment of Chemistry, Boston University, 590 Commonwealth Ave, Boston, MA 02215, USA. E-mail: malikaj@bu.edu
bDivision of Materials Science and Engineering, Boston University, 15 St Mary's St. #117, Boston, MA 02215, USA
cDepartment of Chemistry, University of Toronto, 80 St George St, Toronto, ON M5S 3H6, Canada
dDepartment of Computer Science, University of Toronto, 214 College St, Toronto, ON M5T 3A1, Canada
eInstitute of Nanotechnology, Karlsruhe Institute of Technology, Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
fDepartment of Chemistry, University of Michigan, Ann-Arbor Michigan 48109-1055, USA
First published on 25th November 2022
Abstract
The design of organic light emitting diode (OLED) materials with the potential for exhibiting thermally-activated delayed fluorescence (TADF) is reported. Using computational methods (DFT/TD-DFT) as a guiding tool, six materials with a benzobisoxazole (BBO) core and donor–acceptor–donor architectures were designed by changing the conjugation position of carbazole-substituted phenyl substituents and the type of BBO isomer (cis- vs. trans-). Experimental steady-state and transient absorption spectroscopic techniques were utilized to probe the TADF activity of these molecules. Each material was then used in host–guest OLED devices as either near-UV dopants or host with low singlet-triplet energy differences. The electroluminescent properties show that when used as dopants these materials provide near-UV emission (CIEy < 0.06 and CIEx = 0.16), whereas when used as hosts, these materials show reduced operating voltages and increased performance efficiencies when compared to commercial materials.
Introduction
In recent years, there has been a renewed interest in organic light emitting diodes (OLEDs) due to their potential for the development of next-generation devices such as flexible displays,1 wearables2 and solid-state lighting.3 OLEDs have several advantages over conventional inorganic LEDs including the ability to fabricate them onto either rigid or flexible substrates, to accommodate irregular surfaces. They can also be fabricated almost entirely from solution-processing techniques, eliminating the need for energy-intensive fabrication methods such as thermal evaporation. OLEDs can also be used to make lightweight displays since they do not require backlighting.Purely organic luminophores are promising because, these molecular or polymeric organic semiconductors (OSC)s can be modified through organic synthesis to produce materials with optical and electronic properties tailored for targeted applications.4 OSCs also eliminate the need for toxic, expensive noble metals. However, achieving deep blue emission within the industry standards for color purity: National Television System Committee (NTSC) (CIE: 0.14, 0.08) and European Broadcasting Union (EBU) (CIE: 0.15, 0.06) using a purely organic emitter is challenging.5 Further, since fluorescence OLEDs utilize singlet emission, the theoretical maximum internal quantum efficiencies (IQE) based on these materials is limited to 25% since the ratio of singlets and triplet generated under electroluminescent conditions is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3.6 One solution to overcoming the theoretical limit for fluorescence is through the design of emitters that exhibit thermally activated delayed fluorescence (TADF).7 Such materials can theoretically achieve an IQE of 100% through a process known as reverse intersystem crossing (rISC). This process converts non-emissive excited triplet (T1) excitons into emissive excited singlet state (S1), albeit at a lower rate than intersystem crossing (S1 → T1). By engineering materials with good charge-transfer (CT) character, one can reduce the energetic difference between the S1 and T1 states (ΔEST), increasing the rate of rISC (krISC) thereby inducing TADF.8 This is typically accomplished by spatially separating the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) within a flurophore.9
:3.6 One solution to overcoming the theoretical limit for fluorescence is through the design of emitters that exhibit thermally activated delayed fluorescence (TADF).7 Such materials can theoretically achieve an IQE of 100% through a process known as reverse intersystem crossing (rISC). This process converts non-emissive excited triplet (T1) excitons into emissive excited singlet state (S1), albeit at a lower rate than intersystem crossing (S1 → T1). By engineering materials with good charge-transfer (CT) character, one can reduce the energetic difference between the S1 and T1 states (ΔEST), increasing the rate of rISC (krISC) thereby inducing TADF.8 This is typically accomplished by spatially separating the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) within a flurophore.9
Our group has been developing tunable OSCs based on the benzo[1,2-d:4,5-d′]bisoxazole (BBO) ring system, Fig. 1. This electron-deficient heterocycle can readily be substituted with various aryl substituents in two ways: through the oxazole rings (2,6-axis) and/or the central benzene ring (4,8-axis). As a result, the BBO moiety can be used as a platform to design a multitude of materials with varying opto-electronic properties, using simple, scalable synthons.10–13 Moreover, many aryl-substituted BBOs are highly fluorescent materials that are stable under ambient atmosphere and at high temperatures, making them ideal candidates for use in OLEDs.14,15 Lastly, many aryl-BBOs possess spatially segregated frontier molecular orbitals (FMO)s, which is favorable for achieving a small ΔEST.13–16 Previously, we reported a series of small-molecule BBO dopants which were used in host–guest OLEDs that exhibited deep-blue electroluminescence (EL) (ELλmax ≈ 440 nm; CIEy < 0.10), albeit with external quantum efficiencies (EQE)s less than 3%.14 The low efficiency of the devices was attributed in part to the traditional fluorophore character of the BBO dopants where ∼75% of the excitons created were lost to various non-radiative pathways. Recently, we examined how steric hindrance impacts the opto-electronic properties of a series of aryl-substituted BBOs.12 While theoretical predictions demonstrated that a considerable amount of overlap was observed between the HOMO and LUMO, the materials did show potential for usage as for blue host and emissive materials.
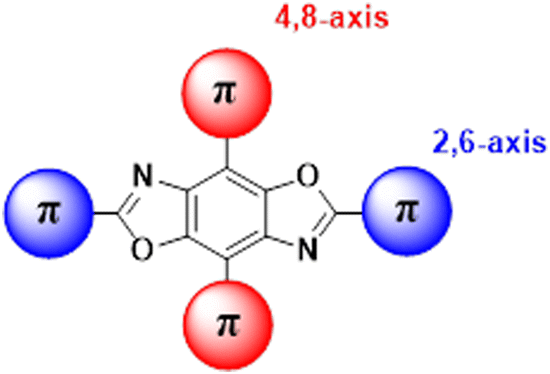 | ||
| Fig. 1 General structure of the BBO core. Conjugation can take part at the 2 and 6 positions (axis) or the 4,8 axis. | ||
In this work, we endeavored to develop TADF emitters by functionalizing the BBO core with electron-rich carbazole pendants. Carbazole was selected as a substituent because of its high triplet energy and good hole-transporting abilities.17 Accordingly, using computational and experimental methods, a series of six BBOs were designed with a donor–acceptor–donor (DAD) architecture to help reduce the ΔEST. Of the six systems, compounds C1, C3, and C5 bore BBO cores with 4-(N-carbazol-9-yl)phenyl- substituents while C2, C4, and C6 contained 3,5-di(N-carbazol-9-yl)phenyl groups. Unfortunately, despite the predictions, experimental results indicate that none of the materials were TADF-active. However, each material was able to be employed into host–guest OLED devices functioning as either fluorescent dopants or ambipolar hosts and are promising precursors for designing BBO-based TADF emitters.
Results and discussion
Computational predictions
The study began by modelling the six materials of interest using DFT/TD-DFT to determine the optimized geometries and predict optical and electronic properties (Table 1). Due to their similarities in structure, compounds C1, C3, and C5 are referred to as the di-substituted carbazole (DC) group while the tetra-substituted carbazole (TC) group includes compounds C2, C4 and C6. By altering the connection and orientation of the carbazole rings relative to the BBO core, we hoped to identify trends leading to materials with small ΔEST.|
|
|||||||
|---|---|---|---|---|---|---|---|
| BBO | Electronic properties | Dihedral angles | |||||
| HOMO (eV) | LUMO (eV) | E optg (eV) | ΔEST (eV) | f | 2,6 (°) | 4,8 (°) | |
| C1 | −5.20 | −1.74 | 3.06 | 0.64 | 8.8 × 10−1 | — | 29.6 |
| C2 | −5.39 | −1.96 | 3.01 | 0.46 | 1.8 × 10−2 | — | 33.7 |
| C3 | −5.39 | −2.01 | 2.96 | 0.59 | 1.5 × 1000 | 0.5 | — |
| C4 | −5.50 | −2.26 | 2.83 | 0.38 | 4.1 × 10−2 | 4.5 | — |
| C5 | −5.42 | −1.93 | 3.06 | 0.47 | 1.3 × 1000 | 3.9 | — |
| C6 | −5.50 | −2.15 | 2.92 | 0.22 | 1.3 × 10−2 | 1.0 | — |
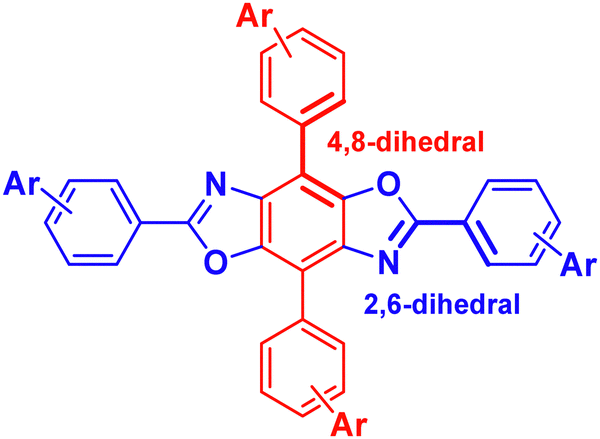
We began the analysis by recognizing structure-property relationships between the HOMO levels of each material. All BBO systems in the DC group were found to have HOMO energies between −5.2–−5.4 eV while those in the TC group are slightly lower between −5.4–−5.5 eV. The similarity in HOMO energies within each group is due to the comparative localization of electron density between the three BBOs (Fig. 2). However, the electron density for the DC group compounds is more delocalized compared the TC group. Furthermore, the HOMO energies of the DC-BBOs C1, C3, and C5 are also computed to be slightly higher than the HOMO of their respective TC group partners. This delocalization and energetic increase can be attributed to the substituent position within the pendant carbazole relative to the BBO core. For the DC group materials, the lone-pair electrons on the carbazole nitrogen are in direct conjugation to the BBO core when conjugated at the para-position while the meta-conjugation of the TC systems force these groups to be electronically orthogonal. Independent of the carbazole substitution, when the aryl substituents are substituted through the 4,8-axis of the BBO core, a slight increase in the HOMO energy is observed compared to those conjugated through the 2,6-axis. According to the computations, there is negligible change in the HOMO energy whether the BBO core is the trans- or cis- isomer.
