
Modulating the microenvironment of AuPd nanoparticles using metal–organic frameworks for selective methane oxidation†
Jianfei
Sui‡
a,
Ming-Liang
Gao‡
a,
Chengyuan
Liu
b,
Yang
Pan
 b,
Zheng
Meng
*a,
Daqiang
Yuan
c and
Hai-Long
Jiang
*a
b,
Zheng
Meng
*a,
Daqiang
Yuan
c and
Hai-Long
Jiang
*a
aHefei National Laboratory for Physical Sciences at the Microscale, Department of Chemistry, University of Science and Technology of China, Hefei, Anhui 230026, P. R. China. E-mail: zhengmeng@ustc.edu.cn; jianglab@ustc.edu.cn
bNational Synchrotron Radiation Laboratory (NSRL), University of Science and Technology of China, Hefei, Anhui 230029, P. R. China
cState Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou 350002, P. R. China
First published on 8th August 2023
Abstract
Direct and selective oxidation of methane (CH4) into methanol (CH3OH) under ambient conditions remains a grand challenge because of the high energy barrier of CH4 activation and the complicated processes involved. Herein, by incorporating AuPd alloy nanoparticles (NPs) into a series of Cu2+-doped metal–organic frameworks (MOFs), namely AuPd@Cu-UiO-66x, efficient and direct catalytic conversion of CH4 to CH3OH can be achieved at mild temperature (70 °C) with H2O2 as an oxidant. The Cu-UiO-66 serving as the microenvironment parameter not only regulates the electronic state of AuPd NPs to improve the CH4 adsorption and activation, but also affects the generation of hydroxyl radicals (˙OH) in H2O2 reaction pathways, which consequently results in a volcano-type dependency of the CH3OH selectivity on the Cu contents. This work represents the first finding on achieving direct catalytic oxidation of CH4 to CH3OH under mild conditions by modulating the microenvironment of catalytic centers based on a MOF platform.
Introduction
The direct conversion of methane, the major component of natural gas, to olefins, aromatics, oxygenates, and other products, has been recognized as a potentially economical and environmentally friendly way to generate value-added chemicals from abundant and inexpensive sources.1–3 Among various routes of methane conversion, the direct and selective oxidation of methane to methanol, known as the ‘Holy Grail’ reaction in catalysis science,4,5 is particularly promising. However, this transformation has long been challenged by the stable and inert C–H bonds (ΔHC–H = 104 kcal mol−1), low polarizability, and negligible electron affinity of methane.4–8 Therefore, energy-intensive routes are conventionally required for CH4 activation.2,3 In addition, the challenges in the CH4 to CH3OH process are also tangled by the fact that CH3OH can be more easily oxidized than CH4, which leads to unexpected peroxidation products.6,7 Therefore, the development of suitable systems for efficient and selective direct conversion of CH4 to CH3OH under mild conditions is highly desirable yet challenging.To achieve selective oxidation of CH4 to CH3OH under mild conditions, environmentally benign hydrogen peroxide (H2O2) has been adopted as the oxidant.4,6,9–11 Unfortunately, this may lead to the formation of methyl-hydroperoxide (CH3OOH) as the main product by the combination of ˙CH3 with dissolved O2 originating from the decomposition of H2O2,9,12 or produce a significant amount of byproducts due to the overoxidation of methanol by ˙OH originating from the homocleavage of H2O2.10,11 Although a few systems, including metal-modified zeolites and metal nanoparticle-based heterostructures,5,11–14 have been employed, the rational control of the catalytic performance is still far from satisfactory to achieve both high selectivity and activity. The above situation is likely rooted in the complex and highly interconnected processes involved in catalytic CH4 oxidation using H2O2, such as the activation of CH4 and H2O2, the degradation of H2O2, overoxidation of methanol, and more.15–17 Evidently, the complicated nature of the H2O2-participated CH4 oxidation requires a precise balance of these processes for achieving high activity and selectivity. A platform that allows the sophisticated control of H2O2 homocleavage by microenvironment modulation is thus highly desirable.
Based on the above analysis, it is assumed that metal–organic frameworks (MOFs), a class of porous crystalline materials featuring high surface areas and tailorable structures,17–20 would be an ideal platform for creating a suitable microenvironment for hosted active centers. Different components involved in catalytic sites and the microenvironment can work synergistically to target optimal catalytic performance. Specifically, the permanent porosity of MOFs can provide congenital advantages for stabilizing active metal nanoparticles (NPs).21–25 The high tailorability of MOFs makes it feasible to introduce extra auxiliary components through various modification strategies, enabling the microenvironment modulation for metal NPs in a rational manner.19
In this work, the representative MOF, UiO-66 with defects in varying degrees, denoted as UiO-66x (x represents the molar ratio of (acetic acid)/(the linker), x = 0, 50, 100, 150, and 200), has been fabricated. Upon furnishing Cu2+ onto the defect sites of Zr-oxo clusters of UiO-66, AuPd NPs are further incorporated to afford AuPd@Cu-UiO-66x composite (Fig. 1). AuPd NPs were utilized in our study for their capability in partial methane oxidation.11 Remarkably, anchoring Cu species to the Zr-oxo clusters around AuPd NPs effectively tunes the selectivity to CH3OH with a volcano-type dependency on the Cu loadings, where the optimized CH3OH selectivity is as high as 80.97%. The regulated selectivity arises from the combined effect of the enhanced CH4 adsorption and the weakened ˙OH formation accompanied by the Cu2+ anchoring. Although MOFs have been reported in the quasi-catalytic CH4 oxidation under high-temperature in the gas phase,26,27 this study, integrating AuPd active sites and the microenvironment modulation by systematically tailoring Cu loadings based on a MOF platform, achieves highly selective CH4 oxidation under mild conditions.