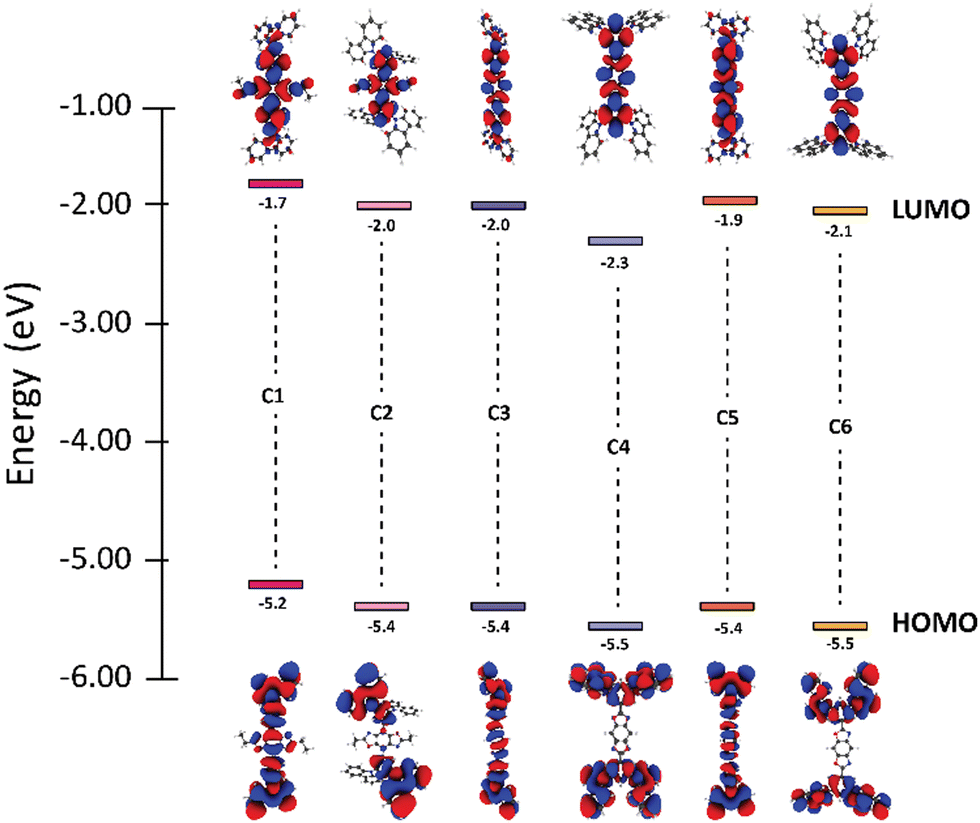 | ||
| Fig. 2 FMOs and electronic energy levels of C1–C6. | ||
Next, trends between the LUMOs of each compound were analysed. The LUMO energy for the DC group is found to range between −1.7–−2.0. The LUMO energy is lowered slightly for the TC group, ranging between −2.0–−2.3 eV. The degree of separation between the HOMO and LUMO is more pronounced in the TC group than the DC group due to the conjugation position of the carbazole units. The axis of conjugation also affects the LUMO energy as C1 is found to be 0.2–0.3 eV higher in energy compared to C3 and C5. A similar result is found comparing C2 to both C4 and C6. Unlike the HOMO, inclusion of the cis-core restricts the conjugation pathway of pi-electrons, thereby increasing the LUMO energy by 0.1–0.2 eV.
The band gaps for all BBOs were found to be suitable for use as deep blue emissive materials or wide bandgap hosts due to their short conjugation pathways (3.1–2.8 eV). Interestingly, the computations show compounds in the TC group have smaller band gaps when compared to their respective DC partners. This can be attributed to the higher CT character of compounds C2, C4, and C6 which has significant impact on lowering the LUMO. As a result, these materials not only have a smaller band gap, but also a smaller ΔEST when compared to their DC partners. However, the S1 oscillator strengths for compounds C2, C4, and C6 are low (<10−1) compared to C1, C3 and C5, indicating the TC materials may not be as luminescent as their DC counterparts. Nonetheless, apart from C1 and C3, all compounds were calculated to exhibit ΔEST < 0.50 eV, which is favorable for TADF. With this knowledge, each compound was then synthesized to determine the experimental optical and electronic properties.
Synthesis and characterization
The BBOs were synthesized using a simple three-step process, minimizing the need for inert atmosphere. The precursors were formed using the pathway outlined in Scheme 1. All precursor materials were formed starting from commercially available fluorinated benzonitrile or bromobenzene derivatives. First, the commercial materials were heated with stoichiometric amounts of carbazole and potassium tert-butoxide in N,N-dimethylformamide under normal atmosphere to produce the carbazole-substituted bromobenzene (1; 86% and 2; 65%) and benzonitrile (3; 78% and 4; 71%) derivatives in moderate yields. Compounds 1 and tert were dissolved in anhydrous tetrahydrofuran (THF), lithiated with n-butyllithium, and quenched with 2-isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane to produce the desired boronic esters 5 (79%) and 6 (60%). Compounds 3 and 4 were hydrolysed under normal atmosphere using an aqueous 6 M sodium hydroxide solution and reagent alcohol to produce nearly quantitative yields of the substituted benzoic acid derivatives 7 (95%) and 8 (89%).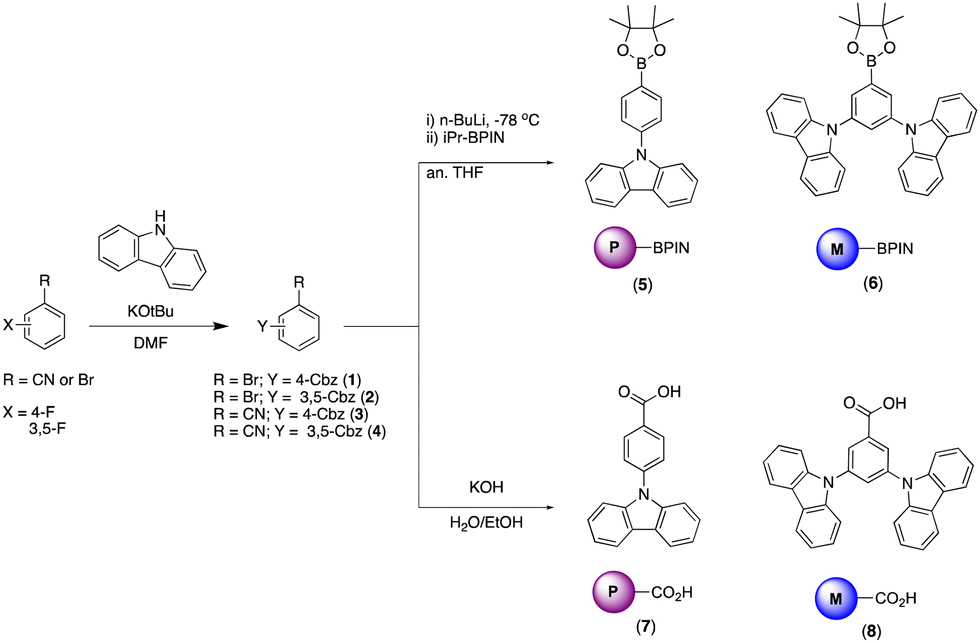 | ||
| Scheme 1 Synthesis of precursor material 1–8. | ||
The final step synthesis is illustrated in Scheme 2. We elected to use high-pressure Suzuki-Miyaura conditions to form the 4,8-conjugated BBOs due to its low toxicity and effectiveness based off previous work.12 Using cesium fluoride as the base and THF as the solvent, C1 and C2 were produced in yields of 60% and 37%, respectively. Lastly, the 2,6-conjugated BBOs were formed through a condensation technique using polyphosphoric acid trimethylsilyl ester (PPSE) as a desiccant with either 2,5-diaminohyroquinone bis-hydrochloride or 4,6-dinitroresorcinol, producing C3–C6 (28–38% yields). All compounds exhibit decent-to-low solubility in chlorinated solvents and where structurally characterized using NMR and high-resolution MS (ESI) techniques.
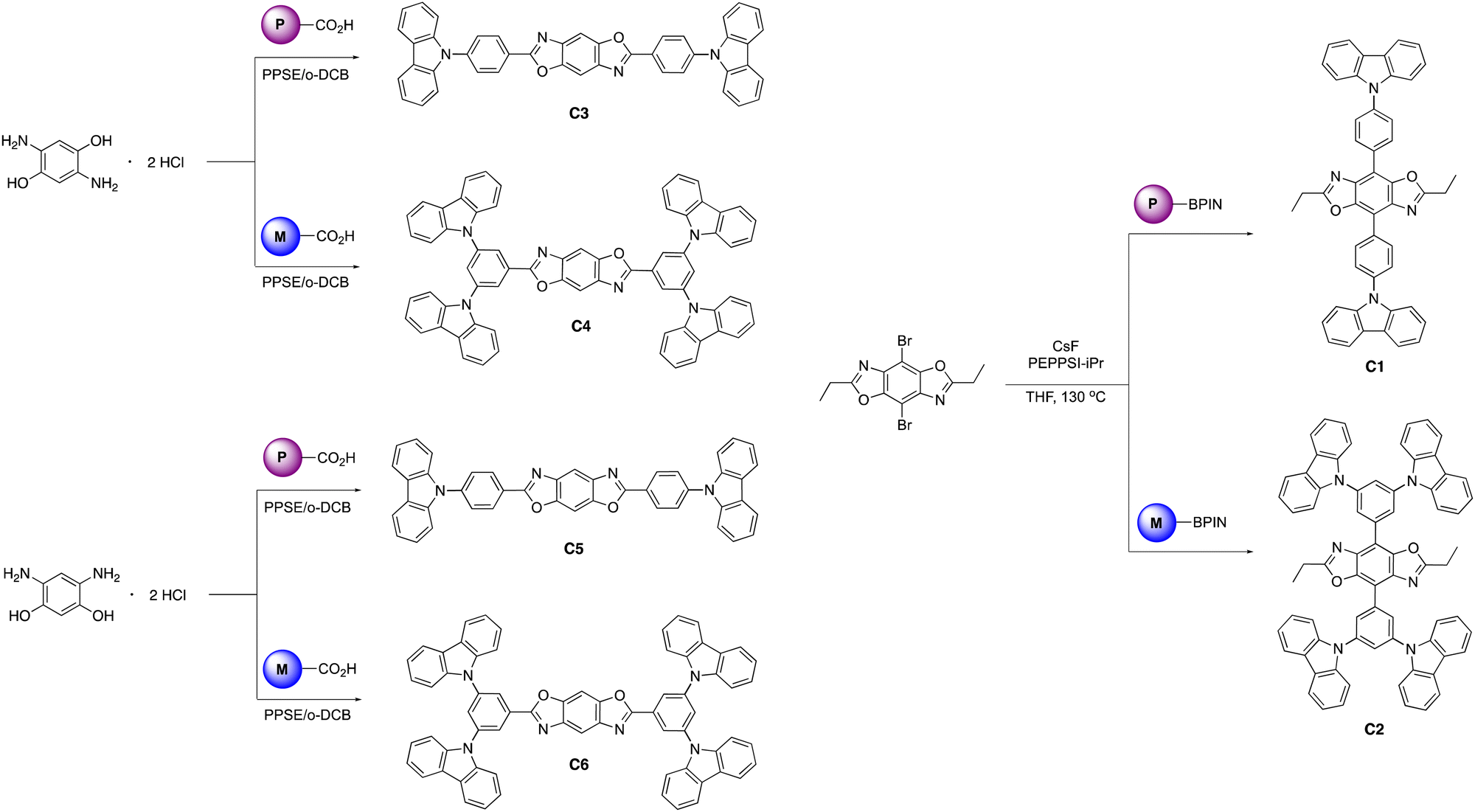 | ||
| Scheme 2 Synthesis of final molecules C1–C6. | ||
Electronic properties
The theoretical and experimental electronic properties of C1–C6 are listed in Table 2. The experimental HOMO energies were calculated using ionization potentials determined from ultraviolet photoelectron spectroscopy (UPS). We chose this technique over cyclic voltammetry due to its ability to measure the work function of materials with high accuracy. Further, many of the BBOs were not soluble in suitable solvents for electrochemistry. The optical HOMO → LUMO transition energies (Eoptg) were determined from the onset of each thin-film UV-Vis spectrum. LUMO levels were then calculated by adding the Eoptg to the experimental HOMO energies.| Optical properties | Electronic properties | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Solution | Thin film | |||||||||
| BBO | ε (104) (M−1 cm−1) | abs λ max (nm) | em λ max (nm) | Φ (%) | ΔEST (eV) | abs λ max (nm) | em λ max (nm) | HOMOa (eV) | LUMOb (eV) | E g (eV) |
| Bold values indicate wavelength of maximum absorbance/emission.a Measured using UPS.b Estimated using the following equation: LUMO = HOMO + Eg.c Calculated from onset of absorbance. | ||||||||||
| C1 | 3.4 | 347 | 415 | 68 | 0.59 | 363 | 424, 445 | −5.9 | −2.9 | 3.0 |
| C2 | 5.7 | 313, 326, 340 | 428 | 10 | 0.22 | 329, 342 | 419 | −6.1 | −2.8 | 3.3 |
| C3 | 6.0 | 372, 388 | 416, 433 | 42 | 0.46 | 380, 401 | 423, 448, 470 | −6.0 | −3.1 | 2.9 |
| C4 | 6.9 | 337, 351, 370 | 448 | 3.9 | 0.47 | 329, 340 | 437 | −5.8 | −2.7 | 3.1 |
| C5 | 5.4 | 363, 377 | 414 | 27 | 0.64 | 369, 388 | 436 | −6.0 | −3.0 | 3.0 |
| C6 | 7.6 | 327, 341, 360 | 440 | 4.8 | 0.38 | 329, 340 | 435 | −5.9 | −2.7 | 3.2 |
In all cases, the HOMO and LUMO levels measured are similar to common commercial hosts/emitters for blue OLEDs. All experimental HOMO energies range from −6.1 to −5.8 eV, which are 0.3–0.7 eV deeper than the computationally predicted values. Similarly, the theoretical LUMO values (−2.3 to −1.7 eV) are calculated to be 0.5–1.2 eV higher in energy than the experimental values (−3.1 to −2.7 eV). The experimental Eoptg suggest excellent potential as host/emitter materials for blue OLEDs as all compounds ranged from 2.9 to 3.3 eV. These values are in good agreement with computational findings (abs. diff. ≤0.3 eV). For BBOs C1, C3, and C5, the Eoptg are lower in energy when compared to their 3,5-disubstituted analogues. This observation is attributed to the para-conjugation of each carbazole to the benzene ring, allowing the non-bonding electron pair of the heteroatom to contribute to the extended pi-system.
Steady-state optical properties
The optical property investigation began by analysing the steady-state absorbance and emission profiles of C1–C6. Each compound was examined as a 0.01 g L−1 solution dissolved in chloroform (Fig. 3) and as a thin film (see ESI†). We first examined the difference in absorption profiles between BBOs in solution. In all cases, two main absorption bands characteristic of chromophores with donor–acceptor configurations, demonstrating their CT nature.18,19 The high energy bands can be ascribed to the localized excitations due to π–π* transition of the moieties, while the low energy bands can be ascribed to the n–π* charge transfer transition among the moieties.18,20 It is interesting to note that the more prominent CT absorption band is obtained from the systems with DC compounds rather than the TC systems. One would expect that C2, C4, and C6 would exhibit higher donor strength and yield more prominent charge transfer features.21 However, the multiple carbazole subunits of the C2, C4, and C6 materials make the localized absorption transition more prominent. Of all materials, the DC group BBOs show bathochromic absorption when compared to the TC systems. This is a direct consequence of the lower Eoptg and substitution pattern of carbazole units for C1, C3, and C5. Compounds which contain the cis-isomeric core exhibit hypsochromic shift in absorption relative to the BBOs with the trans-moiety. When the aryl substituents are conjugated through the 4,8-axis, an enhanced hypsochromic shift is observed compared to the BBOs with 2,6-axis substituents. This is attributed to the increased dihedral angle that exists between the BBO core and aryl substituent, as supported from DFT calculations (Table 1). As thin films, C1, C3, and C5 still demonstrate a bathochromic shift in absorption when compared to their TC group partners. While changing the cis-core to the trans-isomer provides a red-shift in absorbance, this effect is not as intense when compared to the same observation in solution.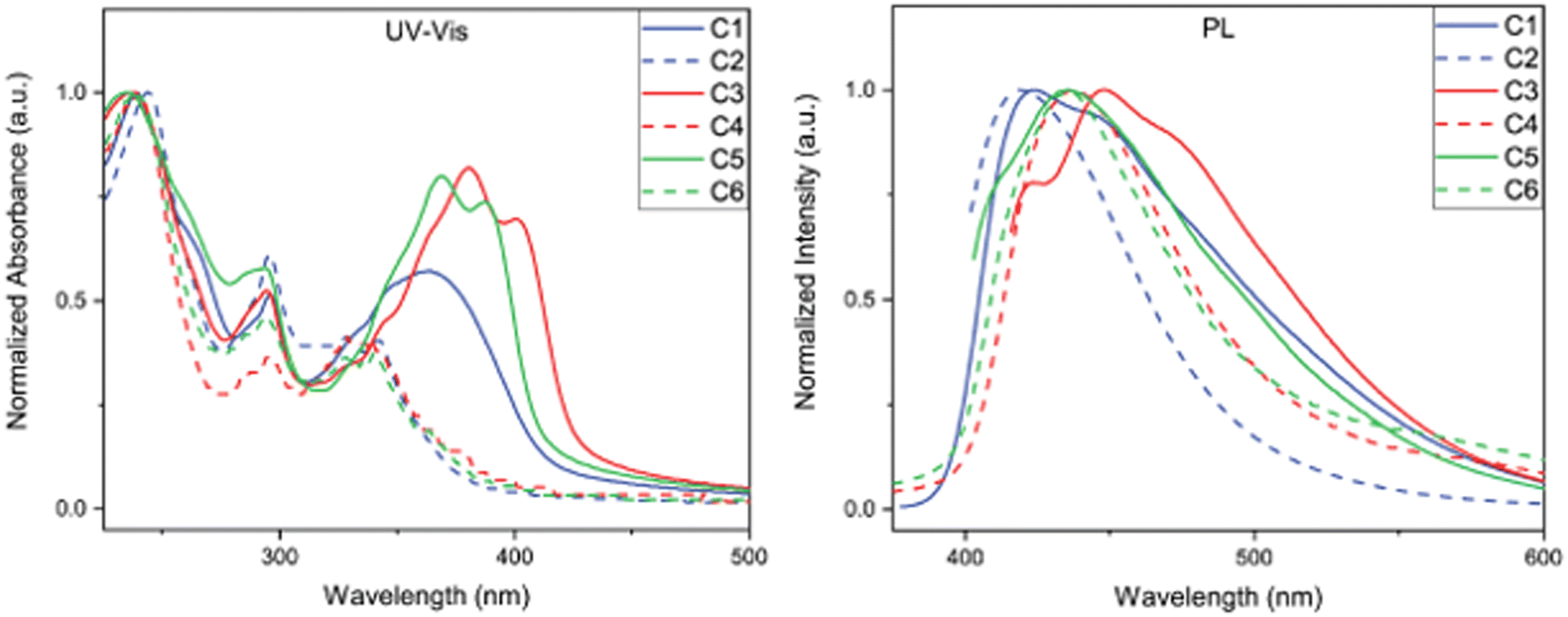 | ||
| Fig. 3 Steady-state absorbance spectra of C1–C6 in chloroform (left) at 0.01 g L−1 and photoluminescence spectra of C1–C6 thin-film (right). | ||
The photoluminescence (PL) spectra were measured to determine emission profiles and quantum yields for each BBO (Fig. 2 and Table 2). In solution, C1, C3, and C5 all produce emission at approximately 415 nm with quantum yields ranging from 27–68%. A secondary peak at 433 nm is observed for C3 but is not observed for the structural isomer, C5. For the TC systems, the experimental observations closely matched the DFT predictions as the emission intensities of these materials are low with broad profiles and quantum yields ranging from 3–10%. When measured as thin films, all compounds were found to exhibit emission maxima between 419–448 nm. The emission profiles for C1, C3, and C5 broaden and produce bathochromic emission compared to their solution-state spectra. These observations are attributed to each compound's ability to undergo π–π stacking readily. Conversely, C2, C4, and C6 undergo slight hypsochromic emission with little change in peak breadth. Due to the bulky nature of these compounds, we believe the pi-systems of each molecule are unable to interact effectively in a macroscopic environment, thereby producing similar emission profiles when compared to their solution spectra. This property is expected to be highly beneficial for bipolar host material that can facilitate energy-transfer without radiative quenching.