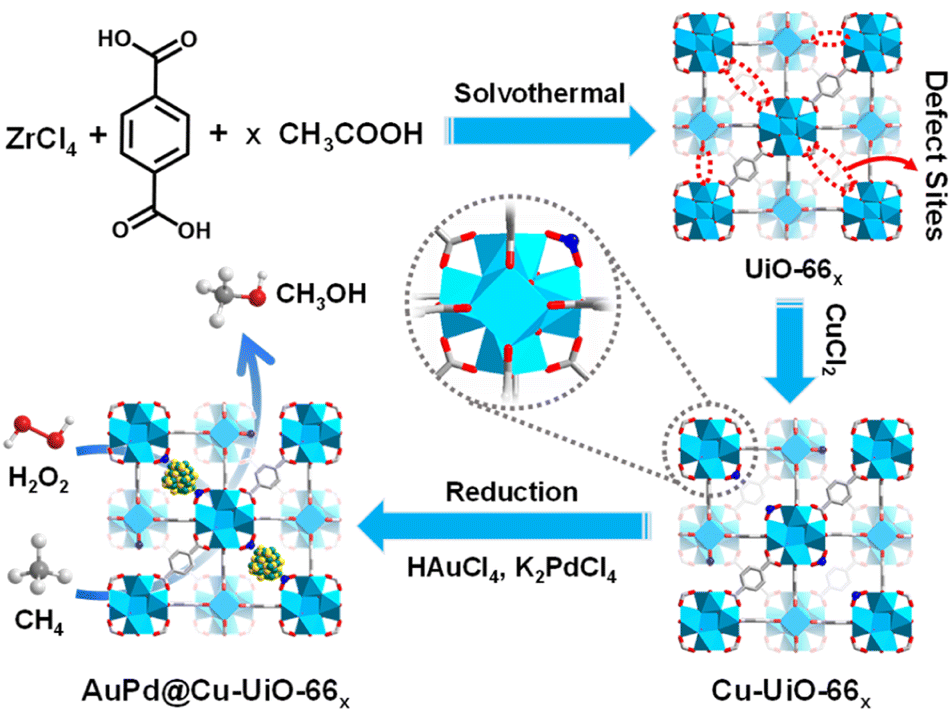 | ||
| Fig. 1 Schematic illustration showing the synthetic process for AuPd@Cu-UiO-66x. | ||
Results and discussion
UiO-66x compounds with different amounts of structural defects were synthesized by reacting ZrCl4 and terephthalic acid with different equivalents of acetic acid as a modulator in DMF. Powder X-ray diffraction (XRD) patterns suggest that all UiO-66x compounds maintain similar high crystallinity to UiO-66 (Fig. S1†). Scanning electron microscopy (SEM) images display that the sizes of MOF particles gradually increase and the morphology changes from intergrown prototypes to octahedral nanocrystals, along with more acetic acid being used (Fig. S2†). N2 sorption isotherms demonstrate that the Brunauer–Emmett–Teller (BET) area for UiO-66x gradually increases from 736 to 1151 m2 g−1 while the pore volume increases from 0.55 to 0.67 cm3 g−1 with increased amounts of acetic acid (Fig. S3†). Cu-UiO-66x were prepared by a microwave-assisted reaction of CuCl2·2H2O and UiO-66x in acetonitrile. Inductively coupled plasma atomic emission spectrometry (ICP-AES) analysis demonstrates that the Cu loadings are approximately 0.41–2.42 wt% in Cu-UiO-66x (Table S1†). To identify the existing form of Cu and its electronic state in the catalysts, Cu-UiO-66200 is chosen considering that its higher Cu content than other catalysts would be beneficial to the characterization precision. The diffuse reflectance infrared Fourier transform (DRIFT) spectra show that the intensities of peaks at 3673 and 2769 cm−1, respectively attributed to the terminal –OH/–OH2 and μ3-OH on the defect sites, are significantly lower in Cu-UiO-66200 than those in UiO-66, which supports the immobilization of Cu atoms to the chelating/defect sites in Cu-UiO-66x (Fig. S4†).28–30 The Raman spectrum for Cu-UiO-66200 clearly showed the Cu–O vibration peak at 150 cm−1, which also supports that Cu is anchored to UiO-66 (Fig. S5†).31 The Cu K-edge X-ray absorption near-edge structure (XANES) spectrum of Cu-UiO-66200 displays an absorption threshold analogous to that of CuO, indicating that the oxidation state of Cu is about +2 (Fig. 2a). The Fourier-transformed extended X-ray absorption fine structure (FT-EXAFS) spectrum of Cu-UiO-66200 shows only one dominant peak at approximately 1.5 Å, confirming the atomically dispersed Cu sites in Cu-UiO-66200 (Fig. 2b).30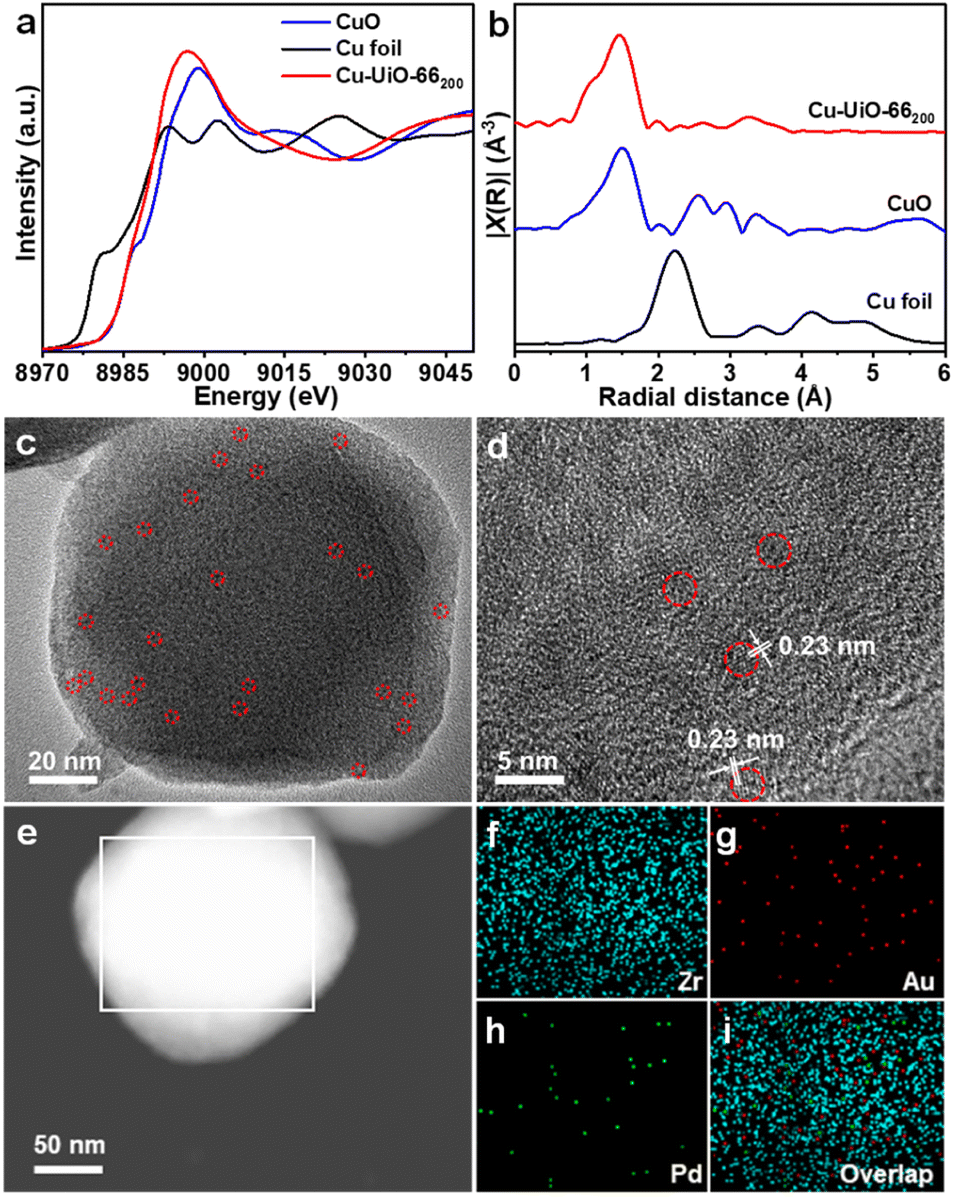 | ||
| Fig. 2 (a) The normalized XANES spectra, (b) FT-EXAFS spectra for Cu-UiO-66200, CuO, and Cu foil. (c) TEM and (d) HRTEM images of AuPd@Cu-UiO-66100. (e–i) High-angle annular darkfield scanning transmission electron microscopy image of AuPd@Cu-UiO-66100, and the corresponding Zr, Au, Pd, and overlapped elemental mapping for the selected area. | ||
To prepare AuPd@Cu-UiO-66x, the mixture of HAuCl4 and K2PdCl4 aqueous solution was impregnated into Cu-UiO-66x and subsequently reduced in a H2/Ar atmosphere at 200 °C. Powder XRD patterns suggest that the crystallinity of Cu-UiO-66x is maintained after the incorporation of AuPd NPs (Fig. S6†). In addition, the comparison shows that the (200), (220), and (222) diffraction peaks of HKUST-1 were absent in AuPd@Cu-UiOx, indicating that there is no formation of HKUST-1 in our work when Cu is added to the MOF and subjected to high temperature (Fig. S6†). The contents of Au and Pd determined by ICP-AES are similar across all AuPd@Cu-UiO-66x samples, which are approximately 3.5 wt% for Au and 1.5 wt% for Pd, respectively (Table S1†). The octahedral shape of UiO-66x remained after the AuPd incorporation, as demonstrated by the SEM images and the selected area electron diffraction (SAED) pattern has been obtained to demonstrate that UiO-66 is stable under the tested condition (Fig. S7†). In