The quantum yields (ϕ) of the investigated compounds were analysed in both unpurged and purged chloroform solutions (see ESI†). In the case of the C5, a ϕ value (27%) was obtained in unpurged solution. An appreciable ϕ enhancement was also obtained for the C5 chromophore after purging oxygen from the solution (33%). It has been suggested that the difference of the ϕ before and after the oxygen purging process is due to excited state dynamics governed by a delayed fluorescence mechanism.22 Contrarily to C5, a very small ϕ (4.8%) was obtained for C6, which suggests a poor fluorescence decay in oxygen-rich environments but is enhanced when oxygen is expelled from the solution (6.2%).18 Such a small enhancement indicates that other decay pathways for the excited states outcompete fluorescence decay. Similar results were obtained for the remaining mono-carbazole donor moiety emitters and their respective bi-carbazole donor moiety counterparts. Based on these results, we sought to further investigated these compounds using time-resolved techniques at the nanosecond timescale.
Nanosecond transient absorbance
Transient absorption (TA) spectra of the investigated compounds were obtained to gain insight into their emissive mechanism (Fig. 4). In each spectrum, positive ΔOD peaks are representative of excited state absorption (ESA) while negative peaks are representative of either ground state bleaching (GSB) or stimulated emission (SE). The lifetimes of each peak were then investigated to aid in determining what emissive mechanisms are accessible by these compounds. Each TA spectrum of the mono-carbazole donor DC emitters display a SE peak that is in good agreement with the steady-state fluorescence results (Table 3). No other peaks are readily observable, indicating that the lifetime of both the GSB and ESA are below the time resolution of this technique. This suggests that fluorescence emission is efficient for these compounds and outcompetes other radiative decay pathways.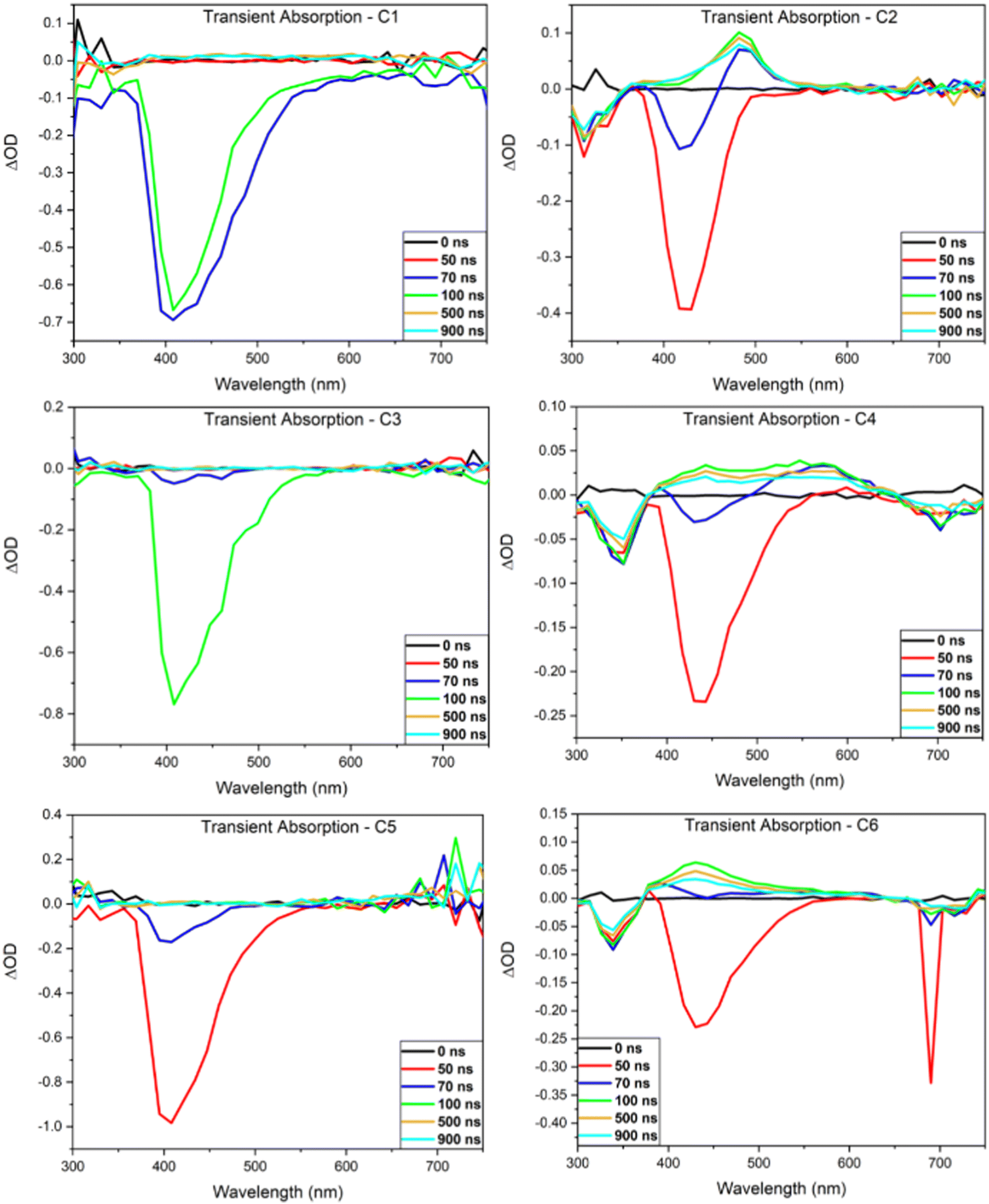 | ||
| Fig. 4 Nanosecond transient absorption spectra of the six BBOs. | ||
| BBO | GSB (nm) | SE (nm) | ESA (nm) | k rISC (s−1) | τ ESA (s) | k ESA (s−1) | τ Ph (s) | k Ph (s−1) |
|---|---|---|---|---|---|---|---|---|
| GSB = ground state bleaching. SE = stimulated emission. ESA = excited state absorbance. krISC = reverse intersystem crossing rate. KESA = ESA rate. KPh = rate of phosphorescence. τESA = ESA lifetime. τPh = phosphorescence lifetime. | ||||||||
| C1 | — | 410 | — | — | — | — | — | — |
| C2 | 325 | 424 | 487 | 4.30 × 104 | 1.16 × 10−6 | 8.59 × 105 | 1.92 × 10−6 | 5.21 × 105 |
| C3 | — | 408 | — | — | — | — | — | — |
| C4 | 350 | 438 | 547 | 5.68 × 103 | 1.41 × 10−6 | 7.10 × 105 | 1.54 × 10−6 | 6.51 × 105 |
| C5 | — | 408 | — | — | — | — | — | — |
| C6 | 340 | 430 | 430 | 1.41 × 104 | 9.92 × 10−7 | 1.01 × 106 | 1.15 × 10−6 | 8.68 × 105 |
Contrary to C1, C3, and C5, the TA spectra of the bi-carbazole donor TC emitters display a GSB, SE, ESA, and a third negative peak. To determine the lifetime of the ESA and third negative peak, transient decay traces were obtained (see ESI†). For C6, the ESA lifetime (430 nm) in unpurged chloroform is 632 ns, which is a longer timescale compared to typical singlet states. However, this lifetime is enhanced with oxygen purging, increasing the timescale just nearly into the microsecond regime (992 ns), indicating that this state has significant triplet character. Similar results are obtained for the third negative peak at ∼700 nm observed after 70 ns (Fig. 4) which has a lifetime of 576 ns in the oxygenated solution. However, this peak is not indicative of GSB or SE as GSB is observed at ∼340 nm and overlaps well with the steady-state absorption spectrum while SE is observed at ∼430 nm and overlaps well with the steady-state emission spectrum. Thus, we determine that this state is representative of phosphorescence emission. The significant enhancement of this state's lifetime with oxygen purging to 1152 ns supports this, as oxygen is known to quench the triplet state. These lifetimes match well with that of the ESA in both the oxygenated and oxygen-purged solutions, suggesting that this emission is the result of the ESA decay. It should be noted that the sharp peak observed at ∼700 nm at 50 ns is the result of the second harmonic of the excitation beam and not emission of C6. Similar results were obtained for C2 and C4 with the most noticeable difference being that both emitters have longer ESA and phosphorescence lifetimes than C6 (Fig. S20, ESI†).
The ESA lifetime results were used to calculate the rate of reverse intersystem crossing (krISC) and determine if any of the emitters are capable of TADF.19,23 No krISC could be determined for the DC emitters, as they possessed no ESA that could be measured by the nanosecond TA technique. Based on the timescale of relaxation, we believe C1, C3, and C5 to be traditional fluorophores. The krISC values calculated for the TC emitters are displayed in Table 3 along with the rate of phosphorescence (kphos). Both krISC and kphos compete for the triplet ESA, where kphos values are an order of magnitude greater than the krISC results, in all cases. Therefore, it is determined that the TC emitters are more efficient phosphorescent compounds than TADF compounds.
Based on the steady-state and transient spectral data, it is evident that C1, C3, and C5 exhibit properties useful as fluorescent dopant materials as seen in their PL spectra and moderate-to-high PLQYs in oxygen-free environments. Unfortunately, none of the BBO materials display any TADF characteristics. Conversely, C2, C4, and C6 show quite narrow ΔEST and long-lived triplet lifetimes before phosphorescence decay (+1 μs). While the rate of krISC is an order of magnitude smaller than the kphos, these materials do not exhibit bright emission and could be useful as potential BBO-based phosphorescent/TADF host materials. Therefore, C1, C3, and C5 were optimized into solution processed devices as active layer materials while C2, C4, and C6 were paired with commercial TADF emitter 2,3,4,5,6-pentakis(3,6-di-tert-butyl-9H-carbazol-9-yl)benzonitrile (5TCzBN) to evaluate preliminary results as ambipolar hosts.
Device properties
We studied OLED characteristics of C1, C3, and C5 as solution-processed dopants in a mixed host matrix. The device architecture consisted of the following: ITO (100 nm)/PEDOT:PSS (35 nm)/Active Layer (mixed host: 5% BBO)/TmPyPB (35 nm)/LiF (1 nm)/Al (100 nm). Shown in Fig. 5 is the band diagram of all active layer materials and efficiency graphs while Table 4 collates the EL properties of the devices using BBO-based dopants.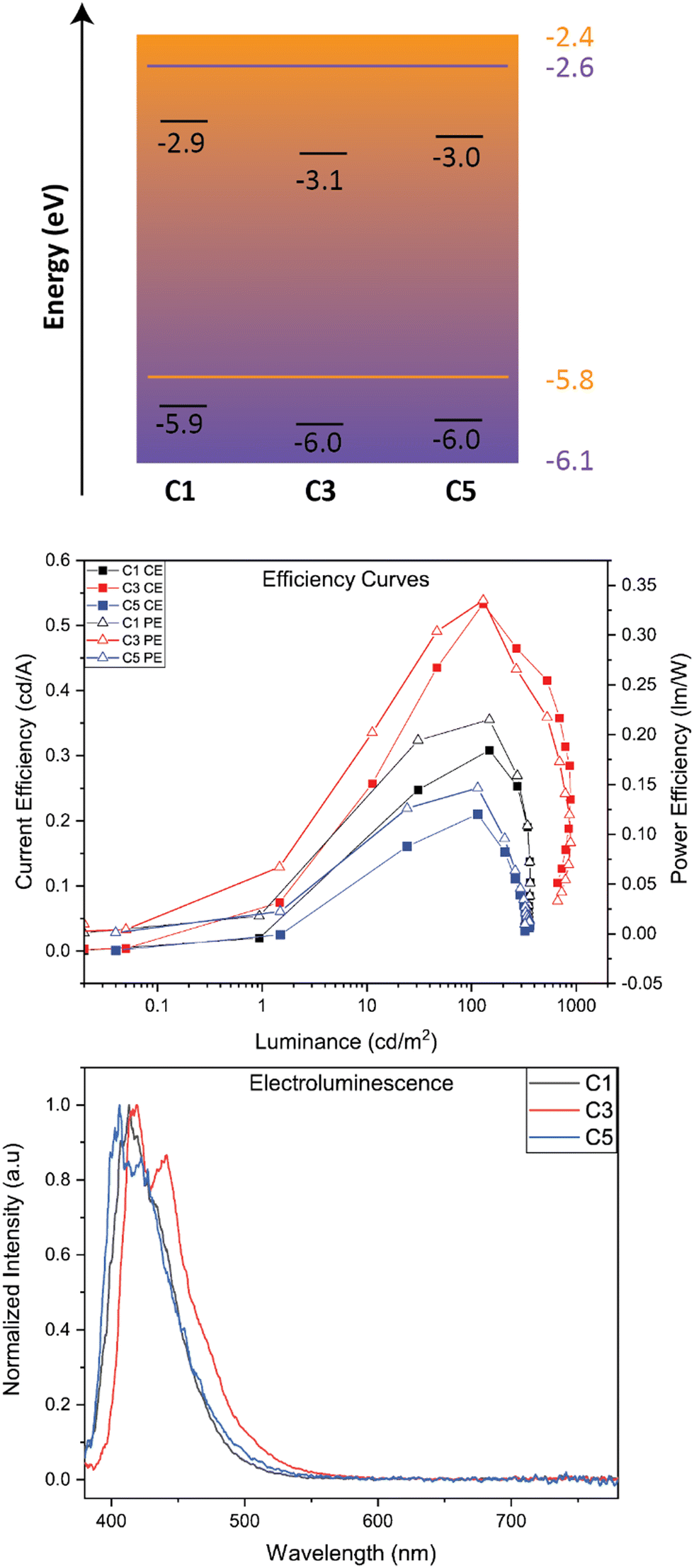 | ||
| Fig. 5 (top) Band diagram of active layer materials using BBO-based dopants. In purple are the HOMO/LUMO levels for 26DCzPPY while in orange is TCTA. (center) Power and current efficiency curves. (bottom) Normalized EL spectra. | ||
| BBO | EL λ max (nm) | V 100 (V) | L Max (cd m−2) | CEMax (cd A−1) | PEMax (lm W−1) | EQEMax (%) | |
|---|---|---|---|---|---|---|---|
| EL λ max = peak maxima of device. V100 = driving voltage at 100 cd m−2. LMax = maximum brightness. CEMax = maximum current efficiency. PEMax = maximum power efficiency. EQEMax = maximum external quantum efficiency. | |||||||
| Dopants | C1 | 413 | 4.3 | 363.4 | 0.308 | 0.215 | 0.874 |
| C3 | 417, 441 | 4.8 | 884.7 | 0.533 | 0.335 | 0.881 | |
| C5 | 405, 424 | 4.4 | 358.6 | 0.210 | 0.147 | 0.538 | |
| Host | mCP | 481 | 8.4 | 1247 | 0.106 | 0.033 | 0.049 |
| C2 | 485 | 4.4 | 1476 | 0.454 | 0.246 | 0.200 | |
| C4 | 484 | 4.5 | 835.0 | 0.487 | 0.306 | 0.864 | |
| C6 | 486 | 5.1 | 1200 | 0.915 | 0.448 | 0.356 | |
The EL spectra of each dopant exhibit a relatively minor red-shift when compared to their solution-state PL spectrum. Of the three dopants, C5 was found to exhibit the most blue-shifted emission of the set at 405 nm, with a second band at 424 nm. C1 and C3 show emission maxima at 413 nm and 417 nm, respectively. The peak widths at half max for all EL spectra were less than 55 nm, indicating good color purity for organic molecules. Furthermore, all three dopants exhibit CIEy values <0.06 and CIEx values of 0.16. While these compounds nearly meet the National Television Systems Committee and European Broadcasting Union's requirements for deep-blue emission, the larger CIEx value indicates near-UV emission, providing a purple hue to the emitted light. Each device demonstrates operating voltages (measured at 100 cd m−2) less than 4.8 V and max luminance between 300–900 cd m−2. The best performing dopant of the set was C3, achieving a current efficiency (CE) of 0.53, power efficiency (PE) of 0.34, and a max EQE of 0.88. While small, the efficiencies of all materials are comparable to other materials which emit near UV emission using identical doping concentrations.24 While specific measurements would be required for support, we attribute the low EQE of these devices to the emission mechanism of the molecule and the traditional host guest architecture of the system. However, two methods of overcoming the latter argument would be to employ these systems into (1) a thermally evaporated device with (2) the use of a TADF-sensitizer to obtain near-UV hyperfluorescence emission. Unfortunately, due to the constraints of the thermal evaporator, this architecture could not be obtained. These architectures will be investigated in the future, when possible.
We next preliminarily evaluated C2, C4, and C6 as a host material. Each system was thermally evaporated with the commercial TADF dopant, 5TCzBN. The device architecture used was ITO (100 nm)/PEDOT:PSS (35 nm)/Active Layer (host: 20% 5TCzBN)/TmPyPB (35 nm)/LiF (1 nm)/Al (100 nm). BBOs C2, C4, and C6 produced more promising results when compared to the device using the commercial mCP host. Demonstrated by the RMS values from AFM measurements (see ESI†), the TC group show very similar active layer morphologies, resulting in hosts which do not alter the emission maxima by more than 5 nm. However, the BBO hosts drastically reduce the operating voltage by as much as 4.0 V when compared to the device using mCP as the host. We hypothesize that this is due to the ambipolar nature of the materials, which can provide better energetic agreement between the LUMO levels of the host and dopant. As a result, the BBO hosts were able to attain higher CE, PE and EQEs while some produced comparable brightness when compared to the mCP device. From these preliminary devices, the BBO materials indicate great potential as hosts due to their improved operating voltages and efficiencies when compared to commercially available mCP.