addition, transmission electron microscopy (TEM) observation of AuPd@Cu-UiO-66x shows that the tiny AuPd NPs with rough sizes of 1.5 nm, matching the pore sizes of defective UiO-66,32,33 are highly dispersed throughout Cu-UiO-66x particles (Fig. 2c, S8–S12†). Although the sizes of the AuPd NPs can be larger than the free volume of the octahedron cages (12 Å) in UiO-66, we hypothesize that large-sized NPs may penetrate multiple adjacent cages through the pore aperture or appear where the defects are present. The direct comparison between the high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and secondary electron STEM (SE-STEM) images acquired at the same location supports that the AuPd NPs are located inside the crystallites of AuPd@Cu-UiO-66100 (Fig. S13†).34–36 Lattice fringes with a d-spacing of ∼0.23 nm are observed by high-resolution TEM (HRTEM) (Fig. 2d), consistent with the (111) plane of AuPd NPs.37 The energy-dispersive X-ray mapping results suggest homogeneous distributions of Au and Pd elements in the MOF particle (Fig. 2e–i), further supporting the well-dispersed feature of AuPd NPs in UiO-66x. N2 sorption isotherms demonstrate that the porous structures of Cu-UiO-66x are all maintained after the incorporation of AuPd NPs, albeit with slightly decreased BET areas due to the mass and pore occupation by AuPd NPs (Fig. S14†). The pore size distributions of the AuPd@Cu-UiO-66x are mostly in the range of 7–15 Å, which are still large enough for CH4, H2O2, and intermediates involved in CH4 oxidation.
The performance of AuPd@Cu-UiO-66x catalysts for direct catalytic oxidation of CH4 to CH3OH has been evaluated in an aqueous H2O2 solution (Fig. 3 and Tables S2, S3†). Without the incorporation of AuPd NPs, UiO-66 or Cu-UiO-66 alone fails to catalyze the oxidation of CH4. The Au@UiO-66 shows good reactivity for CH4 oxidation to give the oxygenated products CH3OH, CH3OOH, and HCOOH, among which CH3OH only takes up a very small portion with a selectivity of 30.19%. Although Pd@UiO-66 shows a moderately high CH3OH selectivity of 50.39%, its reactivity toward CH4 oxidation is very poor since the total oxygenated products are only 39% of those for Au@UiO-66. In contrast to these comparison catalysts, AuPd@UiO-66 gives a reasonable CH3OH selectivity of 53.84% and a much higher CH3OH yield (1.61 μmol) than both Au@UiO-66 (0.93 μmol) and Pd@UiO-66 (0.63 μmol), suggesting AuPd NPs are indispensable for high activity and selectivity in the conversion. Despite this, colloidal AuPd NPs present a low CH3OH selectivity of 23% while CH3OOH is the major species (64%) among all the oxidation products. The significantly lower CH3OH yield of colloidal AuPd NPs (0.58 μmol) than that of AuPd@UiO-66 (1.61 μmol) indicates the critical role of the MOF in this reaction.
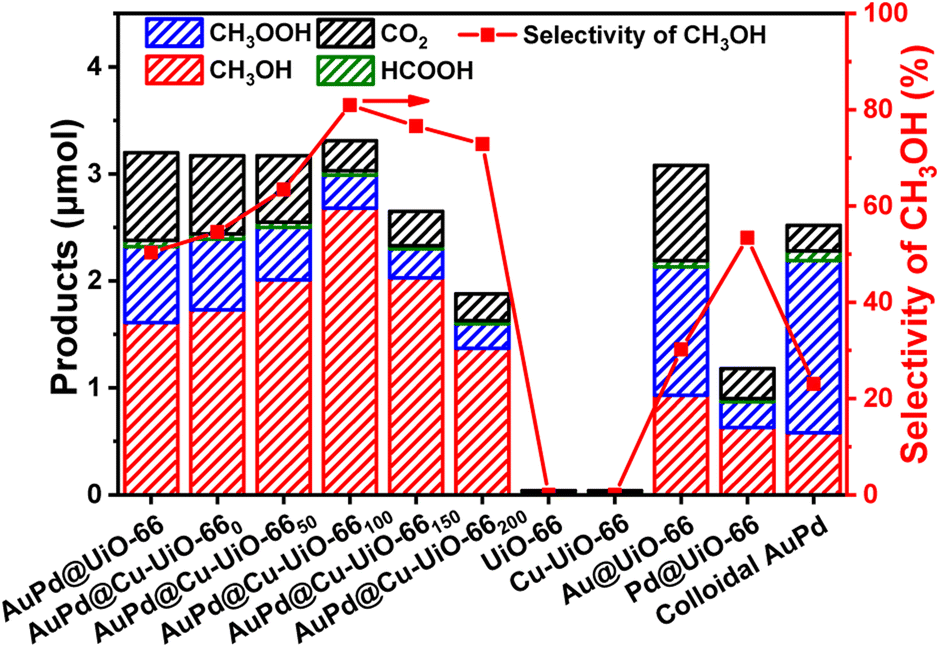 | ||
| Fig. 3 Catalytic performance of AuPd@UiO-66, AuPd@Cu-UiO-66x, UiO-66, Cu-UiO-66, Au@UiO-66, Pd@UiO-66, and colloidal AuPd NPs in the CH4 oxidation (reaction conditions: catalyst 27 mg, CH4 pressure 30 bar, H2O2 0.5 M (aq), 70 °C, reaction time 30 min). | ||
Upon introducing Cu species into AuPd@UiO-66, an enhanced CH3OH selectivity of 54.57% is observed with AuPd@Cu-UiO-660 as the catalyst. With increased Cu loading, the selectivity to CH3OH first exhibits a trend of increase and reaches a maximum of 80.97% with a normalized production rate of 198.52 μmol gcat−1 h−1 using AuPd@Cu-UiO-66100. Further increasing the Cu content leads to a decrease of the selectivity to 76.60% and 72.87% for AuPd@Cu-UiO-66150 and AuPd@Cu-UiO-66200, respectively. The selectivity of CH3OH exhibits a volcano-type dependency on the Cu loadings in AuPd@Cu-UiO-66x (Fig. 3), unambiguously demonstrating that the Cu species effectively regulates the AuPd selectivity