Conclusions
In summary, six BBO materials with a DAD motif were computationally modelled to evaluate their potential as TADF emitters. DFT calculations suggested only three of the six BBOs could potentially exhibit TADF character. To confirm predictions, each material was synthesized in three steps and evaluated using a variety of steady-state and transient absorption spectroscopic techniques to understand their opto-electronic properties. The results indicate twofold. First, meta-conjugation of electron rich aryl substituents on a phenyl spacer conjugated to the BBO core effectively isolates the HOMO and LUMO. This pattern enhances the CT character of the materials and reduces the ΔEST of the molecule, but suppresses photoluminescence, reducing the PLQY of the system. Secondly, para-conjugation of the carbazole moieties result in the opposite effect where the LE character of the molecule is increased, reintroducing fluorescence but sacrificing TADF character. Fortuitously, C1, C3, and C5 demonstrated attractive opto-electronic qualities as near-UV emitting generation 1 dopants while C2, C4, and C6 were repurposed as ambipolar hosts. The solution-processed dopants demonstrated excellent near-UV emission (CIEy < 0.06; CIEx = 0.16) and competitive efficiencies to currently reported near-UV organic emitters. The BBO hosts paired with a commercial TADF emitter demonstrated reduced turn-on voltages and increased efficiencies in comparison to the p-type host, mCP. While these preliminary results show great promise for the candidates in the field of organic electronics, future works include further optimization of both sets of BBO materials using an evaporator with more extensive capabilities to attempt hyperfluorescent architectures (C1, C3, C5) and using the C2, C4, and C6 as sensitizers for triplet recycling materials. Additionally, the TC group scaffold can be further improved to reintroduce fluorescent behaviour towards the production of TADF-active BBO emitters.Experimental
Computational methodology
We generated initial 3D structures from SMILES25 codes using the molconvert tool from ChemAxon. We then used the GFN-xTB26 based conformer search algorithm CREST27 to find the lowest energy conformers of all molecules. DFT based analysis of the molecules was done using the procedure described in Gómez-Bombarelli et al.28 The optimized geometry of the lowest energy conformers was obtained from CREST on a B3-LYP/6-31G* level of theory27,29 in gas phase and performed TDDFT calculations on the same level of theory to obtain HOMO and LUMO levels, permanent dipole moments, S1 and T1 excitation energies as well as S0 to S1 transition dipole moments and oscillator strengths. We computed the TADF rates according to eqn (1) given in Gómez-Bombarelli et al.,28 which depends on the S1 excitation energy, the S1–T1 gap and the oscillator strength for the S0 to S1 transition. We used n = 1 to be consistent with the data reported in Gómez-Bombarelli et al.28 Using a more realistic n would not change the trends. All DFT calculations were done using the QChem package version 5.1.30Materials and characterization
4,8-Dibromo-2,6-diethylbenzobisoxazole,11 2,5-diaminohyroquinone bishydrochloride,31 and 4,6-dinitroresorcinol32 were synthesized according to literature procedures. THF was dried using an Innovative Technologies solvent purification system. All other chemical reagents were purchased from commercial sources and used without further purification unless otherwise noted. Nuclear magnetic resonance (NMR) experiments were carried out in CDCl3 at 500 MHz (1H) AND 125 MHZ (13C). In all spectra, chemical shifts are given in δ relative to residual protonated solvent peak, CHCl3 (7.26 ppm, 1H; 77.16 ppm, 13C). Coupling constants are reported in hertz (Hz). High-resolution mass spectra were recorded on a double-focusing magnetic sector mass spectrometer using ESI. All UV-Vis and fluorescence spectra were obtained using quartz cuvettes with a 10 mm path length in CHCl3 or as spin-casted thin films on a quartz slide (filtered 5 mg mL−1 solution in CHCl3 spun at 1500 rpm). UV-vis spectra were collected on a Shimadzu UV-1800 UV spectrophotometer. Photoluminescence spectra were obtained on a Varian Cary Eclipse spectrophotometer. Absolute solution fluorescence quantum yields were obtained using a HORIBA spectrophotometer Nanolog FL3-2iHR equipped with a Quanta-phi integrating sphere. Ultraviolet photoelectron spectroscopy (UPS) was used to acquire the ionization potentials and approximate the HOMO values for each material. All substrates (positively doped silicon; 10 × 10 mm2) had 40 nm of silver deposited via thermal evaporation. Samples were prepared by dissolution in CHCl3 at a concentration of 5 mg mL−1 and stirred for a minimum of 4 h. Each solution was filtered to remove potential aggregates and sequentially spin-coated under a nitrogen atmosphere. Spectra were then acquired under ultrahigh vacuum at random positions on the formed film. The fluorescence quantum yields (ϕ) of the investigated compounds were calculated using the Williams comparative method.1 A series of increasingly dilute solutions, with optical density starting at 0.10, were utilized for the calculations. The quantum yields were calculated using eqn (1):33| ϕ = ϕSTD (Gradxηx2)/(GradSTDηSTD2) | (1) |
| krISC = kESAϕTADF | (2) |
Synthesis of precursors
9-(4-Bromophenyl)-9H-carbazole (1). White solid. (86%). 1H NMR (500 MHz, CDCl3) δ 8.15 (d, J = 7.8 Hz, 2H), 7.74 (d, J = 8.6 Hz, 2H), 7.49–7.37 (m, 6H), 7.35–7.28 (dd, J = 8.0, 8.0 Hz, 2H).
9,9′-(5-Bromo-1,3-phenylene)bis(9H-carbazole) (2). White solid. (65%). 1H NMR (500 MHz, CDCl3) δ 8.16 (d, J = 7.8 Hz, 4H), 7.87 (d, J = 1.9 Hz, 2H), 7.80 (t, J = 1.9 Hz, 1H), 7.56 (d, J = 8.2 Hz, 4H), 7.51–7.44 (dd, J = 7.7, 1.2 Hz, 4H), 7.34 (t, J = 7.4 Hz, 4H).
4-(9H-Carbazol-9-yl)benzonitrile (3). White solid. (78%). 1H NMR (500 MHz, CDCl3) δ 8.17 (dt, J = 7.8, 1.0 Hz, 2H), 7.89–7.86 (m, 2H), 7.72–7.68 (m, 2H), 7.48–7.45 (m, 4H), 7.37 (ddd, J = 8.0, 5.1, 3.0 Hz, 2H).
3,5-Di(9H-carbazol-9-yl)benzonitrile (4). White solid. (71%). 1H NMR (500 MHz, CDCl3) δ 8.16 (d, J = 7.9 Hz, 4H), 8.07 (t, J = 2.0 Hz, 1H), 7.97 (d, J = 1.8 Hz, 2H), 7.56–7.48 (m, 8H), 7.38 (td, J = 7.4, 6.9, 1.4 Hz, 4H).
9-(4-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)-9H-carbazole (5). Yellow viscous liquid (79%). 1H NMR (500 MHz, CDCl3) δ 8.16 (d, J = 7.7 Hz, 2H), 8.08 (d, J = 8.1 Hz, 2H), 7.62 (d, J = 8.2 Hz, 2H), 7.47 (d, J = 8.2 Hz, 2H), 7.42 (t, J = 7.6 Hz, 2H), 7.30 (t, J = 7.2 Hz, 2H), 1.42 (s, 12H).
9,9′-(5-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3-phenylene)bis(9H-carbazole) (6). White solid. (60%). 1H NMR (500 MHz, CDCl3) δ 8.16 (d, J = 7.8 Hz, 4H), 8.13 (d, J = 2.1 Hz, 2H), 7.89 (t, J = 2.1 Hz, 1H), 7.52 (d, J = 8.2 Hz, 4H), 7.46 (ddd, J = 8.2, 7.1, 1.2 Hz, 4H), 7.32 (ddd, J = 7.5, 1.1 Hz, 4H), 1.38 (s, 12H).
4-(9H-Carbazol-9-yl)benzoic acid (7). White solid. (95%). 1H NMR (500 MHz, CDCl3) δ 8.40 (d, J = 8.1 Hz, 2H), 8.16 (d, J = 7.8 Hz, 2H), 7.76 (d, J = 8.1 Hz, 2H), 7.53 (d, J = 8.2 Hz, 2H), 7.45 (t, J = 7.7 Hz, 2H), 7.34 (t, J = 7.4 Hz, 2H).
3,5-Di(9H-carbazol-9-yl)benzoic acid (8). White solid. (89%). 1H NMR (500 MHz, CDCl3) δ 8.46 (d, J = 2.0 Hz, 2H), 8.17 (d, J = 7.7 Hz, 4H), 8.11 (t, J = 2.0 Hz, 1H), 7.55 (d, J = 8.2 Hz, 4H), 7.47 (t, J = 7.7 Hz, 4H), 7.38 – 7.31 (t, J = 7.8 Hz, 4H).
4,8-Bis(4-(9H-carbazol-9-yl)phenyl)-2,6-diethylbenzo[1,2-d:4,5-d′]bis(oxazole) (C1). Pale-green solid (60%). M.P. > 240 °C. 1H NMR (500 MHz, CDCl3) δ 8.59 (d, J = 8.5 Hz, 4H), 8.19 (d, J = 7.7 Hz, 4H), 7.82 (d, J = 8.5 Hz, 4H), 7.62 (d, J = 8.2 Hz, 4H), 7.47 (ddd, J = 8.2, 7.1, 1.2 Hz, 4H), 7.33 (ddd, J =7.9, 7.0, 1.0 Hz, 4H), 3.15 (q, J = 7.6 Hz, 4H), 1.58 (t, J = 7.6 Hz, 6H). 13C NMR (126 MHz, CDCl3) δ 169.18, 146.24, 140.77, 137.51, 137.17, 131.55, 131.52, 127.00, 126.00, 123.52, 120.33, 120.08, 113.32, 110.04, 77.26, 77.00, 76.75, 22.69, 11.37. HRMS (ESI) m/z: [M + H]+ calcd for C48H35N4O2: 699.2760; found: 699.2778.
4,8-Bis(3,5-di(9H-carbazol-9-yl)phenyl)-2,6-diethylbenzo[1,2-d:4,5-d′]bis(oxazole) (C2). Off-white solid. (37%). M.P. > 240 °C. 1H NMR (500 MHz, CDCl3) δ 8.64 (s, 4H), 8.19 (d, J = 7.7 Hz, 8H), 7.96 (s, 2H), 7.87 (d, J = 8.3 Hz, 8H), 7.49 (t, J = 7.7 Hz, 8H), 7.34 (t, J = 7.5 Hz, 8H), 3.11 (q, J = 7.7 Hz, 4H), 1.58 (t, J = 7.6 Hz, 6H). 13C NMR unable to be obtained. HRMS (ESI) m/z: [M + H]+ calcd for C72H49N6O2: 1029.3917; found: 1029.3954.
Method A – Compounds were passed through a hexanes:CHCl3 plug. The solvent was removed, and crude material was then dissolved in CHCl3 and decolorised with minimal activated charcoal. Filtering off the charcoal, the filtrate was then concentrated onto silica gel for purification via column chromatography (hex:CHCl3 gradient).
Method B – Compounds were recrystallized twice in toluene using decolorizing carbon to remove black color. After filtering, the solid was spun in ample amounts of hot CHCl3 and filtered three times.
2,6-Bis(4-(9H-carbazol-9-yl)phenyl)benzo[1,2-d:4,5-d′]bis(oxazole) (C3). Method A; pale green solid (28%). M.P. > 240 °C. 1H NMR (500 MHz, CDCl3) δ 8.54 (d, J = 8.5 Hz, 4H), 8.17 (d, J = 7.8 Hz, 4H), 8.02 (s, 2H), 7.81 (d, J = 8.5 Hz, 4H), 7.54 (d, J = 8.2 Hz, 4H), 7.46 (ddd, J = 8.2, 7.0, 1.3 Hz, 4H), 7.37–7.32 (ddd, J = 7.8, 6.8, 1.0 Hz, 4H). 13C NMR (126 MHz, CDCl3) δ 163.63, 148.78, 140.96, 140.67, 140.31, 129.27, 127.12, 126.21, 125.51, 123.82, 120.57, 120.47, 109.79, 101.14. HRMS (ESI) m/z: [M + H]+ calcd for C44H27N4O2
:643.2134; found: 643.2139.
2,6-Bis(3,5-di(9H-carbazol-9-yl)phenyl)benzo[1,2-d:4,5-d′]bis(oxazole) (C4). Method B; grey solid (29%). M.P. > 240 °C. 1H NMR (500 MHz, CDCl3) δ 8.63 (s, 4H), 8.18 (d, J = 8.0 Hz, 8H), 8.03 (s, 2H), 7.97 (s, 2H), 7.62 (d, J = 8.0 Hz, 8H), 7.48 (t, J = 7.7 Hz, 8H), 7.35 (t, J = 7.7 Hz, 8H). 13C NMR Unable to be obtained due to limited solubility. HRMS (ESI) m/z: [M + H]+ calcd for C68H41N6O2
:973.3291; found: 973.3318.
2,6-Bis(4-(9H-carbazol-9-yl)phenyl)benzo[1,2-d:5,4-d′]bis(oxazole) (C5). Method A; pale yellow solid (35%). M.P. > 240 °C. 1H NMR (500 MHz, CDCl3) δ 8.53 (d, J = 8.1 Hz, 4H), 8.20 (s, 1H), 8.17 (d, J = 7.8 Hz, 4H), 7.88 (s, 1H), 7.81 (d, J = 8.2 Hz, 4H), 7.54 (d, J = 8.2 Hz, 4H), 7.46 (t, J = 7.6 Hz, 4H), 7.34 (t, J = 7.4 Hz, 4H). 13C NMR (126 MHz, CDCl3) δ 163.14, 149.10, 140.90, 140.32, 140.11, 129.20, 127.13, 126.21, 125.51, 123.81, 120.56, 120.47, 110.11, 109.79, 93.47. HRMS (ESI) m/z: [M + H]+ calcd for C44H27N4O2: 643.2134; found: 643.2135.
2,6-Bis(3,5-di(9H-carbazol-9-yl)phenyl)benzo[1,2-d:5,4-d′]bis(oxazole) (C6). Method B; grey solid (38%). M.P. > 240 °C. 1H NMR (500 MHz, CDCl3) δ 8.62 (s, 4H), 8.18 (d, J = 8.0 Hz, 9H), 8.02 (s, 2H), 7.82 (s, 1H), 7.62 (d, J = 8.0 Hz, 8H), 7.48 (t, J = 7.6 Hz, 8H), 7.35 (t, J = 7.6 Hz, 8H). 13C NMR Unable to be obtained due to limited solubility. HRMS (ESI) m/z: [M + H]+ calcd for C68H41N6O2: 973.3291; found: 973.3293.
Author contributions
David Wheeler: conceptualization, methodology, investigation, writing – original draft, writing – review & editing, and visualization. Pascal Friederich: investigation and data curation. Lloyd Fisher: investigation, writing – original draft, writing – review & editing. Angelar Muthike: investigation, writing – review & editing. Alán Aspuru-Guzik: conceptualization, methodology, investigation, and funding acquisition. Theodore Goodson: investigation, writing – review & editing, visualization and funding acquisition. Malika Jeffries-EL: conceptualization, methodology, investigation, writing – original draft, writing – review & editing, visualization supervision, project administration and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Professor Jeffries-EL thanks the National Science Foundation (CHE-1808402, and CHE-2108810) for financial support of this work. Professor Goodson wishes to acknowledge support from the Department of Energy, Biological and Environmental Research, grant number DE-SC0022118. We would like to thank Dr Ricardo Javier Vásquez for valuable input during discussions. We would also like to thank Dr Paul Ralifo and Dr Norman Lee of the CIC for NMR and HR-MS access and measurements (respectively), and Dr Volodimyr Duzhko, director of the laboratory for electronic materials and devices at the University of Massachusetts, Amherst for guidance and insight of UPS measurements. Computations were performed on the Niagara supercomputer at the SciNet HPC Consortium. SciNet is funded by the Canada Foundation for Innovation; the Government of Ontario; Ontario Research Fund – Research Excellence; and the University of Toronto.Notes and references
- Y.-F. Liu, J. Feng, Y.-G. Bi, D. Yin and H.-B. Sun, Adv. Mater. Technol., 2019, 4, 1800371 CrossRef.
- A. Zhou, H. Wang, Y. Zhang and C. Hu, Fiber Integr. Opt., 2022, 1–25, DOI:10.1080/01468030.2022.2083532; J. Song, H. Lee, E. G. Jeong, K. C. Choi and S. Yoo, Adv. Mater., 2020, 32, 1907539 CrossRef; C. Murawski and M. C. Gather, Adv. Opt. Mater., 2021, 9, 2100269 CrossRef.
- R. Pode, Renewable Sustainable Energy Rev., 2020, 133, 110043 CrossRef CAS; H. Sasabe and J. Kido, Chem. Mater., 2011, 23, 621–630 CrossRef; G. M. Phelan, Inf. Disp., 2018, 34, 10–15 Search PubMed.
- G. Gryn’ova, K.-H. Lin and C. Corminboeuf, J. Am. Chem. Soc., 2018, 140, 16370–16386 CrossRef CAS PubMed; H. Bronstein, C. B. Nielsen, B. C. Schroeder and I. McCulloch, Nat. Rev. Chem., 2020, 4, 66–77 CrossRef; R. M. Pankow and B. C. Thompson, Polymer, 2020, 207, 122874 CrossRef.
- J. Tagare and S. Vaidyanathan, J. Mater. Chem. C, 2018, 6, 10138–10173 RSC; M. Zhu and C. Yang, Chem. Soc. Rev., 2013, 42, 4963–4976 RSC; Z. Xu, B. Z. Tang, Y. Wang and D. Ma, J. Mater. Chem. C, 2020, 8, 2614–2642 RSC; A. Monkman, ACS Appl. Mater. Interfaces, 2022, 14, 20463–20467 CrossRef CAS PubMed.
- M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151 CrossRef CAS.
- X. Cai and S.-J. Su, Adv. Funct. Mater., 2018, 28, 1802558 CrossRef CAS; Y.-Z. Shi, H. Wu, K. Wang, J. Yu, X.-M. Ou and X.-H. Zhang, Chem. Sci., 2022, 13, 3625–3651 RSC.
- C. A. Parker and E. J. Bowen, Proceedings of the Royal Society of London. Series A. Mathematical and Physical Sciences, 1963, 276, 125–135 Search PubMed; H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- Y. Im, M. Kim, Y. J. Cho, J.-A. Seo, K. S. Yook and J. Y. Lee, Chem. Mater., 2017, 29, 1946–1963 CrossRef CAS; I. Zoh, M. Imai-Imada, J. Bae, H. Imada, Y. Tsuchiya, C. Adachi and Y. Kim, J. Phys. Chem. Lett., 2021, 12, 7512–7518 CrossRef PubMed.
- B. C. Tlach, A. L. Tomlinson, A. Bhuwalka and M. Jeffries-El, J. Org. Chem., 2011, 76, 8670–8681 CrossRef CAS.