in the partial oxidation of CH4 to CH3OH. The comprehensive normalized conversions for all the C1 products have also been provided (Table S2†). It is worth noting that by simply increasing the Cu content from 0.4 wt% in AuPd@Cu-UiO-660 to 1.4 wt% in AuPd@Cu-UiO-66100, a 48% improvement of the methanol selectivity can be achieved. Since Cu in AuPd@Cu-UiO-66x is anchored to the defect sites of UiO-66, the defect sites exposed in AuPd@Cu-UiO-66x catalysts will be negligible. Therefore, with other chemical parameters kept the same in AuPd@Cu-UiO-66x, the observed difference of the selectivity provided by AuPd@Cu-UiO-66x should be ascribed to their different Cu loadings. Considering that Cu actually accounts for only very small mass percentages in the catalysts, the dependency of selectivity on Cu loading highlights the importance of the precise control of the microenvironment to the optimization of catalytic performance. In addition, a recent report on the use of AuPd NPs encapsulated by ZIF-8 gave a selectivity of 21.9% for CH3OH.38 Compared with AuPd@Cu-UiO-66100, the sharp performance contrast is another sign of the significance of the microenvironment modulation.
To examine the source of the products, the isotope trace experiment by using 13CH4 has been conducted. Synchrotron radiation photoionization mass spectrometry (SR-PIMS) for the reaction mixture gives peaks at m/z = 33 and m/z = 47 at a photon energy of 11.8 eV, which are respectively corresponding to 13CH3OH and H13COOH (Fig. 4a). The results unambiguously confirm that CH4 is the exclusive carbon source of CH3OH and HCOOH products, which also demonstrate the successful implementation of catalytic CH4 oxidation over the AuPd@Cu-UiO-66100 catalyst. Interestingly, a peak at m/z = 31 assigned to H13CHO is also observed (Fig. 4a). HCHO has been confirmed as the primary and active intermediate in the oxidation of CH3OH.39,40 However, the low concentration of HCHO in the reaction and the overlap of its chemical shift with solvent make its detection extremely challenging. In our system, the signal of HCHO becomes more apparent when CH3OH is adopted as the reactant (Fig. S15†). The in situ SR-PIMS test demonstrates the appearance of HCHO after 10 min in the CH3OH oxidation, prior to the observation of HCOOH (Fig. S16†). HCOOH can be detected when HCHO is used as the reactant (Fig. S17†). These results support that CH4 is first converted to the desired product CH3OH, and further overoxidized into HCOOH through the formation of intermediate HCHO (Fig. 4b).
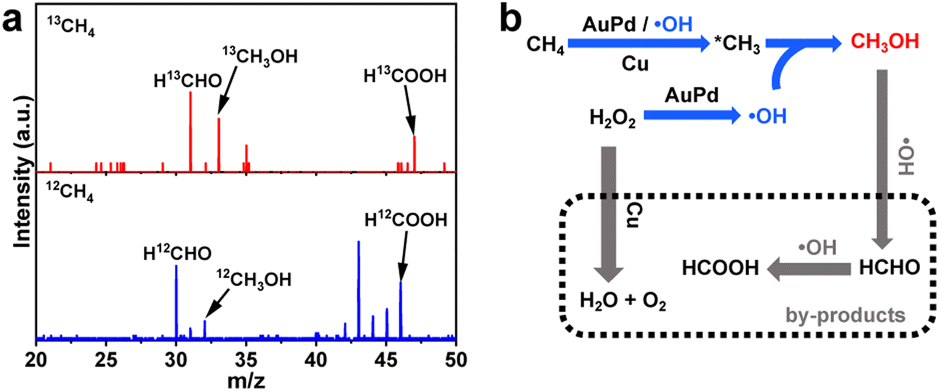 | ||
| Fig. 4 (a) Photoionization mass spectrum of the products using 13CH4 and 12CH4 as the reactant. (b) Proposed reaction path for methane oxidation over AuPd@UiO-66100. | ||
N2 sorption isotherms (Fig. S18†) and powder XRD pattern of AuPd@Cu-UiO-66100 after catalysis manifest that the porosity and crystalline feature of the MOF is maintained (Fig. S19†). No identifiable peak assigned to AuPd NPs can be observed in powder XRD pattern, reflecting that AuPd NPs are not agglomerated during the catalysis, in line with the TEM observation (Fig. S20†). The metal loadings are maintained after the reaction based on ICP-AES results, suggesting the absence of metal leaching during the reaction (Table S4†). The FT-IR, 1H NMR spectra, and elemental analysis of AuPd@Cu-UiO-66100 before and after the reaction are nearly identical, implying that the organic components in the catalysts keep intact during the reaction (Fig. S21, S22, and Table S4†). Furthermore, no apparent activity and selectivity loss occur to AuPd@Cu-UiO-66100 in three consecutive cycles (Fig. S23†), demonstrating its good reusability. All the above results evidence the stability of the catalysts under the tested conditions.