- B. C. Tlach, A. L. Tomlinson, A. G. Ryno, D. D. Knoble, D. L. Drochner, K. J. Krager and M. Jeffries-El, J. Org. Chem., 2013, 78, 6570–6581 CrossRef CAS.
- D. L. Wheeler, A. V. Diodati, A. L. Tomlinson and M. Jeffries-El, ACS Omega, 2020, 5, 12374–12384 CrossRef CAS PubMed.
- A. A. Burney-Allen, J. Shaw, D. L. Wheeler, L. Diodati, V. Duzhko, A. L. Tomlinson and M. Jeffries-El, Asian J. Org. Chem., 2021, 10, 215–223 CrossRef CAS.
- R. Chavez III, M. Cai, B. Tlach, D. L. Wheeler, R. Kaudal, A. Tsyrenova, A. L. Tomlinson, R. Shinar, J. Shinar and M. Jeffries-El, J. Mater. Chem. C, 2016, 4, 3765–3773 RSC.
- D. Wheeler, S. Tannir, E. Smith, A. Tomlinson and M. Jeffries-El, Mater. Adv., 2022, 3, 3842–3852 RSC; X. Yin, T. Zhang, Q. Peng, T. Zhou, W. Zeng, Z. Zhu, G. Xie, F. Li, D. Ma and C. Yang, J. Mater. Chem. C, 2015, 3, 7589–7596 RSC.
- Y. Sagara, K. Shizu, H. Tanaka, H. Miyazaki, K. Goushi, H. Kaji and C. Adachi, Chem. Lett., 2015, 44, 360 CrossRef CAS; J. Lim, T. A. Albright, B. R. Martin and O. Š. Miljanić, J. Org. Chem., 2011, 76, 10207–10219 CrossRef.
- J. Luo, S. Gong, Y. Gu, T. Chen, Y. Li, C. Zhong, G. Xie and C. Yang, J. Mater. Chem. C, 2016, 4, 2442–2446 RSC.
- R. J. Vázquez, H. Kim, P. M. Zimmerman and T. Goodson, J. Mater. Chem. C, 2019, 7, 4210–4221 RSC.
- R. J. Vázquez, J. H. Yun, A. K. Muthike, M. Howell, H. Kim, I. K. Madu, T. Kim, P. Zimmerman, J. Y. Lee and T. G. Iii, J. Am. Chem. Soc., 2020, 142, 8074–8079 CrossRef PubMed.
- R. J. Vázquez, H. Kim, B. M. Kobilka, B. J. Hale, M. Jeffries-El, P. Zimmerman and T. Goodson, J. Phys. Chem. C, 2017, 121, 14382–14392 CrossRef.
- B. Carlotti, Z. Cai, H. Kim, V. Sharapov, I. K. Madu, D. Zhao, W. Chen, P. M. Zimmerman, L. Yu and T. Goodson, Chem. Mater., 2018, 30, 4263–4276 CrossRef CAS; Z. Cai, R. J. Vázquez, D. Zhao, L. Li, W.-Y. Lo, N. Zhang, Q. Wu, B. Keller, A. Eshun, N. Abeyasinghe, H. Banaszak-Holl, T. Goodson and L. Yu, Chem. Mater., 2017, 29, 6726–6732 CrossRef; B. Keller, Z. Cai, A. K. Muthike, P. K. Sahu, H. Kim, A. Eshun, P. M. Zimmerman, D. Zhang and T. Goodson, J. Phys. Chem. C, 2018, 122, 27713–27733 CrossRef.
- S. Hirata, Y. Sakai, K. Masui, H. Tanaka, S. Y. Lee, H. Nomura, N. Nakamura, M. Yasumatsu, H. Nakanotani, Q. Zhang, K. Shizu, H. Miyazaki and C. Adachi, Nat. Mater., 2015, 14, 330 CrossRef CAS PubMed.
- L. Fisher, R. J. Vázquez, M. Howell, A. K. Muthike, M. E. Orr, H. Jiang, B. Dodgen, D. R. Lee, J. Y. Lee, P. Zimmerman and T. Goodson, Chem. Mater., 2022, 34, 2161–2175 CrossRef.
- J. Tagare, R. Boddula, S. S. Sudheendran, D. K. Dubey, J.-H. Jou, S. Patel and S. Vaidyanathan, J. Mater. Chem. C, 2020, 8, 16834–16844 RSC.
- D. Weininger, J. Chem. Inf. Comput. Sci., 1988, 28, 31–36 CrossRef CAS.
- S. Grimme, C. Bannwarth and P. Shushkov, Journal of Chemical Theory and Computation, 2017, 13, 1989–2009 CrossRef CAS PubMed; C. Bannwarth, S. Ehlert and S. Grimme, J. Chem. Theory Comput., 2019, 15, 1652–1671 CrossRef.
- P. Pracht, F. Bohle and S. Grimme, Phys. Chem. Chem. Phys., 2020, 22, 7169–7192 RSC.
- R. Gomez-Bombarelli, J. Aguilera-Iparraguirre, T. D. Hirzel, D. Duvenaud, D. Maclaurin, M. A. Blood-Forsythe, H. S. Chae, M. Einzinger, D. G. Ha, T. Wu, G. Markopoulos, S. Jeon, H. Kang, H. Miyazaki, M. Numata, S. Kim, W. Huang, S. I. Hong, M. Baldo, R. P. Adams and A. Aspuru-Guzik, Nat. Mater., 2016, 15, 1120 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- Y. Shao, Z. Gan, E. Epifanovsky, A. T. B. Gilbert, M. Wormit, J. Kussmann, A. W. Lange, A. Behn, J. Deng, X. Feng, D. Ghosh, M. Goldey, P. R. Horn, L. D. Jacobson, I. Kaliman, R. Z. Khaliullin, T. Kuś, A. Landau, J. Liu, E. I. Proynov, Y. M. Rhee, R. M. Richard, M. A. Rohrdanz, R. P. Steele, E. J. Sundstrom, H. L. Woodcock, P. M. Zimmerman, D. Zuev, B. Albrecht, E. Alguire, B. Austin, G. J. O. Beran, Y. A. Bernard, E. Berquist, K. Brandhorst, K. B. Bravaya, S. T. Brown, D. Casanova, C.-M. Chang, Y. Chen, S. H. Chien, K. D. Closser, D. L. Crittenden, M. Diedenhofen, R. A. DiStasio, H. Do, A. D. Dutoi, R. G. Edgar, S. Fatehi, L. Fusti-Molnar, A. Ghysels, A. Golubeva-Zadorozhnaya, J. Gomes, M. W. D. Hanson-Heine, P. H. P. Harbach, A. W. Hauser, E. G. Hohenstein, Z. C. Holden, T.-C. Jagau, H. Ji, B. Kaduk, K. Khistyaev, J. Kim, J. Kim, R. A. King, P. Klunzinger, D. Kosenkov, T. Kowalczyk, C. M. Krauter, K. U. Lao, A. D. Laurent, K. V. Lawler, S. V. Levchenko, C. Y. Lin, F. Liu, E. Livshits, R. C. Lochan, A. Luenser, P. Manohar, S. F. Manzer, S.-P. Mao, N. Mardirossian, A. V. Marenich, S. A. Maurer, N. J. Mayhall, E. Neuscamman, C. M. Oana, R. Olivares-Amaya, D. P. O’Neill, J. A. Parkhill, T. M. Perrine, R. Peverati, A. Prociuk, D. R. Rehn, E. Rosta, N. J. Russ, S. M. Sharada, S. Sharma, D. W. Small, A. Sodt, T. Stein, D. Stück, Y.-C. Su, A. J. W. Thom, T. Tsuchimochi, V. Vanovschi, L. Vogt, O. Vydrov, T. Wang, M. A. Watson, J. Wenzel, A. White, C. F. Williams, J. Yang, S. Yeganeh, S. R. Yost, Z.-Q. You, I. Y. Zhang, X. Zhang, Y. Zhao, B. R. Brooks, G. K. L. Chan, D. M. Chipman, C. J. Cramer, W. A. Goddard, M. S. Gordon, W. J. Hehre, A. Klamt, H. F. Schaefer, M. W. Schmidt, C. D. Sherrill, D. G. Truhlar, A. Warshel, X. Xu, A. Aspuru-Guzik, R. Baer, A. T. Bell, N. A. Besley, J.-D. Chai, A. Dreuw, B. D. Dunietz, T. R. Furlani, S. R. Gwaltney, C.-P. Hsu, Y. Jung, J. Kong, D. S. Lambrecht, W. Liang, C. Ochsenfeld, V. A. Rassolov, L. V. Slipchenko, J. E. Subotnik, T. Van Voorhis, J. M. Herbert, A. I. Krylov, P. M. W. Gill and M. Head-Gordon, Mol. Phys., 2015, 113, 184–215 CrossRef CAS.
- M. Inbasekaran and R. Strom, OPPI Briefs, 1994, 23, 447–450 Search PubMed.
- R. J. Schmitt, D. S. Ross, J. R. Hardee and J. F. Wolfe, J. Org. Chem., 1988, 53, 5568–5569 CrossRef CAS.
- A. T. R. Williams, S. A. Winfield and J. N. Miller, Analyst, 1983, 108, 1067–1071 RSC.
- I. B. Berlman, in Handbook of Fluorescence Spectra of Aromatic Molecules, ed. I. B. Berlman, Academic Press, 2nd edn, 1971, pp. 67–95 DOI:10.1016/B978-0-12-092656-5.50009-5.
- A. K. Sharma, K. Gowdahalli, J. Krzeminski and S. Amin, J. Org. Chem., 2007, 72, 8987–8989 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectroscopy, theoretical Cartesain coordinates, etc. See DOI: https://doi.org/10.1039/d2tc03190f |
| This journal is © The Royal Society of Chemistry 2023 |