To disclose the role of Cu in catalysis, X-ray photoelectron spectroscopy (XPS) has been conducted to investigate the oxidation state of Au and Pd species (Fig. S24†). The AuPd@UiO-66 presents two sets of peaks: one set of peaks are at binding energies of 83.65 and 87.35 eV, corresponding to Au0 4f7/2 and Au0 4f5/2, respectively; the other set of peaks at 335.22 and 340.61 eV are ascribed to 3d5/2 and 3d3/2 of Pd0, respectively.41–45 Compared to monometallic Au0 and Pd0, both Au 4f and Pd 3d peaks in AuPd@UiO-66 shift to lower binding energies. The electronic state change of Au and Pd indicates the formation of AuPd alloy, which is supported by the previous reports.37,41–45 With increased Cu loadings, the Au 4f and Pd 3d peaks gradually shift to higher binding energies in AuPd@Cu-UiO-66x. These changes may be ascribed to the electron-withdrawing effect of Cu2+, leading to electron transfer from AuPd to Cu2+.29 To further probe the modulated electronic state of AuPd NPs in AuPd@Cu-UiO-66x, the carbon monoxide (CO) adsorption analysis using diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) has been carried out (Fig. S25†). Both adsorption bands of linearly adsorbed CO on Au (2150–2000 cm−1) and twofold bridge-bonded CO on Pd (2000–1900 cm−1) shift to higher wavenumbers with the change of wavenumber >20 cm−1 when creasing Cu loadings, which further supports enhanced electron transfer from AuPd to the MOFs with increased Cu loadings.46,47 This phenomenon can be ascribed to the electron-withdrawing effect of Cu2+ attached to the Zr-oxo clusters of UiO-66, leading to the electron transfer from Au and Pd to Cu. With the increase of Cu content, the electron-withdrawing effect becomes stronger, resulting in a gradual increase of the binding energy. Taking all these spectroscopic results together, it is suggested that the Cu2+ incorporation effectively modulates the AuPd electronic state, which may be responsible for the observed different CH3OH selectivity of AuPd@Cu-UiO-66x.14,48
It is generally believed that CH4 oxidation using H2O2 as an oxidant involves at least three key steps, the adsorption and activation of CH4 to form active *CHx species on the surface of catalysts13 or through the attack of CH4 by ˙OH,49 the homolytic cleavage of H2O2 to generate ˙OH, and the combination of *CHx and ˙OH (Fig. 4b).5 To investigate the adsorption of CH4 on catalysts, CH4 temperature-programmed desorption coupled with mass spectrometric analysis (TPD-MS) has been carried out. The results show that the desorption temperature of CH4 from AuPd@Cu-UiO-66100 is approximately 184 °C, in comparison to 158 °C for AuPd@UiO-66 (Fig. 5a, S26†), revealing stronger CH4 adsorption on AuPd@Cu-UiO-66100. The CH4 sorption displays the higher CH4 adsorption capacity (17.6 cm3 g−1) of AuPd@Cu-UiO-66100 than AuPd@UiO-66 (13.5 cm3 g−1) at 1 bar and 298 K (Fig. S27†). From AuPd@UiO-66 to AuPd@Cu-UiO-66100, the increase of CH4 adsorption is 30%, which is higher than the magnitude of the increase of the BET area, supporting the enhanced CH4 adsorption of the latter. This enhanced CH4 adsorption would be beneficial to its activation on AuPd sites and competition with the CH3OH adsorption to avoid overoxidation,50 giving rise to enhanced activity and selectivity toward CH3OH.
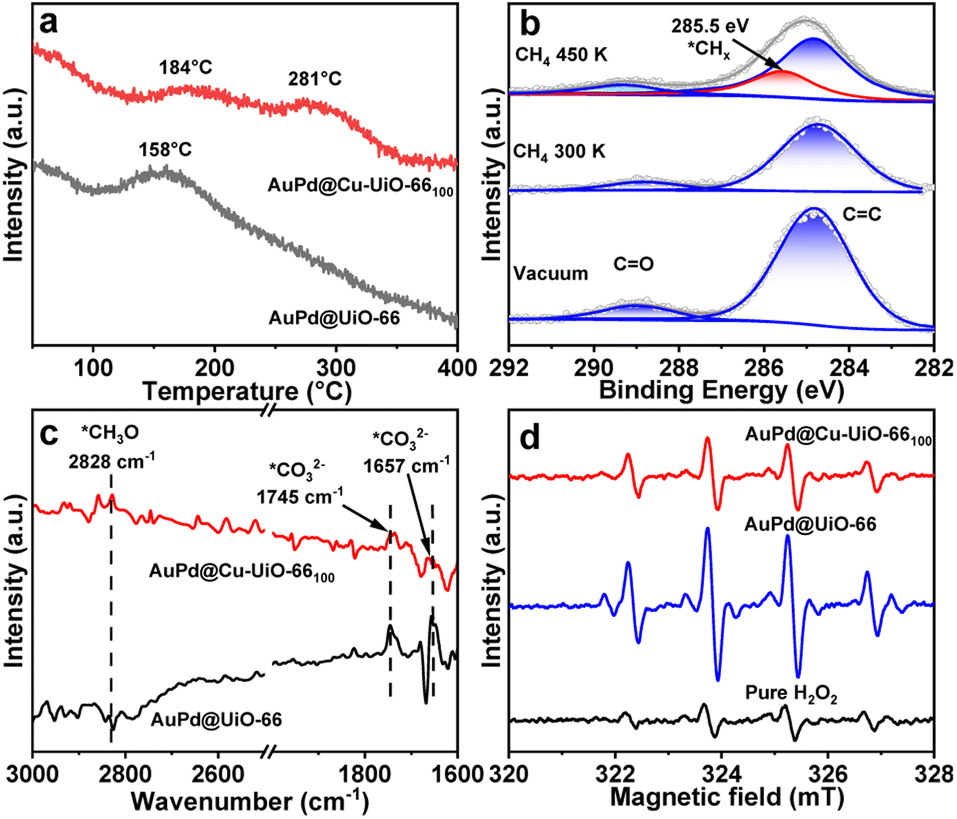 | ||
| Fig. 5 (a) TPD-MS profiles (m/z = 15) of CH4 on AuPd@UiO-66 and AuPd@UiO-66100. (b) The C 1s XPS spectra of the AuPd@UiO-66100 in ultrahigh vacuum and after introducing 1 mbar of CH4 at different temperatures. (c) DRIFT spectra of CH4 adsorbed AuPd@UiO-66 and AuPd@UiO-66100 after being purged with Ar gas for 30 min. (d) EPR trapping experiment over AuPd@UiO-66 and AuPd@UiO-66100 with H2O2 as the oxidant and DMPO as the radical trapping agent. | ||
To identify the possible behavior of CH4 after its adsorption, near-ambient pressure XPS (NAP-XPS) has been conducted. Before CH4 exposure, the C 1s spectrum of AuPd@Cu-UiO-66100 displays two peaks at 284.8 and 288.8 eV attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C–C, C–H, and CO bonds in the MOF linkers (Fig. 5b).51,52 The subsequent exposure of the sample to 1 mbar of CH4 at 300 K doesn't result in additional peaks, probably due to the overlap of signals between CH4 and CC, C–C, and C–H bonds of the MOF. When the temperature is raised to 450 K, deconvolution of the C 1s spectrum gives a new peak at 285.5 eV (Fig. 5b). This peak can be assigned to the *CHx species, which may include CH3, CH2, and CH, inferring the activation of CH4 on the catalyst.50In situ DRIFTS has been further conducted to investigate the transformation of CHx species on different catalyst surfaces in the presence of H2O2. When CH4 is exposed to the catalyst for 30 minutes, peaks at 1305 and 3015 cm−1 assigned to the typical vibration of absorbed CH4 are observed (Fig. S28†).53,54 After the physically adsorbed CH4 is removed by flushing Ar flow through an H2O2 aqueous solution, the peaks in the ranges of 1500–1800 cm−1 and 2700–3000 cm−1 appear (Fig. 5c). The peaks at 1657 and 1745 are assigned to *CO32−, and the signal at 2828 cm−1 is ascribed to the *CH3O intermediate.53–55 The appearance of these peaks indicates the transformation of *CHx to oxygen-containing species on the catalyst with the participation of H2O2. Compared to those in AuPd@UiO-66, the intensity of the peak at 2828 cm−1 (*CH3O) increases and the intensities at 1657 and 1745 cm−1 (*CO32−) decrease in the presence of AuPd@Cu-UiO-66100, in line with its enhanced CH3OH selectivity. The above results demonstrate that AuPd NPs play several important roles in the reaction, in which they not only affect CH4 adsorption but also influence the activation and transformation of CH4. The modulated electronic state of AuPd by the incorporated Cu2+ will, thus, have a profound effect on the CH3OH selectivity.
C, C–C, C–H, and CO bonds in the MOF linkers (Fig. 5b).51,52 The subsequent exposure of the sample to 1 mbar of CH4 at 300 K doesn't result in additional peaks, probably due to the overlap of signals between CH4 and CC, C–C, and C–H bonds of the MOF. When the temperature is raised to 450 K, deconvolution of the C 1s spectrum gives a new peak at 285.5 eV (Fig. 5b). This peak can be assigned to the *CHx species, which may include CH3, CH2, and CH, inferring the activation of CH4 on the catalyst.50In situ DRIFTS has been further conducted to investigate the transformation of CHx species on different catalyst surfaces in the presence of H2O2. When CH4 is exposed to the catalyst for 30 minutes, peaks at 1305 and 3015 cm−1 assigned to the typical vibration of absorbed CH4 are observed (Fig. S28†).53,54 After the physically adsorbed CH4 is removed by flushing Ar flow through an H2O2 aqueous solution, the peaks in the ranges of 1500–1800 cm−1 and 2700–3000 cm−1 appear (Fig. 5c). The peaks at 1657 and 1745 are assigned to *CO32−, and the signal at 2828 cm−1 is ascribed to the *CH3O intermediate.53–55 The appearance of these peaks indicates the transformation of *CHx to oxygen-containing species on the catalyst with the participation of H2O2. Compared to those in AuPd@UiO-66, the intensity of the peak at 2828 cm−1 (*CH3O) increases and the intensities at 1657 and 1745 cm−1 (*CO32−) decrease in the presence of AuPd@Cu-UiO-66100, in line with its enhanced CH3OH selectivity. The above results demonstrate that AuPd NPs play several important roles in the reaction, in which they not only affect CH4 adsorption but also influence the activation and transformation of CH4. The modulated electronic state of AuPd by the incorporated Cu2+ will, thus, have a profound effect on the CH3OH selectivity.
Reactive oxygen species originating from H2O2, such as superoxide anion radicals (O2˙−) and hydroxyl radicals (˙OH), are the other group of important intermediates in CH4 oxidation by H2O2. Electron paramagnetic resonance (EPR) experiments have been conducted to detect the radical species involved in the reaction. With 5,5-dimethyl-pyrroline-N-oxide (DMPO) as a radical trapping agent, EPR spectra exhibit a characteristic signal of DMPO-OH (Fig. 5d). This result suggests that ˙OH is the main active species in methane oxidation. Compared with AuPd@Cu-UiO-66, the intensity of EPR signals in AuPd@Cu-UiO-66100 is lower, indicating its reduced generation of ˙OH. No DMPO-CH3 peak is observed likely attributed to the fast combination of as-generated ˙CH3 with the relatively abundant ˙OH.56,57 When CH4 is replaced by CH3OH, products HCOOH and CO2 are detected (Fig. S29†), implying that ˙OH generated from H2O2 also participates in the overoxidation process. Generally, H2O2 has two competitive pathways in CH4 oxidation, the generation of O2 and H2O through decomposition and the formation of ˙OH from homolytic cleavage.15,46–49 It is found that the decomposition rate of H2O2 increases with increased Cu content in AuPd@Cu-UiO-66x (Fig. S30†), probably owing to the catalytic effect of Cu in H2O2 decomposition through the CuI/CuII redox couple.16 Therefore, with increased Cu loadings in AuPd@Cu-UiO-66x, the generation of ˙OH will be more or less suppressed due to the enhanced decomposition of H2O2. The introduction of Cu at too low loadings will result in the formation of excess ˙OH and lead to over-oxidation during CH4 oxidation. On the other hand, a too-high loading of Cu2+ will reduce the concentration of ˙OH around the catalytic sites, leading to inhibition of oxidation. Therefore, only a suitable content of Cu2+ will allow optimal catalytic performance.58
The above-mentioned characterization studies and in situ technique shed light on the mechanism of the oxidation of CH4 to CH3OH mediated by anchored Cu regarding the activation of CH4 and the formation of CH3OH as shown in Fig. 4b, in which the Cu species plays multiple roles as the microenvironment factors for AuPd sites. Firstly, the incorporation of Cu modulates the AuPd electronic state, giving rise to enhanced adsorption of CH4 on AuPd NPs. Secondly, the presence of Cu regulates H2O2 reaction pathways. With increased Cu loadings, the generation of ˙OH is reduced, which weakens both the CH3OH formation and its overoxidation processes.
Conclusions
In summary, AuPd alloy NPs incorporated into Cu-modified MOF, namely AuPd@Cu-UiO-66x, have been fabricated and exhibit excellent activity and selectivity in the direct oxidation of CH4 to CH3OH in aqueous solution with H2O2 as an oxidant under mild conditions. The selectivity presents a volcano-type trend with increased Cu amount in the catalysts, among which AuPd@Cu-UiO-66100, featuring an optimized Cu2+ content, possesses the best CH3OH selectivity of 80.97%. A slight change of Cu content from 0.4 wt% in AuPd@UiO-660 to 1.4 wt% in AuPd@UiO-66100 significantly boosts the CH3OH selectivity by 48%. Mechanistic investigations reveal that the auxiliary Cu2+ on the MOF skeleton, acting as the microenvironment, has a profound influence on AuPd NPs for CH4 oxidation, where it not only modulates the AuPd electronic state to improve CH4 adsorption but also regulates the formation of ˙OH in H2O2 reaction pathways. This work provides significant insights into the intricate processes involved in CH4 oxidation toward optimized methanol selectivity, which would open an avenue to microenvironment modulation of active sites for enhanced catalysis.Author contributions
H.-L. J. conceived the idea and supervised the project. J. S. and M.-L. G. performed the experiments and collected the data. C. L. and Y. P. conducted the synchrotron radiation photoionization mass spectrometry experiments. D. Y. conducted the CH4 sorption isotherm experiments. H.-L. J., Z. M., and J. S. co-wrote the manuscript. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2021YFA1500402), the NSFC (U22A20401, 22161142001, and 22101269), the International Partnership Program of CAS (123GJHZ2022028MI), and the Fundamental Research Funds for the Central Universities (WK3450000007 and WK2060000038). We thank the 1W1B station for XAFS measurement at the Beijing Synchrotron Radiation Facility (BSRF).References
- P. Schwach, X. Pan and X. Bao, Chem. Rev., 2017, 117, 8497–8520 CrossRef CAS PubMed.
- P. Tang, Q. Zhu, Z. Wu and D. Ma, Energy Environ. Sci., 2014, 7, 2580–2591 RSC.
- D. Saha, H. A. Grappe, A. Chakraborty and G. Orkoulas, Chem. Rev., 2016, 116, 11436–11499 CrossRef CAS PubMed.
- P. Tomkins, M. Ranocchiari and J. A. van Bokhoven, Acc. Chem. Res., 2017, 50, 418–425 CrossRef CAS PubMed.
- Z. Jin, L. Wang, E. Zuidema, K. Mondal, M. Zhang, J. Zhang, C. Wang, X. Meng, H. Yang, C. Mesters and F. S. Xiao, Science, 2020, 367, 193–197 CrossRef CAS PubMed.
- X. Meng, X. Cui, N. P. Rajan, L. Yu, D. Deng and X. Bao, Chem, 2019, 5, 2296–2325 CAS.
- Z. Liu, E. Huang, I. Orozco, W. Liao, R. M. Palomino, N. Rui, T. Duchon, S. Nemsak, D. C. Grinter, M. Mahapatra, P. Liu, J. A. Rodriguez and S. D. Senanayake, Science, 2020, 368, 513–517 CrossRef CAS PubMed.
- Y. Zhou, L. Zhang and W. Wang, Nat. Commun., 2019, 10, 506 CrossRef CAS PubMed.
- M. Ravi, V. L. Sushkevich, A. J. Knorpp, M. A. Newton, D. Palagin, A. B. Pinar, M. Ranocchiari and J. A. van Bokhoven, Nat. Catal., 2019, 2, 485–494 CrossRef CAS.
- C. Hammond, M. M. Forde, M. H. Ab Rahim, A. Thetford, Q. He, R. L. Jenkins, N. Dimitratos, J. A. Lopez-Sanchez, N. F. Dummer, D. M. Murphy, A. F. Carley, S. H. Taylor, D. J. Willock, E. E. Stangland, J. Kang, H. Hagen, C. J. Kiely and G. J. Hutchings, Angew. Chem., Int. Ed., 2012, 51, 5129–5133 CrossRef CAS PubMed.
- S. J. Freakley, N. Dimitratos, D. J. Willock, S. H. Taylor, C. J. Kiely and G. J. Hutchings, Acc. Chem. Res., 2021, 54, 2614–2623 CrossRef CAS PubMed.
- N. Agarwal, S. J. Freakley, R. U. McVicker, S. M. Althahban, N. Dimitratos, Q. He, D. J. Morgan, R. L. Jenkins, D. J. Willock, S. H. Taylor, C. J. Kiely and G. J. Hutchings, Science, 2017, 358, 223–227 CrossRef CAS PubMed.
- S. Bai, Y. Xu, P. Wang, Q. Shao and X. Huang, ACS Catal., 2019, 9, 6938–6944 CrossRef CAS.
- C. Williams, J. H. Carter, N. F. Dummer, Y. K. Chow, D. J. Morgan, S. Yacob, P. Serna, D. J. Willock, R. J. Meyer, S. H. Taylor and G. J. Hutchings, ACS Catal., 2018, 8, 2567–2576 CrossRef CAS.
- Y. Xing, Z. Yao, W. Li, W. Wu, X. Lu, J. Tian, Z. Li, H. Hu and M. Wu, Angew. Chem., Int. Ed., 2021, 60, 8889–8895 CrossRef CAS PubMed.
- R. Serra-Maia, F. M. Michel, Y. Kang and E. A. Stach, ACS Catal., 2020, 10, 5115–5123 CrossRef CAS.
- H. C. Zhou and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5415–5418 RSC.
- B. Li, H. M. Wen, Y. Cui, W. Zhou, G. Qian and B. Chen, Adv. Mater., 2016, 28, 8819–8860 CrossRef CAS PubMed.
- L. Jiao, J. Wang and H.-L. Jiang, Acc. Mater. Res., 2021, 2, 327–339 CrossRef CAS.
- L. Z. Dong, L. Zhang, J. Liu, Q. Huang, M. Lu, W. X. Ji and Y. Q. Lan, Angew. Chem., Int. Ed., 2020, 59, 2659–2663 CrossRef CAS PubMed.
- A. Aijaz, A. Karkamkar, Y. J. Choi, N. Tsumori, E. Ronnebro, T. Autrey, H. Shioyama and Q. Xu, J. Am. Chem. Soc., 2012, 134, 13926–13929 CrossRef CAS PubMed.
- X. Li, T. W. Goh, L. Li, C. Xiao, Z. Guo, X. C. Zeng and W. Huang, ACS Catal., 2016, 6, 3461–3468 CrossRef CAS.
- Q. Yang, Q. Xu and H.-L. Jiang, Chem. Soc. Rev., 2017, 46, 4774–4808 RSC.
- F. Chen, K. Shen, J. Chen, X. Yang, J. Cui and Y. Li, ACS Cent. Sci., 2019, 5, 176–185 CrossRef CAS PubMed.
- D. Chen, W. Yang, L. Jiao, L. Li, S. H. Yu and H.-L. Jiang, Adv. Mater., 2020, 32, e2000041 CrossRef PubMed.
- J. Baek, B. Rungtaweevoranit, X. Pei, M. Park, S. C. Fakra, Y. S. Liu, R. Matheu, S. A. Alshmimri, S. Alshehri, C. A. Trickett, G. A. Somorjai and O. M. Yaghi, J. Am. Chem. Soc., 2018, 140, 18208–18216 CrossRef CAS PubMed.
- J. Zheng, J. Ye, M. A. Ortuno, J. L. Fulton, O. Y. Gutierrez, D. M. Camaioni, R. K. Motkuri, Z. Li, T. E. Webber, B. L. Mehdi, N. D. Browning, R. L. Penn, O. K. Farha, J. T. Hupp, D. G. Truhlar, C. J. Cramer and J. A. Lercher, J. Am. Chem. Soc., 2019, 141, 9292–9304 CrossRef CAS PubMed.
- J. Sui, H. Liu, S. Hu, K. Sun, G. Wan, H. Zhou, X. Zheng and H.-L. Jiang, Adv. Mater., 2022, 34, e2109203 CrossRef PubMed.
- C. Xu, Y. Pan, G. Wan, H. Liu, L. Wang, H. Zhou, S. H. Yu and H.-L. Jiang, J. Am. Chem. Soc., 2019, 141, 19110–19117 CrossRef CAS PubMed.
- A. M. Abdel-Mageed, B. Rungtaweevoranit, M. Parlinska-Wojtan, X. Pei, O. M. Yaghi and R. J. Behm, J. Am. Chem. Soc., 2019, 141, 5201–5210 CrossRef CAS PubMed.
- Y. Deng, A. D. Handoko, Y. Du, S. Xi and B. S. Yeo, ACS Catal., 2016, 6, 2473–2481 CrossRef CAS.
- Y. Jiao, Y. Liu, G. Zhu, J. T. Hungerford, S. Bhattacharyya, R. P. Lively, D. S. Sholl and K. S. Walton, J. Phys. Chem. C, 2017, 121, 23471–23479 CrossRef CAS.
- F. Ahmadijokani, R. Mohammadkhani, S. Ahmadipouya, A. Shokrgozar, M. Rezakazemi, H. Molavi, T. M. Aminabhavi and M. Arjmand, Chem. Eng. J., 2020, 399, 125346 CrossRef CAS.
- P. Wang, B. Wang, Y. Liu, L. Li, H. Zhao, Y. Chen, J. Li, S. F. Liu and K. Zhao, Angew. Chem., Int. Ed., 2020, 59, 23100–23106 CrossRef CAS PubMed.
- L. Lin, J. Liu, X. Liu, Z. Gao, N. Rui, S. Yao, F. Zhang, M. Wang, C. Liu, L. Han, F. Yang, S. Zhang, X. D. Wen, S. D. Senanayake, Y. Wu, X. Li, J. A. Rodriguez and D. Ma, Nat. Commun., 2021, 12, 6978 CrossRef CAS PubMed.
- H. Wang, X. Liu, W. Yang, G. Mao, Z. Meng, Z. Wu and H.-L. Jiang, J. Am. Chem. Soc., 2022, 144, 22008–22017 CrossRef CAS PubMed.
- J. Long, H. Liu, S. Wu, S. Liao and Y. Li, ACS Catal., 2013, 3, 647–654 CrossRef CAS.
- G. Xu, A. Yu, Y. Xu and C. Sun, Catal. Commun., 2021, 158, 106338 CrossRef CAS.
- W. Wen, S. Yu, C. Zhou, H. Ma, Z. Zhou, C. Cao, J. Yang, M. Xu, F. Qi, G. Zhang and Y. Pan, Angew. Chem., Int. Ed., 2020, 59, 4873–4878 CrossRef CAS PubMed.
- Y. Liu, F. M. Kirchberger, S. Muller, M. Eder, M. Tonigold, M. Sanchez-Sanchez and J. A. Lercher, Nat. Commun., 2019, 10, 1462 CrossRef PubMed.
- J. Xu, T. White, P. Li, C. He, J. Yu, W. Yuan and Y. F. Han, J. Am. Chem. Soc., 2010, 132, 10398–10406 CrossRef CAS PubMed.
- X. Zhu, Q. Guo, Y. Sun, S. Chen, J. Q. Wang, M. Wu, W. Fu, Y. Tang, X. Duan, D. Chen and Y. Wan, Nat. Commun., 2019, 10, 1428 CrossRef PubMed.
- W. Guan, Y. Zhang, C. Yan, Y. Chen, Y. Wei, Y. Cao, F. Wang and P. Huo, ChemSusChem, 2022, 15, e202201041 CrossRef CAS PubMed.
- W. Zhan, Q. He, X. Liu, Y. Guo, Y. Wang, L. Wang, Y. Guo, A. Y. Borisevich, J. Zhang, G. Lu and S. Dai, J. Am. Chem. Soc., 2016, 138, 16130–16139 CrossRef CAS PubMed.
- Y. Qiu, L. Xin, Y. Li, I. T. McCrum, F. Guo, T. Ma, Y. Ren, Q. Liu, L. Zhou, S. Gu, M. J. Janik and W. Li, J. Am. Chem. Soc., 2018, 140, 16580–16588 CrossRef CAS PubMed.
- N. E. Kolli, L. Delannoy and C. Louis, J. Catal., 2013, 297, 79–92 CrossRef CAS.
- E. K. Gibson, A. M. Beale, C. R. A. Catlow, A. Chutia, D. Gianolio, A. Gould, A. Kroner, K. M. H. Mohammed, M. Perdjon, S. M. Rogers and P. P. Wells, Chem. Mater., 2015, 27, 3714–3720 CrossRef CAS.
- M. H. Ab Rahim, R. D. Armstrong, C. Hammond, N. Dimitratos, S. J. Freakley, M. M. Forde, D. J. Morgan, G. Lalev, R. L. Jenkins, J. A. Lopez-Sanchez, S. H. Taylor and G. J. Hutchings, Catal. Sci. Technol., 2016, 6, 3410–3418 RSC.
- R. Serra-Maia, F. M. Michel, T. A. Douglas, Y. Kang and E. A. Stach, ACS Catal., 2021, 11, 2837–2845 CrossRef CAS.
- Y. Fan, W. Zhou, X. Qiu, H. Li, Y. Jiang, Z. Sun, D. Han, L. Niu and Z. Tang, Nat. Sustain., 2021, 4, 509–515 CrossRef.
- H. Prats, R. A. Gutierrez, J. J. Pinero, F. Vines, S. T. Bromley, P. J. Ramirez, J. A. Rodriguez and F. Illas, J. Am. Chem. Soc., 2019, 141, 5303–5313 CrossRef CAS PubMed.
- Z. Liu, P. Lustemberg, R. A. Gutierrez, J. J. Carey, R. M. Palomino, M. Vorokhta, D. C. Grinter, P. J. Ramirez, V. Matolin, M. Nolan, M. V. Ganduglia-Pirovano, S. D. Senanayake and J. A. Rodriguez, Angew. Chem., Int. Ed., 2017, 56, 13041–13046 CrossRef CAS PubMed.
- X. Chen, Y. Li, X. Pan, D. Cortie, X. Huang and Z. Yi, Nat. Commun., 2016, 7, 12273 CrossRef CAS PubMed.
- H. Song, X. Meng, S. Wang, W. Zhou, S. Song, T. Kako and J. Ye, ACS Catal., 2020, 10, 14318–14326 CrossRef CAS.
- Q.-L. Zhu and Q. Xu, Chem, 2016, 1, 220–245 CAS.
- X. Cui, H. Li, Y. Wang, Y. Hu, L. Hua, H. Li, X. Han, Q. Liu, F. Yang, L. He, X. Chen, Q. Li, J. Xiao, D. Deng and X. Bao, Chem, 2018, 4, 1902–1910 CAS.
- W. Zhao, Y. Shi, Y. Jiang, X. Zhang, C. Long, P. An, Y. Zhu, S. Shao, Z. Yan, G. Li and Z. Tang, Angew. Chem., Int. Ed., 2021, 60, 5811–5815 CrossRef CAS PubMed.
- A. Barnes, R. J. Lewis, D. J. Morgan, T. E. Davies and G. J. Hutchings, Catalysts, 2022, 12, 1396 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials and instrumentation, experimental section, and supplemental figures and tables. See DOI: https://doi.org/10.1039/d3ta03712f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |