DOI:
10.1039/D3TA03055E
(Paper)
J. Mater. Chem. A, 2023,
11, 18740-18754
An interfacially stacked covalent porous polymer on graphene favors electronic mobility: ensuring accelerated oxygen reduction reaction kinetics by an in situ study†
Received
23rd May 2023
, Accepted 15th August 2023
First published on 16th August 2023
Abstract
The oxygen reduction reaction (ORR) is largely influenced by material conductivity as well as electron transfer mobility. Usually, covalent porous polymers are fascinating in terms of the surface area and availability of abundant functionalities serving as active sites. However, this class of materials largely suffers from electronic conductivity issues, which limits their extensive application in electrocatalytic reactions. To overcome this long-standing issue, herein, we have developed a metallo [Fe(II)]-porphyrin-pyrene based pi-conjugated porous polymer (FePP), which was further modified with electrophoretically exfoliated graphene (FePP@G30/3/7). The interfacing of these two units via π–π interaction introduces the flexibility of >C–C< bond rotation resulting in-plane flipping of the bridging –Ph ring from out of plane orientation. The ring flipping-induced co-planarity was investigated through experimental and computational studies. In situ FTIR and operando Raman studies reveal that the ORR process with the FePP@G30/3/7 catalyst follows a 4e− reduction pathway. This phenomenon drives axial as well as equatorial charge mobility within the system influencing the active FeN4 site toward lowering the overpotential for the ORR.
Introduction
Metalloporphyrin plays a vital role in energy conversion, electron transfer and O2 activation in nature leading to the oxygen reduction reaction (ORR) to produce water and energy.1–3 To mimic the biological process, the scientific community has been trying to perform O2-activation utilizing synthetic Fe-porphyrins (FePs) and derivative-based heterogeneous catalysts.4–7 However, the sluggish reaction kinetics of the ORR is the main gridlock of energy conversion and storage devices like fuel cells and metal–air batteries (MABs).8,9 Polymeric network/framework based molecular heterogeneous electrocatalysts are considered to have the potential to replace Pt-based noble metal catalysts, and other Pt-rich materials that are used as electrocatalysts in fuel cells and MABs.10 A few obstructions of metalloporphyrin-based porous electrocatalysts like covalent organic frameworks (COFs), metal–organic frameworks (MOFs), porous organic polymers (POPs), hyper crosslinked polymers (HCPs), conjugated microporous polymers (CMPs) and conjugated porous polymers (CPPs) are (1) impoverished stability for a long run, (2) metal leaching from the framework in the electrolyte, (3) poor electrical conductivity and thus less availability of true active sites for the catalysis and (4) immobilization of the catalyst to the secondary conductive element.11,12 To address these issues, several strategies have been taken into consideration in the recent past. They include (a) making composites with carbon black or other nanocarbon materials,13 (b) charge transport through redox hooping,14 (c) non-covalent functionalization via π-stacking,15,16 and (d) chemical functionalization in a conductive matrix.17,18
In nature, FeP heme sites act as a catalyst for O2 binding and its activation, followed by reduction. However, although the binding site is the heme unit, the activity of the catalyst is governed by the axial ligands and surrounding environments. Therefore, synthetic FeP-based catalysts can be systematically engineered based on the peripheral substituents, which can play an important role in regulating the electronic modulation, charge transport and proton relays during the ORR process. Recently, Cao and co-workers have shown that due to the imidazole moiety that is coordinated to the ‘Fe’ center of FeP-based catalysts from the axial position, an electron surge occurs over Fe from the axial direction, which facilitates O2 activation.14 Similar electronic modulation could be made dominant along the equatorial axis via push–pull electronic interactions of π–electrons through a planar π-conjugated network where one of the axial positions over the metal active site remains vacant to interact with O2.19 To address this challenge, the pyrene unit can be functionalized with a FeP unit as it contains highly delocalized pi–electron cloud that can act as a potential electron donor.20 Therefore, FeP moieties together with pyrene (FePP: Fe(II)-porphyrin-pyrene) containing pi-conjugated materials have the proficiency to increase the electron density on the metal site through push–pull interaction, which may play a significant role in the ORR.
In the recent past, several methodologies have been adopted to convert an FeP-based molecular catalyst to a heterogeneous catalyst including (a) drop-casting of the catalyst into a conductive support, (b) integration of the molecular catalyst into framework/polymer materials and (c) grafting of the catalyst on conductive carbon materials.21–27 Electrophoretic exfoliation is a facile, in situ and environmentally friendly technique to produce highly stable and easily grafted catalysts onto graphene layers. Conventional techniques, like chemical vapour deposition (CVD), chemical method, and mechanical exfoliation are multi-step processes, where in the first step, graphene/reduced graphene oxide has been synthesized followed by loading the catalyst on the graphene surface. Furthermore, these methods have several limitations such as requiring high temperature, long time, being difficult to scale up, low yield, and sometimes involving hazardous reagents. In contrast, electrophoretic exfoliation is an in situ synthesis technique, where functionalization of graphene sheets can be obtained via a single-step method by exfoliating graphite in the presence of a catalyst by π–π interaction in aqueous medium. Moreover, this process can be easily scaled up, and minimize the oxygen content in the hybrid material and the pristine structure is retained to realize on-demand properties in the desired materials.28–30
Bearing all these parameters in mind during the engineering and designing of the material, in this work, we focused not only on the improvement of electrical conductivity, but also on the role of charge-transfer modulation through push–pull interaction via a very facile green synthetic approach and cost-effective protocol for sustainable heterogeneous electrocatalysis. Herein, we have synthesized a functionalized π-conjugated Fe(II)-porphyrin-pyrene (FePP) based 2D porous polymer material through the Suzuki cross coupling reaction and further anchored the material on a graphene backbone via in situ electrophoretic exfoliation of graphite for an economically viable electrocatalyst for the ORR. In this case, the pyrene unit in the network plays a dual role. First, it can act as a donor and provides pi–electron density towards the FeP unit via push–pull electronic interaction when O2 binds to the heme unit during the ORR. Secondly, the introduction of the pyrene unit into the FeP-based networks increases the efficiency of 2D–2D π–π interactions of the matrix with graphene. More interestingly, the pi-conjugated extended polymeric network contains a freely rotating –Ph ring that attains co-planarity via ring-flipping when modified with graphene through 2D–2D π–π interaction and accelerates the ORR kinetics. The FePP catalyst after grafting with electrophoretically exfoliated graphene, FePP@G30/3/7, is able to simultaneously regulate the charge transport and electron mobility as well as generating favorable active sites for oxygen reduction. An electrochemical ORR study was exclusively performed using both the materials (FePP and FePP@G30/3/7) along with a commercial Pt/C catalyst and the improvement in the performance of half-wave potential, current density, reaction kinetics, impedance, stability and methanol tolerance was explored. This study further explored some interesting properties of the as synthesized electrocatalyst, like 2D–2D π–π stacking interaction, floating of the free rotation of the –Ph ring inducing a symmetric reorientation of the perpendicularly aligned p-orbitals for an extended pi-conjugation among donor–acceptor units, superior conductivity of the catalyst and the axial position stacked with graphene sheets pushing electrons towards metal active sites. Both the equatorial and axial positions play a significant role in the ORR, which is well proven through experimental as well as theoretical support.
Experimental details
Catalyst preparation
A donor–acceptor palladium (0) catalyzed Suzuki cross coupling reaction between 5,10,15,20-(tetra-4-bromo-phenyl) porphyrin Fe(II) (FeP) and 1,3,6,8 tetrakis-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl) pyrene (PyTBE) was employed to synthesize a conjugated microporous polymer (CPP) namely FePP. All experimental procedures for the synthesis of different monomers, associated materials and electrochemical techniques are described in Section 1 of the ESI.† Also, the NMR study revealing the step-wise advancement of the reaction and the formation of monomers PhTBE, Br-porphyrin and FeP was described and highlighted in the synthesis part of the ESI and is shown in Fig. S1–S3,† respectively. Thereafter, the as synthesized FePP was further modified with graphene through a facile electrophoretic exfoliation technique (details schematically presented in Fig. S4†) under acidic media to obtain the electrocatalyst, namely FePP@Gmg/V/t (here mg, V and t denote the amount of FePP (mg), voltage (V) and time (t) used for the exfoliation).31 The striking advantage of the underlying graphene support with the FePP material has been brought to the front by several experimental techniques, which was finally corroborated with the electrocatalytic performances of the catalysts, as discussed in the Results and discussion part.
Formula used for conversion to standard electrode potential.
All the experiments were performed using a three electrode system, a GC/RDE/RRDE as a working electrode, a graphite rod as a counter electrode and an Ag/AgCl (3 M KCl) reference electrode and potentials were converted to standard RHE (reversible hydrogen electrode) potential using (eqn (1)) as reported in this manuscript. For 0.1 M KOH (pH > 13), the following equation was considered:| | | ERHE (V) = EAg/AgCl (3 M KCl) (in V) + (0.059 × pH) + 0.210 V | (1) |
The other formulae for calculation of the number of electrons and H2O2 (% yield) involved during the ORR in alkaline medium are briefly discussed in the ESI File.† The electrochemical surface area calculation equation is also discussed in ESI eqn (7).†
Computational details
All the calculations are performed in the Vienna Ab initio Simulation Package (VASP) using non-spin polarized density functional theory. The Projected Augmented Wave (PAW) method is used to describe the potentials of the atoms with Generalized Gradient Approximation (GGA) considered for exchange and correlation effects at the Perdew–Burke–Ernzerhof (PBE) level. A plane wave cut-off energy of 450 eV is used for the calculations. A gamma centered 3 × 3 × 1 K-point grid is used for the Brillouin zone sampling. Structural optimization was done until the total energy converged to less than 10−5 eV per atom and the maximum force converged to less than 0.01 eV Å−1. Grimme's DFT-D2 method was used for van der Waals dispersion correction. Henkelman's Bader charge analysis is used for estimating the charges on the atoms. The Fe-porphyrin-pyrene (FePP) moiety is modelled with the ends of the carbon atoms terminated with hydrogen atoms. A 9 × 9 graphene supercell and graphene flakes with 10 hexagonal rings are taken as a substrate for FePP. For simplicity and computational cost, two layers of graphene are taken. To avoid any periodic interaction along the z-axis, a vacuum of ∼15 Å is taken. The projected density of states (PDOS) of the d-orbital of FePP, FePP on a single graphene sheet (Ph-ring out of plane) and FePP between a fixed graphene sheet and flake (FePP@G30/3/7) is discussed in ESI Section 3 and Fig. S27.†
Results and discussion
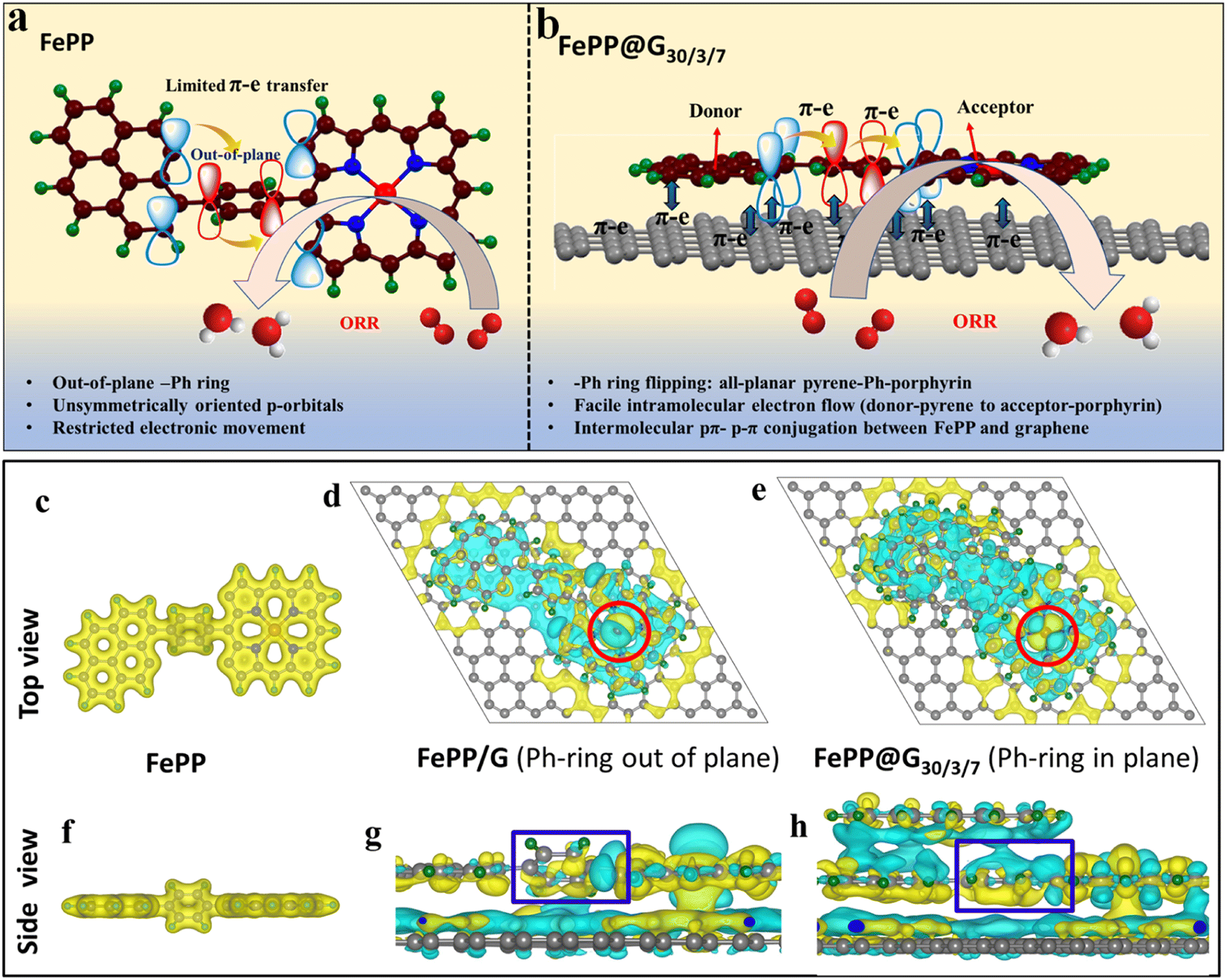
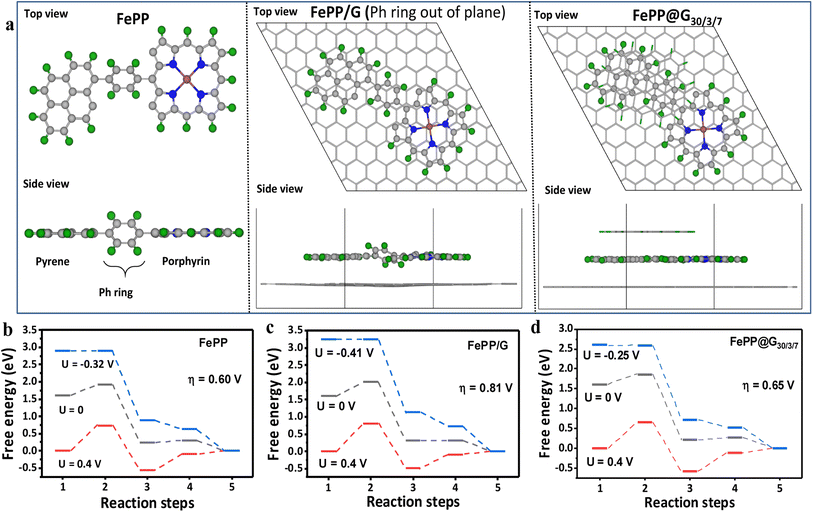
This work mainly aims to establish the electronic mobility of the electrocatalyst and electronic transition operating at the active sites from the molecular orbital perspective, which benignly participates in an improved ORR performance of FePP@G30/3/7. It is however known that the electrocatalytic processes require an electron sufficiency, which could be provided by the adsorbents for the subsequent reduction reaction. Fe-porphyrin is immensely important for the ORR. Despite having an FeN4 active unit, it lags in performance owing to the conductivity issue. To overcome this, we have adopted an extremely facile approach by conjugating the FePP system with a graphene support. The pi-conjugated graphene framework induces –Ph ring flipping of the FePP unit to achieve an all-planar 2D porous material. This causes the re-orientation of the perpendicularly aligned vacant p-orbitals of the constituting elements adopting an all-symmetric orientation. As a consequence, a continuous electron flow is achieved along the equatorial plane of FePP along with an appropriate π–π conjugation axially, between graphene and FePP as shown in Fig. 1a and b. In order to check the interaction between graphene and the FePP moiety, a charge density distribution study was conducted and the charge density difference analysis for FePP and FePP@G30/3/7 is demonstrated in Fig. 1c–hvia top and side views. The charge density distribution plots of FePP top and side views shown in Fig. 1c and f explained that when the –Ph ring was out of plane, the Fe atom has less charge on top. After exfoliation, we assumed two models FePP/G (where we kept the –Ph ring out of plane) and FePP@G30/3/7 (where we obtained an energy optimized structure with the –Ph ring flipping to an in-plane orientation). In FePP/G, when the –Ph ring was out of plane, the Fe atom has a less charge accumulated on FeN4 between graphene sheets, which suggested that the orientation of FePP did not support the axial charge transfer between the FePP and graphene moieties. Meanwhile for FePP@G30/3/7, there was more charge accumulated on FeN4 between graphene sheets, indicating better charge transfer and thus binding. Secondly, when the Ph ring was out of plane the charge on the Fe atom is depleted and for the in-plane case there is alternate charge accumulation and depletion lobes present on the Fe atom (red circle region in Fig. 1d and e). Also, the charge on the –Ph ring from graphene is more when the ring is in-plane (blue box regions in Fig. 1g and h). These analyses thus showed the importance of the in/out of plane orientation of the Ph ring for better charge transfer characteristics of the material and thereby improved catalytic activity. It can be inferred that, when the Ph ring is planar better charge transfer occurred that could aid in better binding of the OOH* intermediates to the charge sufficient Fe atom, which is usually the PDS and therefore it is expected to initiate the ORR at a much less overpotential.32–34
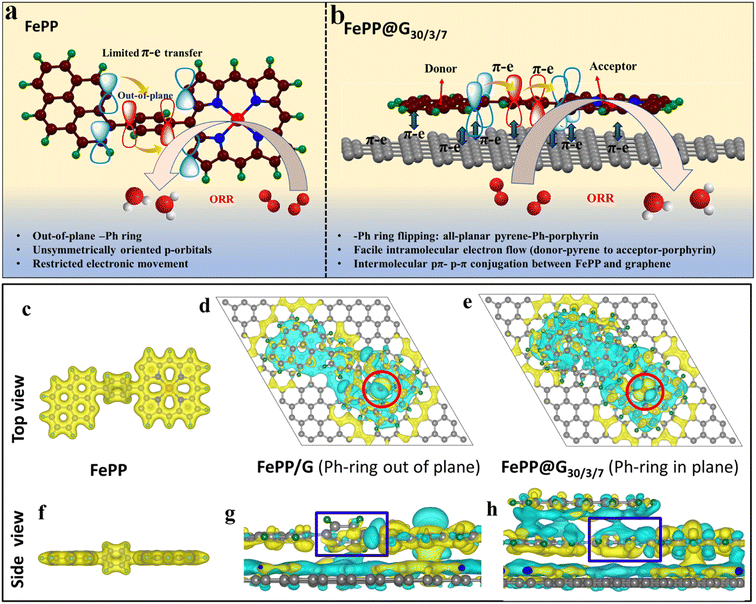 |
| | Fig. 1 Schematic representation of the limitations with (a) FePP and (b) FePP–graphene interaction induced electron mobility in FePP@G30/3/7. Charge density and charge density difference plots of (c) an FePP flake covering pyrene and the Ph ring with the Ph ring out of plane, (d) FePP on a single graphene sheet (Ph-ring out of plane), and (e) FePP between a fixed graphene sheet and flake, where the Ph ring is completely in-plane. (Top view) and (f–h) the same from the side view of FePP, FePP on a single graphene sheet (Ph-ring out of plane), and FePP between a fixed graphene sheet and flake, where the Ph ring is completely in-plane, respectively. Isosurface value is 1.6 × 10−4 e Bohr−3. Yellow colored lobe indicates the charge-accumulated region and blue colored lobes indicate the charge-depleted region. Green, grey, blue and brown colored spheres represent hydrogen, carbon, nitrogen and iron atoms respectively. | |
Physical characterization of electrode materials
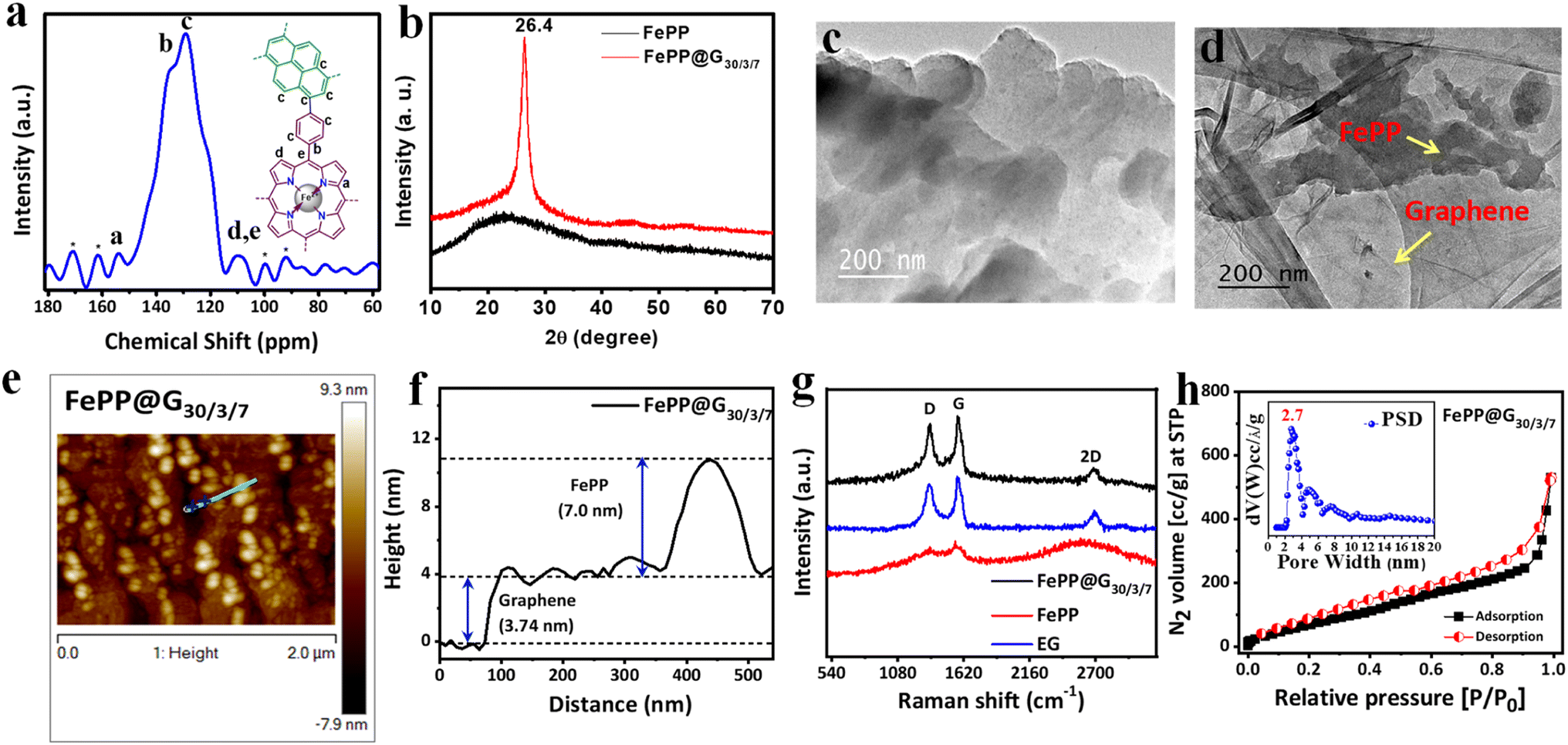
The formation of the conjugated porous polymer material (FePP) was confirmed through solid state 13C-nuclear magnetic resonance (NMR) analysis by assigning different chemical environments of carbon atoms present in the polymer network (Fig. 2a). The solid-state NMR peak for pyrrolic ‘C’ adjacent to the ‘N’ atom (a) in the porphyrin ring appeared at 154.2 ppm. NMR signals for carbon atoms (b and c) from the phenyl ring connected with the porphyrin ring and pyrene ring adjacent to the phenyl ring along with other aromatic carbons were shown at 135 and 129 ppm, respectively. The NMR peaks for another pyrrolic and aliphatic carbon atom of FePP designated as (d) and (e) appeared at 109 ppm.35 Furthermore, one-pot electrophoretic exfoliation of graphene from graphite in acidic medium was carried out in the presence of the FePP material to obtain the desired conductive material FePP@G30/3/7. To investigate the structural confirmation and amorphous or crystalline nature of the materials (FePP and FePP@G30/3/7), the powder X-ray diffraction (PXRD) pattern was recorded in the 2θ value range of 10 to 70° (Fig. 2b). In the PXRD pattern, a broad peak appeared for FePP in the range of 15 to 30°, which indicated the amorphous nature of the material. Meanwhile, FePP@G30/3/7 showed a peak with maxima at 26.4°, clearly indicating the merging of two PXRD peaks with each other, which established the formation of an FePP-loaded graphene-based 2D hybrid material FePP@G30/3/7.36 The morphology of both the electrocatalysts FePP and FePP@G30/3/7 has been studied using high-resolution transmission electron microscopy (HRTEM) analysis, presented in Fig. 2c and d, respectively, where a 2D sheet-like morphology was observed for FePP. In the HRTEM images of FePP@G30/3/7, a clear visualization of FePP on the graphene sheet confirmed the successful grafting of the porous polymer material into the exfoliated thin graphene sheet. The tapping-mode atomic force microscopy (AFM) technique was used to characterize the FePP@G30/3/7 catalyst, as illustrated in Fig. 2e. As expected, it has been observed that the FePP material is stacked onto the graphene sheets, which is shown in Fig. 2e. We have also measured the thickness of 2D sheet-like FePP@G30/3/7 and found it to be ∼3.74 nm for the graphene sheet and ∼7 nm for the FePP material as shown in Fig. 2f. Raman spectroscopic analysis was conducted to confirm the two-dimensional nature of the electrocatalysts, namely FePP@G30/3/7, FePP and exfoliated graphene (EG), as shown in Fig. 2g. In the Raman spectrum of FePP, D and G bands appeared at 1336 and 1574 cm−1 along with a broad peak in between 2200 and 3000 cm−1 with a maximum at 2698 cm−1, which revealed the 2D nature of the extended pi-conjugated polymer network. It is worth mentioning that the D and G bands had similar intensity with a calculated ID/IG ratio of 0.93. This indicated disorderness in the material, attributed to the presence of the FeN4 unit within the carbon framework of the porphyrin unit. The Raman spectrum of exfoliated graphene (EG) was also obtained and the ID/IG ratio was calculated to be 0.89 due to the distortions in the basal carbon framework. The Raman spectra of the FePP@G30/3/7 hybrid material resembled that of FePP with a sharp 2D band at 2600 cm−1 along with the very familiar D and G bands at 1341 and 1574 cm−1, respectively. A decrease in the ID/IG ratio to 0.87 in FePP@G30/3/7 compared to EG (0.89) indicated the proper grafting of FePP onto the graphene backbone to achieve a planar 2D sheet like architecture through pi–pi stacking between the FePP and exfoliated graphene sheets. A slightly lowering of defects in the hybrid FePP@G30/3/7 materials might be due to some structural transformation to achieve planarity from a non-planar phenyl ring connected with pyrene and iron-porphyrin moieties.37 These results are also summarized in Table T1 in the ESI.† However, the specific surface area and porosity of both the electrocatalysts were investigated through a volumetric N2 adsorption/desorption study at 77 K (Fig. 2h and ESI, S5†). According to the IUPAC (International Union of Pure and Applied Chemistry) classification, the obtained Brunauer–Emmett–Teller (BET) results of FePP and FePP@G30/3/7 belongs to Type-I isotherm followed by Type-IV isotherm. The specific surface area of FePP and FePP@G30/3/7 was 840 and 350 m2 g−1, respectively. After the exfoliation, the surface area might be decreased because of the strong interaction between the polymer network and graphene sheet in the hybrid materials. The pore size distribution (PSD) was calculated employing the NLDFT (non-local density functional theory) method on the N2 adsorption/desorption isotherm where the PSD is found to be 1.7 for FePP and 2.7 nm for FePP@G30/3/7, which are displayed in the inset of Fig. 2h and ESI S5.† The obtained average pore diameter of both the materials indicates that the FePP material possesses both micro- and meso-pores in its polymeric network, whereas the FePP@G30/3/7 material is mainly mesoporous in nature. The presence of highly accessible mesoporosity is very suitable for facile ion transportation that makes it a superior electrocatalyst. The mesoporosity with a moderately high surface area together with the metal coordinated to electronegative N atoms with extensive pi-conjugation and grafted on the conducting graphene sheet can make the material a worthy electrocatalyst for the ORR.
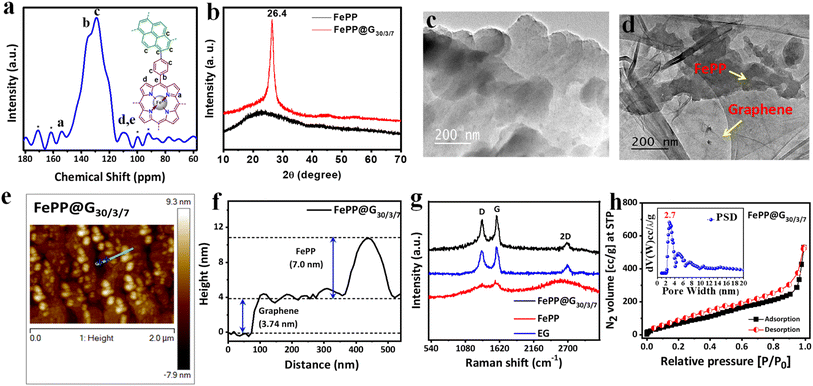 |
| | Fig. 2 (a) Solid state 13C-NMR spectra of the FePP catalyst. (b) XRD pattern of FePP and FePP@G30/3/7 catalysts. (c) HRTEM image of FePP and (d) FePP@G30/3/7 catalysts. (e) AFM image and (f) height profile of FePP@G30/3/7. (g) Raman spectra of FePP@G30/3/7, FePP and EG catalysts. (h) N2 adsorption/desorption isotherm with pore size distribution (inset) of the FePP@G30/3/7 catalyst. | |
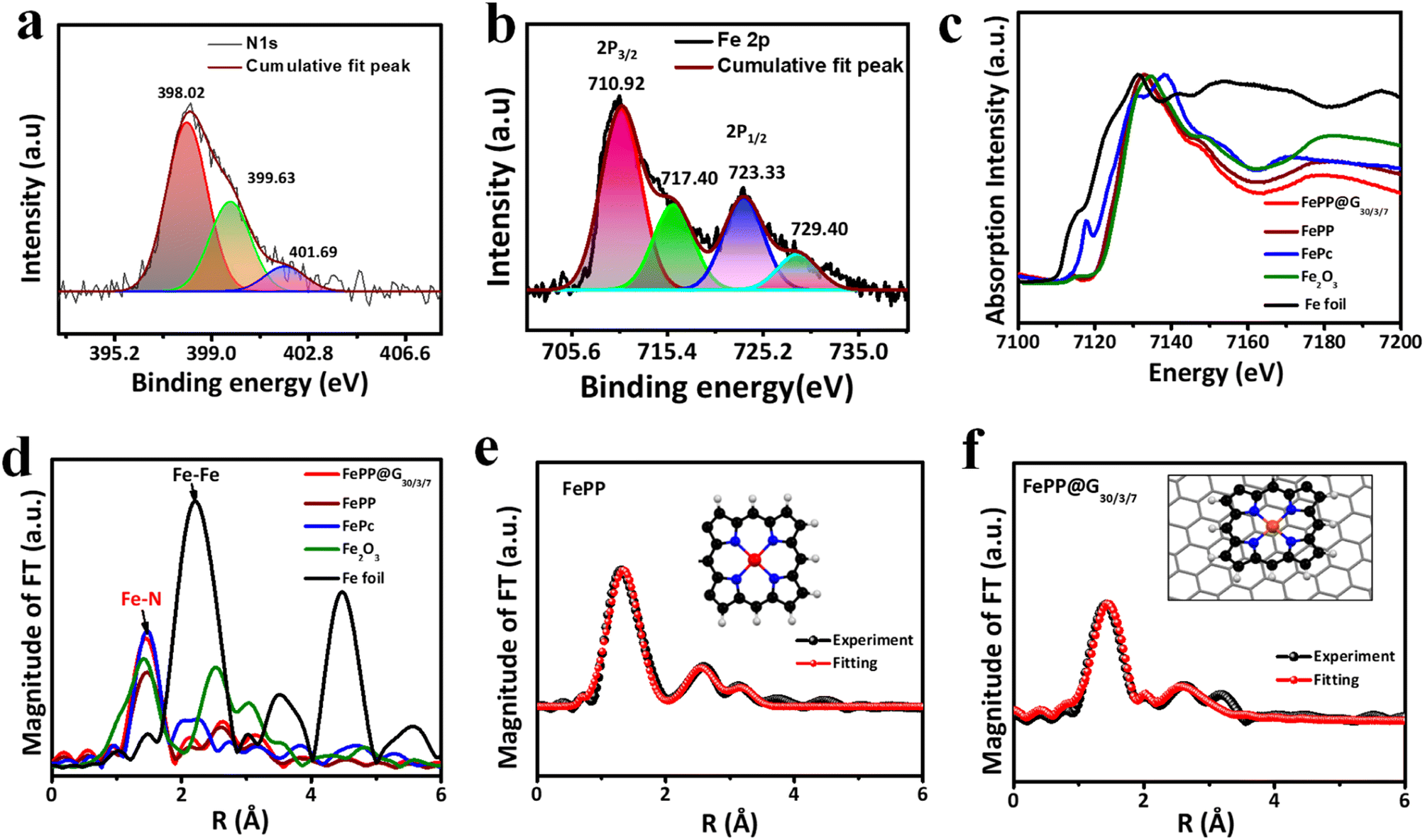
An insight into the surface chemical composition and valence states in both the materials (FePP and FePP@G30/3/7) was attained from X-ray photoelectron spectroscopy (XPS) analysis (Fig. 3a and b, ESI, S6 and S7†). The comparative survey spectra of FePP and FePP@G30/3/7 reveal that the carbon content in the latter is expectedly more than that of the former, owing to the underlying graphene support. The elemental composition of FePP@G30/3/7 and FePP catalysts is included in ESI, Table T2.† The deconvoluted peaks of C 1s spectra of both the materials FePP and FePP@G30/3/7 appeared at almost the same positions of 284.14 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C/C–C), 285.02 (CN) and 286.09 eV (ESI, Fig. S6a and S7b†). The deconvoluted N 1s spectra of both the materials also displayed similar peak positions at 398.01, 399.7, and 401.7 and 398.02, 399.63, and 401.69 eV for Fe–N, pyrrolic N, oxidized N in FePP and FePP@G30/3/7, respectively (Fig. 3a and ESI, S7c†). For the high-resolution Fe 2p spectra of FePP, intense peaks appeared at 711 and 723.6 eV along with a satellite peak (ESI, Fig. S7d†) corresponding to Fe 2p3/2 and Fe 2p1/2, respectively. Meanwhile for the FePP@G30/3/7 material two signals for the respective Fe 2p3/2 and Fe 2p1/2 appeared slightly shifted at 710.92 and 723.33 eV, respectively, along with the characteristic satellite peak (Fig. 3b).38 This confirms that the chemical composition of FePP remains unaffected after exfoliation, while the peak shifting again proved the pi–pi stacking of the FePP onto the graphene moiety suggesting that its valence electrons increased.
C/C–C), 285.02 (CN) and 286.09 eV (ESI, Fig. S6a and S7b†). The deconvoluted N 1s spectra of both the materials also displayed similar peak positions at 398.01, 399.7, and 401.7 and 398.02, 399.63, and 401.69 eV for Fe–N, pyrrolic N, oxidized N in FePP and FePP@G30/3/7, respectively (Fig. 3a and ESI, S7c†). For the high-resolution Fe 2p spectra of FePP, intense peaks appeared at 711 and 723.6 eV along with a satellite peak (ESI, Fig. S7d†) corresponding to Fe 2p3/2 and Fe 2p1/2, respectively. Meanwhile for the FePP@G30/3/7 material two signals for the respective Fe 2p3/2 and Fe 2p1/2 appeared slightly shifted at 710.92 and 723.33 eV, respectively, along with the characteristic satellite peak (Fig. 3b).38 This confirms that the chemical composition of FePP remains unaffected after exfoliation, while the peak shifting again proved the pi–pi stacking of the FePP onto the graphene moiety suggesting that its valence electrons increased.
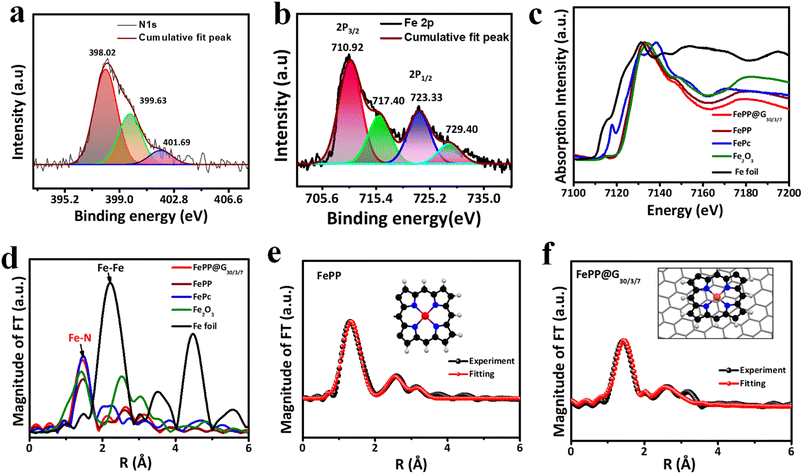 |
| | Fig. 3 High resolution deconvoluted (a) N 1s and (b) Fe 2p XPS spectra of the FePP@G30/3/7 catalyst. Structural characterization of Fe atoms in the FePP and FePP@G30/3/7 catalysts. The normalized XANES spectra and the Fourier transform of EXAFS spectra at the (c and d) Fe K-edge of the FePP and FePP@G30/3/7 catalysts and the reference materials. Corresponding EXAFS fitting of FePP and FePP@G30/3/7 catalysts at r space spectra in (e and f), and the inset shows a schematic structure of FeN4: Fe (red), N (blue), H (grey), and C (black). | |
To gain further insight into the noteworthy details of the chemical state and coordination environment of Fe atoms in the framework matrix, X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) spectroscopy were performed. The XANES spectra at the Fe K-edge for FePP@G30/3/7, FePP, and references, such as iron phthalocyanine (FePc) and Fe2O3, were collected to analyze their coordination states (Fig. 3c). The XANES spectra of FePP@G30/3/7 and FePP catalysts at the Fe K-edge were compared with those of the reference materials indicating that the absorption edge line position of both the catalysts were located between those of Fe foil (Fe(0)) and Fe2O3 (Fe3+) samples, suggesting that the Fe in FePP@G30/3/7 and FePP was in a +2 oxidation state, matching with the results obtained from the XPS analysis.39,40 The EXAFS of the Fe K-edge shows that the magnitude of the Fourier Transform (FT) spectra of the FePP and FePP@G30/3/7 samples also closely resembles that of the original FePc reference curve. The amplitude and shape of the first strong absorption peak located at 1.52 Å in the FT plot, which was equivalent to Fe–N scattering, indicate that the bonding environment in the first shell of FePP@G30/3/7 and FePP catalysts was the same as that of FePc suggesting that one Fe atom connects with four N atoms. In addition, there was no peak observed corresponding to Fe–Fe at 2.2 Å in both synthesized catalysts (Fig. 3d), reflecting the nonexistence of any iron particles or clusters in FePP and FePP@G30/3/7 catalysts. To gain better understanding, quantitative EXAFS fitting was performed to study the local chemical coordination environment around the Fe atom in FePP and FePP@G30/3/7 catalysts and fitting parameters, including atomic distances and coordination numbers, are arranged in ESI Table T3.† The first coordination shell fitting results for FePP (Fig. 3e) and FePP@G30/3/7 (Fig. 3f) indicate that the Fe–N shells have average distances in R-space of 2.04 and 1.98 Å, which correspond to the coordination numbers of 3.99 and 4.08, respectively. The fitting k3 space results of FePP and FePP@G30/3/7 catalysts are also shown in ESI Fig. S8 and S9.† These results are in agreement with the experimental data, and similar to Fe–N coordination environment present in the catalysts, where the Fe atom is coordinated with four nitrogen atoms in both FePP and FePP@G30/3/7 catalysts as shown in the insets of Fig. 3e and f.41,42
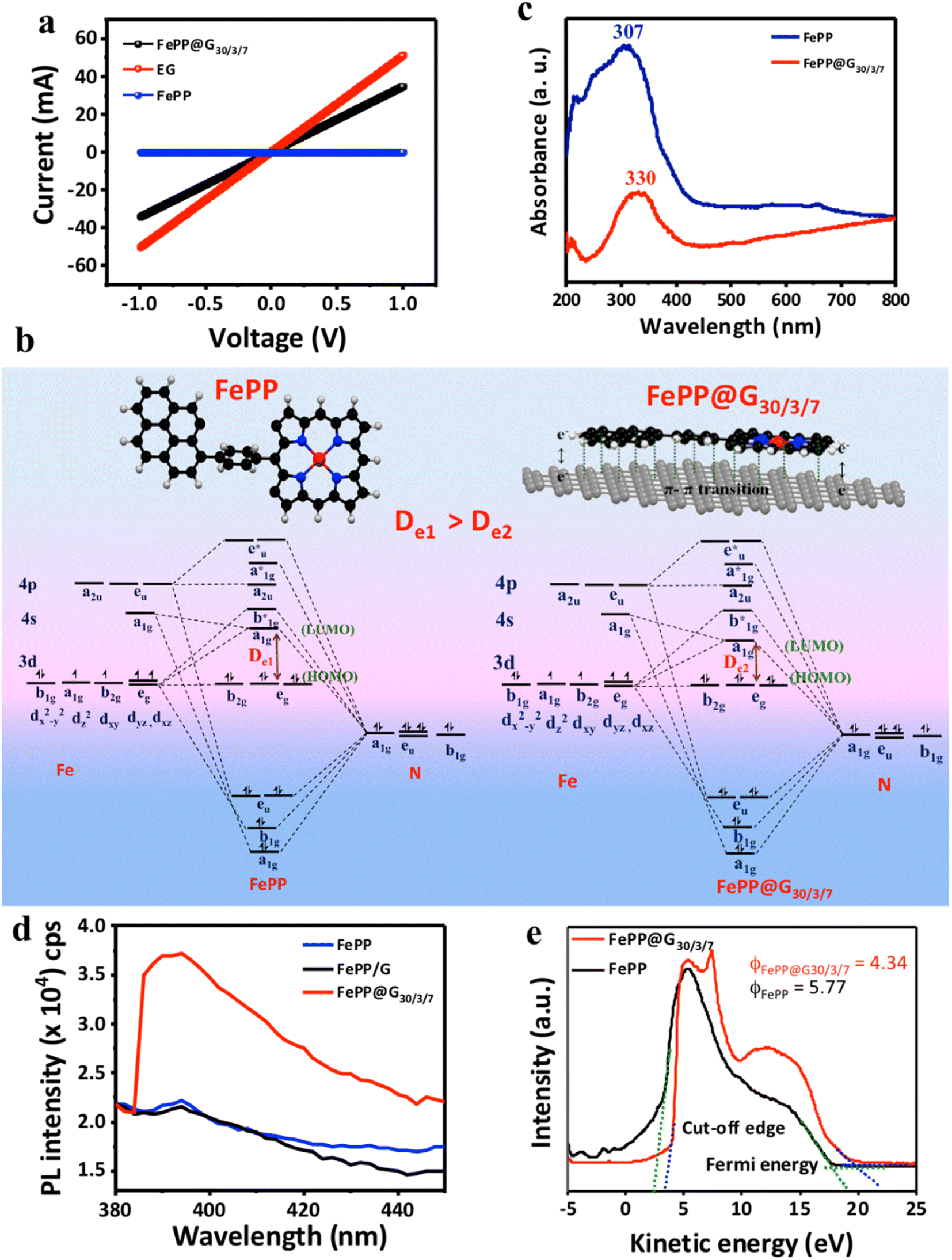
Furthermore, the electronically well-connected 2D framework of FePP@G30/3/7 was expected to be conductive enough as compared to EG. In order to check that, comparative sheet resistance was measured with all synthesized materials using the 2-probe technique by changing the bias from −1 to +1 volt in the device.43 The current–voltage (I–V) characteristics of FePP@G30/3/7 and EG show linear plots, indicating a higher slope as compared to the slope of the FePP material (Fig. 4a). The electrical sheet resistance of FePP@G30, EG and FePP catalysts is 28.90 Ω, 19.63 Ω and 1.976 × 107 Ω respectively. This impressively low resistance of FePP@G30/3/7 could result from an unattenuated and easy electronic transition throughout the conductive catalyst system (FePP@G30/3/7) with a low energy difference (De: delocalization energy) between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) (De1 > De2 as evident for FePP and FePP@G30/3/7, respectively) as demonstrated in Fig. 4b.44–46 This inference accorded well with the red-shifted peak at 330 nm corresponding to the π to π* transition in the ultra violet-visible (UV-vis) absorption spectrum of FePP@G30/3/7, while FePP displayed a peak at 307 nm (Fig. 4c).37 This red-shifted peak position in FePP@G30/3/7 could also be attributed to the partial recovery of conjugation with the graphene network and also a coupling effect of different functional groups on the surface of the graphene framework and FePP polymeric material. The –Ph ring flipping after stacking with graphene sheets produced the opportunity to intra- (from pyrene to FeP) and inter- (graphene–FePP) layer charge distribution and increased the number of active sites. Further confirmation of co-planarity and proper heterostructure formation of the FePP with graphene in the FePP@G30/3/7 catalyst was obtained with photoemission spectroscopy measurement. Solid-state photoluminescence (ss-PL) spectroscopy of FePP, FePP/G and FePP@G30/3/7 was performed as shown in Fig. 4d. All the materials were excited at 310 nm and emission was recorded in the range of 380–450 nm with 200 nm min−1 rates. The ss-PL spectrum of the FePP material shows very less emission at 395 nm, whereas upon introducing FePP into the graphene sheet (Fe-PP@G30/3/7) the emission intensity increases up to 36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 counts per second. This effect is ascribed to aggregation-induced emission (AIE) in the hybrid material.47,48 Upon formation of Fe-PP@G30/3/7via in situ electrophoretic exfoliation of graphene, the FePP unit sits between the graphene layers by maintaining planarity. This restricts the intermolecular rotation (RIR) of the introduced phenyl ring of FePP by increasing π–π stacking, resulting in inducing emission caused by aggregation. On the other hand, we have also checked the ss-PL of FePP/G material; the stacking of the phenyl ring was improper causing a rotated phenyl ring in the case of FePP/G. Upon exciting this material at the same wavelength, it shows very less PL activity may be due to the improper composite formation, which restricts the rotation of the phenyl ring. The observed results are well corroborated with UV-visible data. The π–π stacking interaction between graphene and FePP induces co-planarity in the FePP system, which has been thus confirmed from UV-visible, Raman and photoluminescence spectroscopy. Owing to this co-planarity of the –Ph ring, the π–electron mobility gets enhanced from the pyrene ring to the porphyrin ring through push–pull interaction, which is responsible for extended pi–electron delocalization through the polymeric network as well as increasing the electron density on the electroactive metal center (FeN4), which is further beneficial for the electrocatalytic performance of the catalyst. Therefore, the charge transfer mobility dramatically enhances the electrical conductivity of the system, or in other words lowers the electrical resistivity, providing accessible charge carriers, which has also been confirmed from the lowered work function of the FePP@G30/3/7 system (Φ = 4.34) as compared to FePP (Φ = 5.77) (Fig. 4e). The work function (Φ) values were analyzed by ultraviolet photoelectron spectroscopy (UPS) upon applying a −10 V bias voltage to obtain a sharp cut-off edge of the secondary electrons. Thus, our final material with a lowered work function is more prone to adsorb O2 on its surface with better accessible free charge carriers, which eventually lead to a low overpotential for the catalytic process. These factors directly impacted the kinetics of the ORR, which have been vividly discussed in the following section.
000 counts per second. This effect is ascribed to aggregation-induced emission (AIE) in the hybrid material.47,48 Upon formation of Fe-PP@G30/3/7via in situ electrophoretic exfoliation of graphene, the FePP unit sits between the graphene layers by maintaining planarity. This restricts the intermolecular rotation (RIR) of the introduced phenyl ring of FePP by increasing π–π stacking, resulting in inducing emission caused by aggregation. On the other hand, we have also checked the ss-PL of FePP/G material; the stacking of the phenyl ring was improper causing a rotated phenyl ring in the case of FePP/G. Upon exciting this material at the same wavelength, it shows very less PL activity may be due to the improper composite formation, which restricts the rotation of the phenyl ring. The observed results are well corroborated with UV-visible data. The π–π stacking interaction between graphene and FePP induces co-planarity in the FePP system, which has been thus confirmed from UV-visible, Raman and photoluminescence spectroscopy. Owing to this co-planarity of the –Ph ring, the π–electron mobility gets enhanced from the pyrene ring to the porphyrin ring through push–pull interaction, which is responsible for extended pi–electron delocalization through the polymeric network as well as increasing the electron density on the electroactive metal center (FeN4), which is further beneficial for the electrocatalytic performance of the catalyst. Therefore, the charge transfer mobility dramatically enhances the electrical conductivity of the system, or in other words lowers the electrical resistivity, providing accessible charge carriers, which has also been confirmed from the lowered work function of the FePP@G30/3/7 system (Φ = 4.34) as compared to FePP (Φ = 5.77) (Fig. 4e). The work function (Φ) values were analyzed by ultraviolet photoelectron spectroscopy (UPS) upon applying a −10 V bias voltage to obtain a sharp cut-off edge of the secondary electrons. Thus, our final material with a lowered work function is more prone to adsorb O2 on its surface with better accessible free charge carriers, which eventually lead to a low overpotential for the catalytic process. These factors directly impacted the kinetics of the ORR, which have been vividly discussed in the following section.
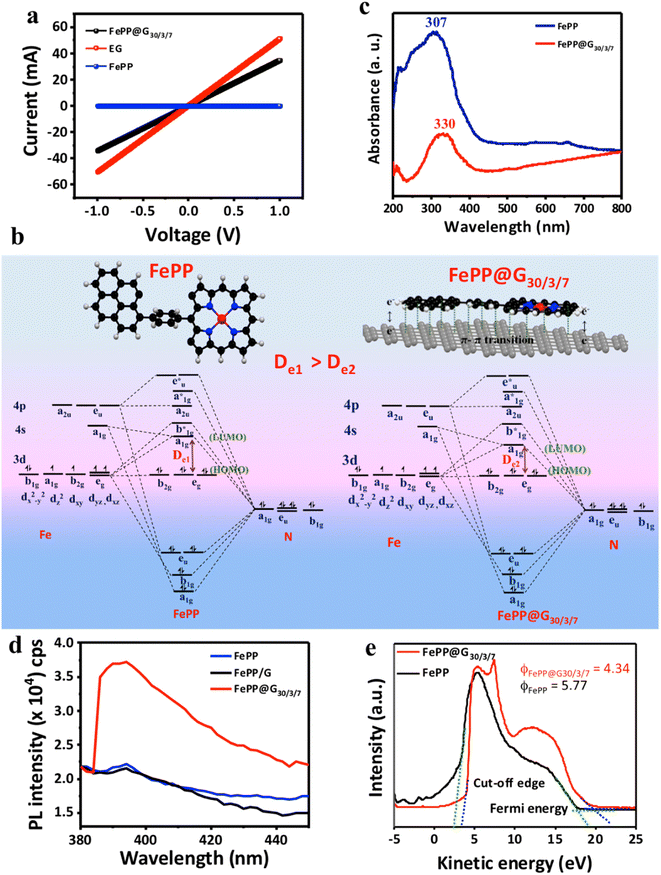 |
| | Fig. 4 (a) Electrical sheet resistance measurement (i–v) curve of FePP@G30/3/7, FePP and EG catalysts. (b) Molecular orbital diagram of FePP and FePP@G30/3/7 catalysts. (c) UV-visible absorbance spectra of FePP@G30/3/7 and FePP catalysts. (d) Solid-state photoluminescence spectra of FePP, FePP/G and FePP@G30/3/7. (e) The work function (Φ) of FePP@G30/3/7 and FePP catalysts. | |
These experimental results due to the –Ph ring flipping through 2D–2D interaction led to different kinds of changes of the structural and electrochemical properties of the materials, which are further supported by quantum chemical calculations for the lowering of overpotential towards the ORR. In this case, the coplanarity of the 2D layered FePP is gained with the co-planar graphene sheet through the rotation of the –Ph ring resulting in better electron mobility throughout the catalyst surface.
Electrocatalytic ORR study
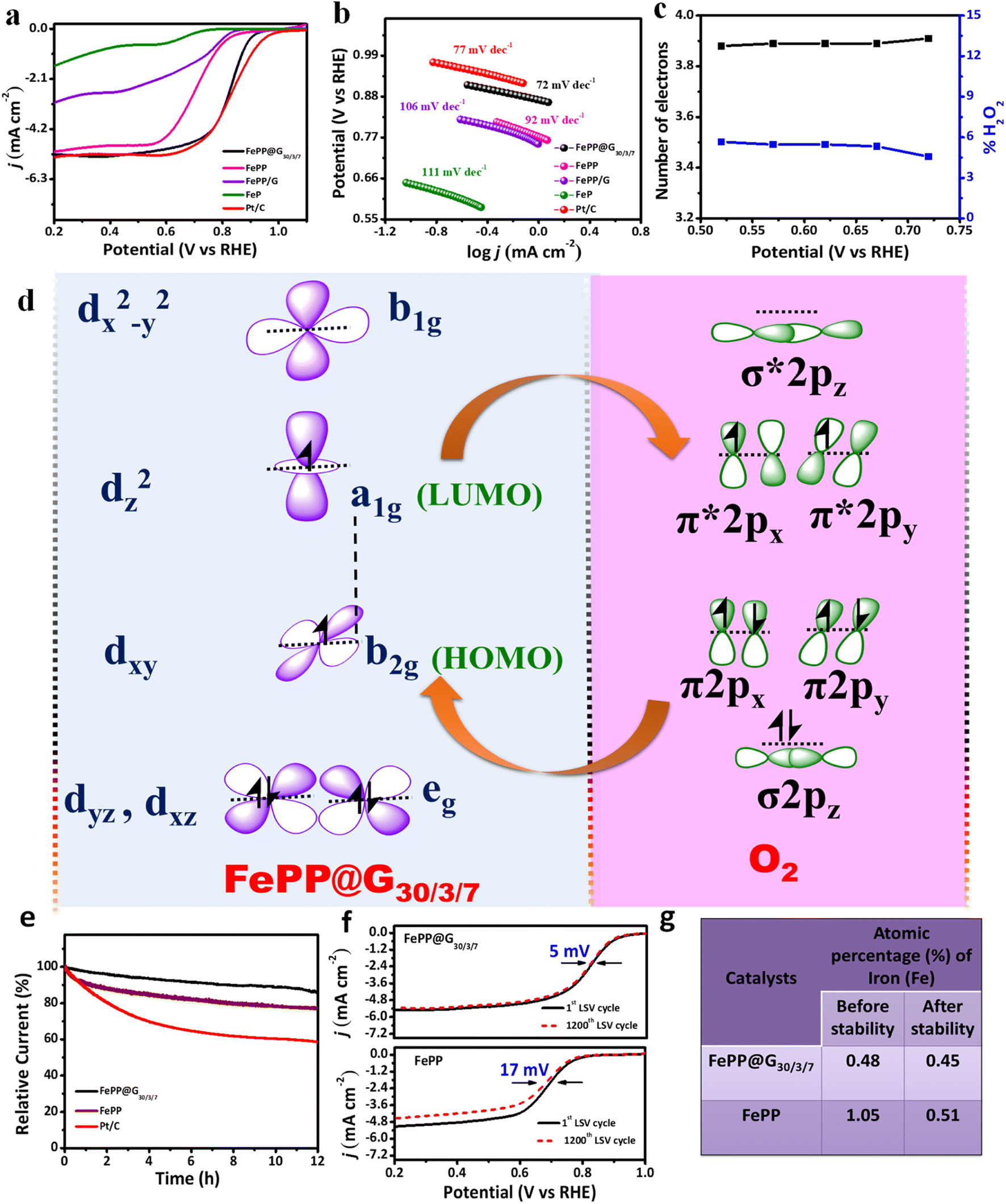
To evaluate the oxygen reduction reaction activity of the as synthesized electrocatalysts, electrochemical measurements with a rotating disk electrode (RDE) were carried out in 0.1 M KOH electrolyte solution in a standard 3-electrode electrochemical cell. First, the cyclic voltammetry (CV) technique was employed with the as-synthesized FePP and FePP@G30/3/7 and commercially available Pt/C (20 wt%) in Ar- and O2-saturated electrolyte solutions at a scan rate of 10 mV s−1. As shown in Fig. S10a–c (ESI),† a quasi-rectangular shape voltammogram appeared in the Ar-saturated solution, while a well distinguished cathodic peak appeared in the O2-saturated electrolyte solution which indicates the efficiency of the FePP@G30/3/7 catalyst towards O2 reduction. Linear sweep voltammetry (LSV) was carried out with FeP, FePP, FePP@G30/3/7, FePP/G and Pt/C at 1600 rpm with a 10 mV s−1 scan rate as shown in Fig. 5a. The FePP@G30/3/7 catalyst exhibits excellent ORR catalytic activity with a better positive half-wave (E1/2) potential of 0.84 V vs. RHE out performing all the control catalysts (the E1/2 of FePP was found to be 0.71 V). This dramatic improvement of O2 reduction potential was observed due to modification of FePP with the graphene material where the conformational changes of FePP through –Ph ring flipping towards in plane geometry caused flawless electron mobility over the FePP system and FeP moieties together with pyrene containing pi-conjugated materials have the proficiency to increase the electron density on the Fe-metal site through push–pull interaction. For the optimisation of catalytic activity, LSV studies have been done with all the as-synthesized materials like FePP@G20/3/7, FePP@G30/3/7, and FePP@G40/3/7 containing different amounts (20 mg, 30 mg and 40 mg respectively) of FePP with exfoliated graphene, while retaining all other experimental conditions (ESI, Fig. S11†). The achieved half-wave potential for FePP@G30/3/7 outperformed that of the other control samples, probably owing to the lowered active site content in FePP@G20/3/7 and improper stacking interaction in the case of FePP@G40/3/7. The electrochemical ORR activity of the composites of FePP with commercially available activated carbon, such as XC-72 and SuperP (1:1), has been determined for comparison with the catalyst. The ORR performance for other controlled samples, such as FePP@G30/1.5/7, FePP@G30/5/7, FePP@G30/3/5, FePP@G30/3/10, FePP@G20/3/7, and FePP@G40/3/7, was checked. The electrochemical response of all the controlled samples is presented in ESI Fig. S11 and S12,† and the results are also summarized in ESI Table T4 and T5.† Thus FePP@G30/3/7 has been considered as the optimized material with respect to E1/2 and JL and further used for all the electrochemical measurements. To evaluate the standard of performance of our active material, it is crucial to have a comparative study with the state-of-the-art catalyst (Pt/C). The commercial Pt/C (20 wt%) showed an E1/2 value of 0.86 V, which is comparable to that of FePP@G30/3/7. Additionally, the FePP@G30/3/7 catalyst shows a higher diffusion-limiting current density (JL, 5.2 mA cm−2) and kinetic current density (JK) of 56.62 mA cm−2 (calculated by using eqn (2), ESI†) at 0.62 V. All the electrochemical parameters of the controlled samples are listed in Table T4, ESI.† We have also compared our results with those of other Fe-porphyrin based materials, iron oxides and metal nanoparticles in the literature (Table T6, ESI†), and our results were comparable to those of other reported ORR catalysts. The electrochemical surface area (ECSA) of the as synthesized material was calculated by using eqn (7) (ESI).† To calculate the ECSA of both the catalysts (FePP and FePP@G30/3/7), CV analysis was performed in the non-Faradic region with different scan rates from 20 to 150 mV s−1 in alkaline solution as shown in ESI, Fig. S13.† The double layer capacitance (Cdl) values of 57 μF and 9.57 μF, respectively, were calculated from the slope of the current vs. scan rate plots of FePP@G30/3/7 and FePP, respectively (ESI, Fig. S13†). The electrochemically active surface area (ECSA) values were calculated, taking specific capacitance (Cs) as 40 μF cm−2.49 The obtained ECSA values of FePP@G30/3/7 and FePP were 1.42 and 0.24 cm2, respectively. It is worth mentioning here that the obtained higher Cdl value for the FePP@G30/3/7 catalyst as compared to the FePP catalyst is mainly due to the higher electrical conductivity, mesoporosity, and increased electrochemically active sites in FePP@G30/3/7, which proves the higher ECSA value of FePP@G30/3/7 as compare to the FePP catalyst. It is clear that the higher ECSA of FePP@G30/3/7 as compare to FePP helps to achieve better electrocatalytic activity of FePP@G30/3/7 towards the ORR. The Nyquist plots obtained from the electrochemical impedance spectroscopy (EIS) study suggest that the FePP@G30/3/7 catalyst has a smaller semicircle diameter than the FePP catalyst (ESI, Fig. S14†), which indicates a lower charge transfer resistance (RCT), consistent with its more favourable charge transfer process. Our claim of the facile electron mobility throughout the catalyst and an electron sufficiency over the mesoporous active surface of the material reinforces the charge transfer rate at the electrode–electrolyte interface, as reflected by the lower RCT value.50 In addition, all LSV plots were recorded at a slow scan rate of 1 mV s−1 for Tafel slope measurements. The Tafel slope of the FePP@G30/3/7 catalyst was obtained as 72 mV dec−1, which was smaller than that of the Pt/C catalyst (77 mV dec−1) as well as FePP (92 mV dec−1). The Tafel slope of other controlled samples (FePP/G and FeP) is shown in Fig. 5b, and summarized in ESI Table T4.† The faster reaction kinetics of FePP@G30/3/7 than that of FePP as well as Pt/C also suggested the more accessible active catalytic sites of the mesoporous catalytic surface along with facile electron transfer towards metal centre through the electron push effect of the pyrene ring as shown in Fig. 5b.51 To assess the ORR pathway of the FePP@G30/3/7 catalyst, RDE measurements at various rotational speeds (625 to 4900 rpm) were carried out (ESI, Fig. S15†). As observed, the reduction current increases with increase in the rotational speed due to a convectional phenomenon and the shortening of the formed diffusion layer on the electrode surface. The Koutecky–Levich (K–L) equation52 (ESI, eqn (4)†) and the corresponding parallel K–L plots (ESI, Fig. S16†) suggested that the FePP@G30/3/7 catalyst follow the first order reaction kinetics towards the concentration of dissolved O2 gas.53,54 Moreover, the rotating ring disk electrode (RRDE) measurements (ESI, Fig. S17†) reveal that the undesirable H2O2 yield of the FePP@G30/3/7 catalyst remains below 5% in the applied potential range (0.72 to 0.42 V vs. RHE), corresponding to a high electron transfer number of 3.91 calculated by using eqn (5) and (6) in the ESI† (Fig. 5c). The H2O2 yield and electron transfer number demonstrate that the FePP@G30/3/7 catalyst possesses a 4e-reaction path for the alkaline ORR. LSVs at all rotations, K–L plots, the number of electrons and %H2O2 of FePP are shown in ESI, Fig. S18a–c,† which show a lower ORR activity as compare to the FePP@G30/3/7 catalyst. As discussed from the above results, the enhanced performance of the FePP@G30/3/7 catalyst can be explained by the presence of pyrrolic N, which is coordinated with the Fe atom in the form of FeN4 as determined from the XPS and EXAFS analyses. A high JK value of the catalyst indicates substantial spin density and density of states near the Fermi level. The abundant FeN4 active sites and highly conducting nature of catalyst is responsible for the 4e− pathway during the ORR. The high content of pyrrolic N and –Ph ring flipped in a plane with pyrene and porphyrin units strongly encouraged an extended pi-conjugation followed by push–pull
interaction with graphene in the catalyst. The electron transfer during oxygen reduction on the FeN4 active site from oxygen to metal and back donation of the electrons from metal to the vacant orbital of oxygen are represented in Fig. 5d, which is responsible for excellent ORR activity in the alkaline medium.
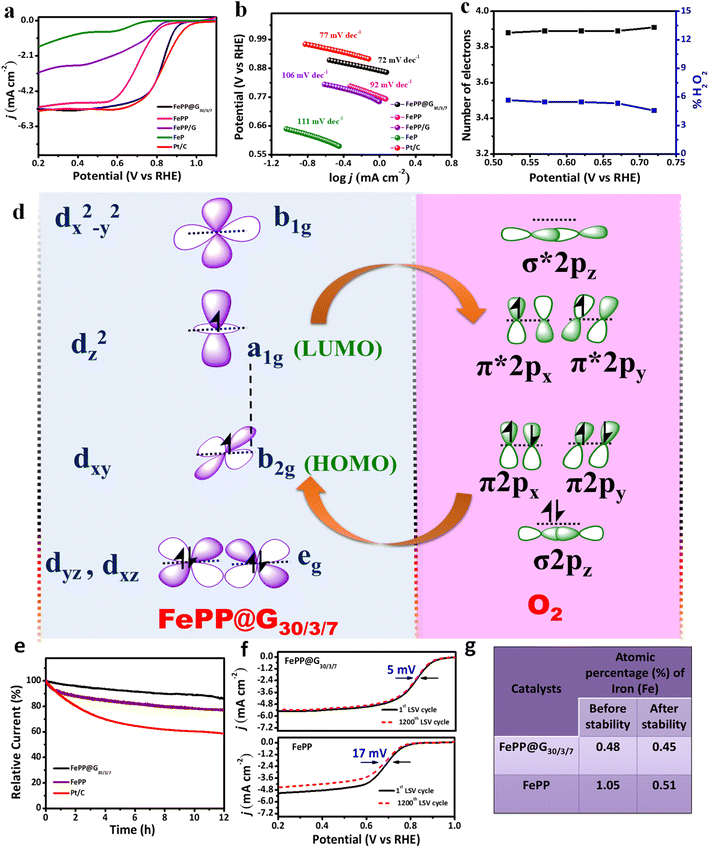 |
| | Fig. 5 (a) LSV curves of the as synthesized electrocatalysts and Pt/C catalyst in O2-saturated alkaline electrolyte solution. (b) Tafel plots of all catalysts and comparison with the Pt/C catalyst. (c) Number of electrons involved and %H2O2 yield formed during the ORR with the FePP@G30/3/7 catalyst in alkaline solution. (d) Diagrammatic representation of molecular orbital interaction between FePP@G30/3/7 and O2. (e) Chronoamperometry curve of FePP@G30/3/7 and FePP and comparison with the Pt/C catalyst. (f) Linear sweep voltammetry polarization curve before and after the LSV cycling stability test of FePP@G30/3/7 and FePP catalysts in O2-saturated 0.1 M KOH electrolyte solution. (g) Atomic percentage of iron before and after stability measurement. | |
Apart from the ORR activity, the long-time durability and methanol resistance ability of the synthesized catalyst are other essential concerns for the applications of device fabrication. In this current study, the chronoamperometric (CA) technique was used in O2-saturated alkaline electrolyte solution for the study of long-term durability at 0.62 V constant potential. The FePP@G30/3/7 catalyst was found to be highly stable for 12 hours, retaining 87% of the initial current density, and FePP retaining 77% of initial current, while that of the Pt/C catalyst degraded up to 57% (Fig. 5e). Further for better understanding the stability of the as synthesized catalysts, LSV cycling was also performed at 1600 rpm with a 10 mV s−1 scan rate and it was found that there was very less negative shift (5 mV vs. RHE) in the half-wave potential after 1200 cycles in the case of the FePP@G30/3/7 catalyst (Fig. 5f), which indicates that the catalyst was highly stable in the harsh alkaline environment during the ORR, while comparatively a higher shift of negative potential (17 mV vs. RHE) was observed in the FePP catalyst as shown in Fig. 5f. The shifting in the potential in the FePP catalyst might be due to metal leaching during the cycling test in alkaline media. In order to verify the metal leaching from the porphyrin ring, XPS analysis has been performed for FePP@G30/3/7 and FePP catalysts after the stability test as shown in Fig. S19 and S20, ESI.† High resolution XPS analysis of deconvoluted C 1s, N 1s, and Fe 2p peaks for both the FePP@G30/3/7 and FePP catalysts obtained after stability revealed similar results in comparison to their before stability results. The elemental analysis results from XPS measurement before and after the stability experiment are summarized in Table T2 in the ESI,† which reveals that the atomic % of Fe is drastically reduced (1.05 to 0.51) in the FePP catalyst after the stability test unlike the FePP@G30/3/7 catalyst, where there are insignificant changes in the elemental composition noticed as shown in Fig. 5g. These data suggested that due to π–π stacking of the catalyst in between the graphene sheets, the metal has not leached out, which helps to improve the stability of the FePP@G30/3/7 catalyst. To check the viability of the catalyst to be used in direct methanol fuel cells, chronoamperometric measurement was conducted at a constant potential of 0.62 V, where after 700 s, 1 M CH3OH was added into the catalytic solution. The current density of the FePP@G30/3/7 catalyst remained constant till continuous 3600 s of CA run; moreover the CA curve of the Pt/C catalyst drops rapidly when methanol was added into the electrochemical cell (Fig. S21, ESI†). Additionally, the CV response in the presence and absence of 1 M CH3OH in O2-saturated alkaline solution demonstrated no noticeable change in the reduction current of the FePP@G30/3/7 catalyst in terms of onset potential and E1/2 but in the case of the Pt/C catalyst the CV curve drastically changed in the presence of methanol (ESI, Fig. S22 and S23†). Therefore, the stability of the FePP@G30/3/7 catalyst in comparison to the commercially available Pt/C catalyst towards the methanol resistance ability shows the acceptability of the catalyst for practical applications in zinc–air batteries and direct methanol fuel cells. So, employing the exfoliation technique the modified catalyst FePP@G30/3/7 gained an improvement of various electrochemical ORR parameters like E1/2, overpotential, reaction kinetics and ECSA. These enhancements of catalytic efficiency occurred mainly due to the electronic mobility modulation from pyrene to the metalloporphyrin centre along with interlayer electron transfer among FePP and the conducting surface of graphene.
Reaction kinetics and mechanism of the ORR on the catalyst surface
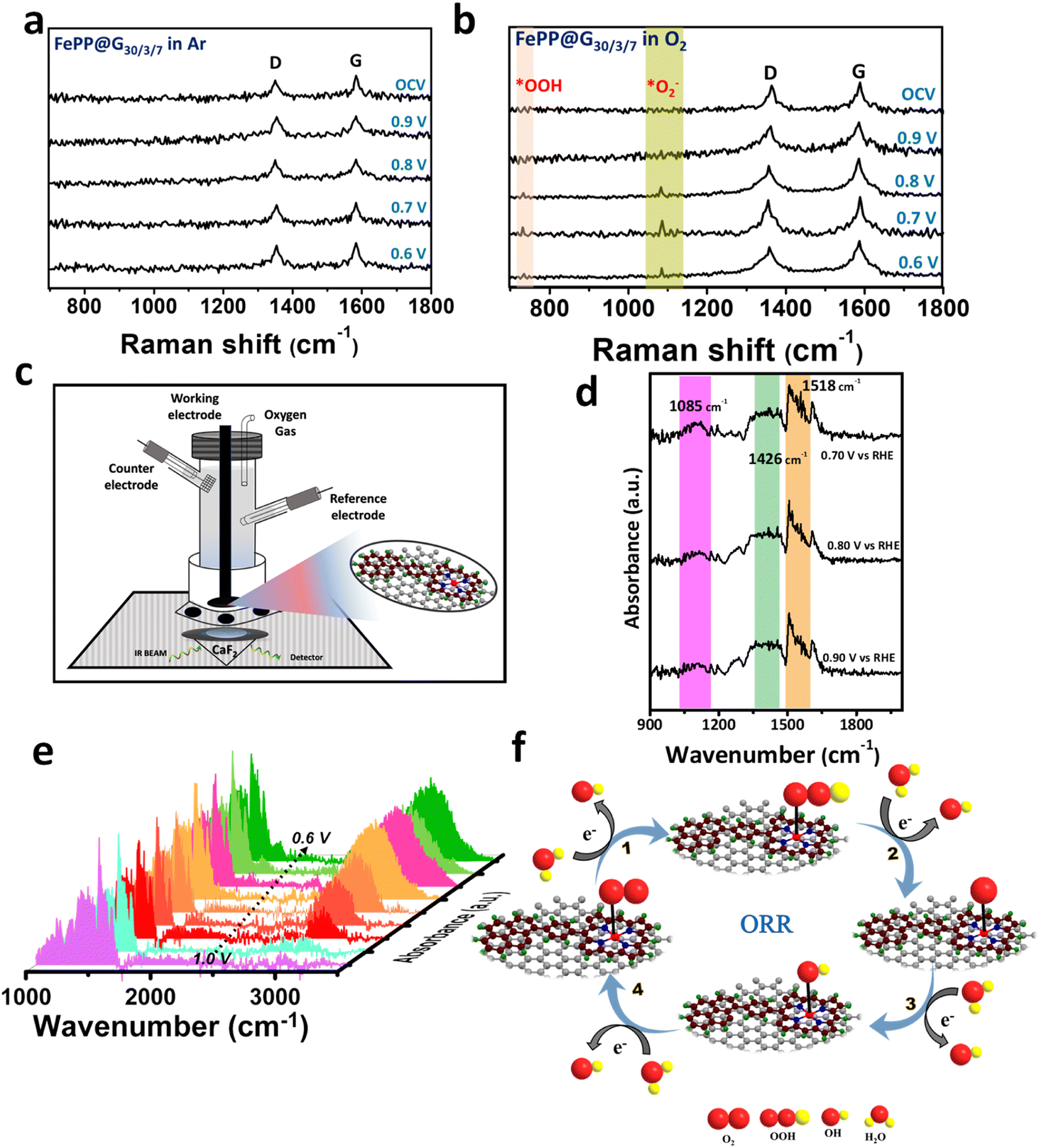
Although effective catalysts for the ORR have been found, the fundamental obstacle is still the selectivity of the reaction mechanism that proceeds via the 4e-transfer path generating water. Strong adsorption on the active site and charge polarization on the active material surface, which can easily polarize the electron cloud of the stable and neutral molecule, will enable O–O bond breaking, which is necessary for the creation of water from molecular oxygen. If there is less O–O cleavage, the ORR uses the 2e-route to produce hydrogen peroxide. The RRDE experiment already confirmed the ORR process following the 4-electron path using the FePP@G30/3/7 catalyst. This prompted us to provide mechanistic insights to shed light on the catalytic processes. For better understanding the mechanistic path, we have performed the electrochemical and operando Raman analysis of the FePP@G30/3/7 catalyst in Ar- and O2-saturated alkaline electrolyte solutions. The Raman setup for the operando study is shown in ESI, Fig. S24.† We observed D and G bands in the Ar-saturated alkaline electrolyte media at positions 1337 and 1576 cm−1, respectively (Fig. 6a) and there are no other peaks found on holding potential in chronoamperometry for 10 min each. The Raman data are recorded at various potentials in the O2-saturated electrolyte, which is shown in Fig. 6b. We observed strong peaks at 1087 cm−1 and 734 cm−1 with D and G bands which confirmed the formation of *O2− and *OOH intermediates respectively,55,56 and following the 4e-path for the ORR catalyzed over the FePP@G30/3/7 catalyst in alkaline medium. The operando Raman data also confirm that Fe–Nx works as the active site during the ORR in alkaline media. Furthermore, the attenuated total reflection Fourier transform infrared (ATR-FTIR) spectroscopic analysis was carried out in alkaline media to identify the corresponding intermediates formed during the ORR process (Fig. 6c). There is no peak in the Ar-saturated electrolyte solution (ESI, Fig. S25†), which reflects that all peaks that appear in O2-saturated solution are attributed to the ORR intermediates formed during the ORR. Fig. 6d shows peaks corresponding to the O–O stretching mode of adsorbed O2 as an *O2− intermediate on the FePP@G30/3/7 catalyst surface at 1085 cm−1 and 1426 cm−1 during the ORR and H–O–H bending at 1518 cm−1.57–59 Further we have run chronoamperometry for 10 min and recorded spectra at various potentials 1.0, 0.95, 0.90, 0.85, 0.80, 0.75, 0.70, 0.65 and 0.60 V vs. RHE, and found the enhancement of the peaks at 1518 cm−1 and 3000–3400 cm−1, which confirmed the production of water during the ORR process as shown in Fig. 6e. The mechanistic representation of ORR kinetics is shown in Fig. 6f, which illustrate the formation of the different intermediates during ORR process. Therefore, operando Raman and in situ FTIR coupled with an electrochemical study confirmed that the ORR process on the FePP@G30/3/7 catalyst follows the four-electron pathway and Fe–Nx works as the active site.
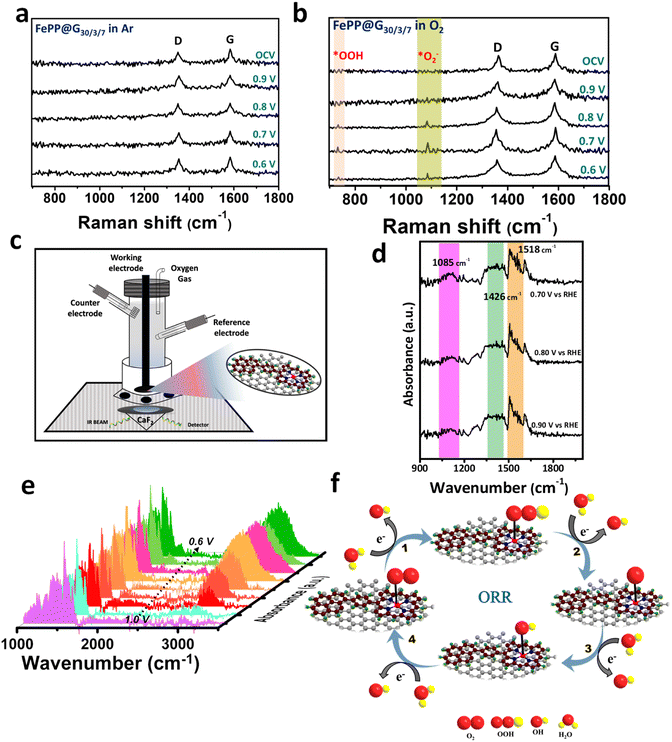 |
| | Fig. 6 (a) In situ Raman spectra of FePP@G30/3/7 in argon saturated 0.1 M KOH electrolyte solution. (b) In situ Raman spectra of FePP@G30/3/7 in oxygen saturated 0.1 M KOH electrolyte solution at different potentials. (c) The in situ experimental setup of FT-IR. (d) In situ FT-IR spectra of FePP@G30/3/7 in oxygen saturated 0.1 M KOH electrolyte solution at various potentials (V vs. RHE) and (e) in situ FTIR spectra showing the water formation during the ORR study. (f) Proposed dynamic mechanistic of the ORR at the FePP@G30/3/7 catalyst surface. | |
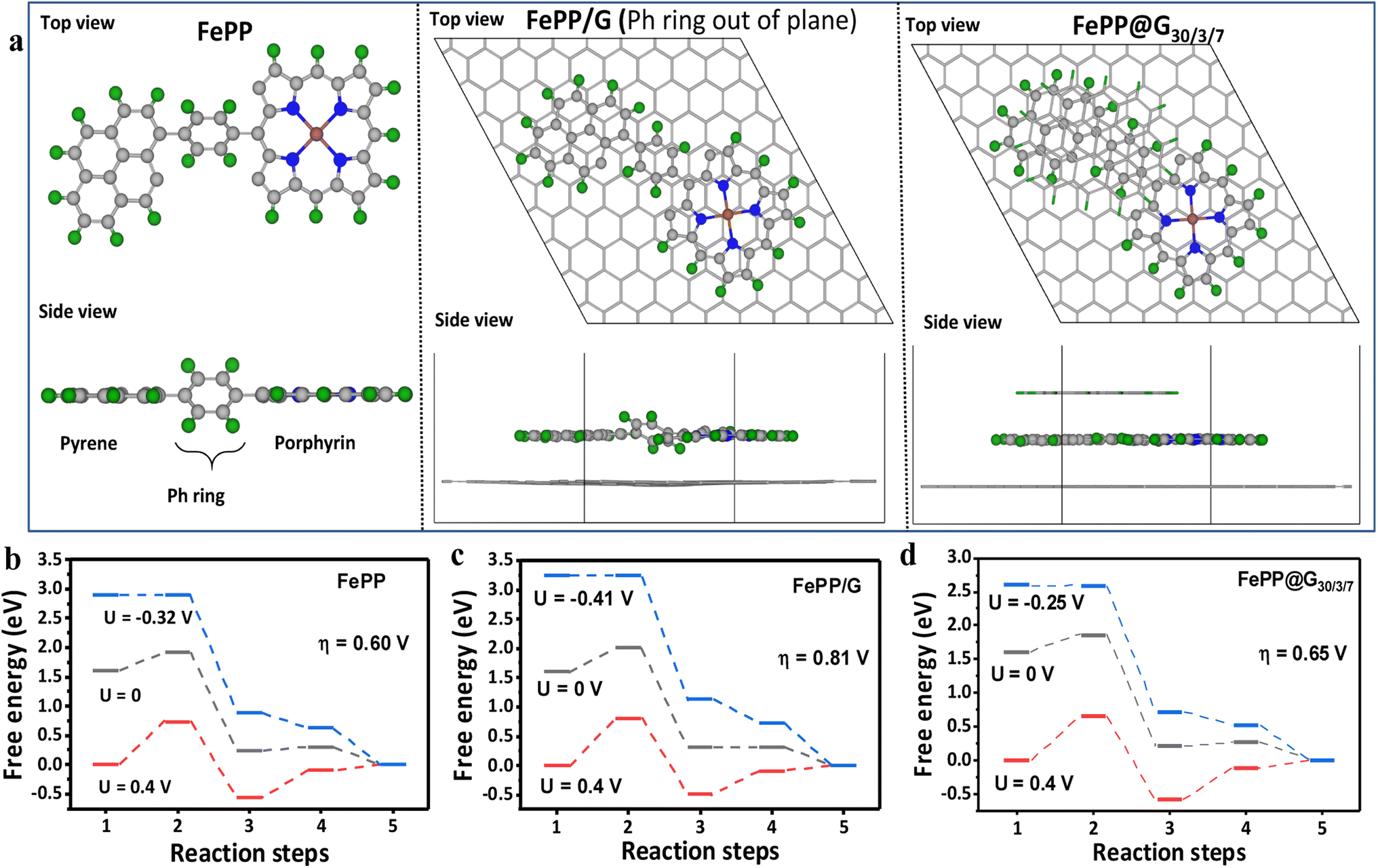
The drastic improvement in the ORR performance of the FePP@G30/3/7 catalyst was attributed to the Ph ring flipping to an all-planar orientation. To acquire more insight into the extent of ring flipping and the energy optimized structures and to deduce the mechanistic pathway for the ORR, we sought the help of a density functional theory (DFT) based study. Top and side views of the Fe-porphyrin-pyrene (FePP) moiety with the Ph ring out of plane at 45°, FePP on a single graphene sheet (Ph-ring out of plane) and FePP@G30/3/7, and FePP between a fixed graphene sheet and flake, where the Ph ring is completely in-plane are shown in Fig. 7a. In all the cases, Fe is taken as the active site for estimating the ORR activity. ORR free energy steps under alkaline conditions are as follows
| | | ΔG1 = O2 + 2H2O + 4e− → OOH* + H2O + OH− + 3e− | (2) |
| | | ΔG2 = OOH* + H2O + OH− + 3e− → O* + 2OH− + H2O + 2e− | (3) |
| | | ΔG3 = O* + 2OH− + H2O + 2e− → OH* + 3OH− + e− | (4) |
| | | ΔG4 = OH* + 3OH− + e− → * + 4OH− | (5) |
Here * represents the active site on the surface. The structural models of adsorbed intermediates and their corresponding binding energies are given in ESI Fig. S26.
† It was found that, in the case of free FePP, the Ph ring is out of the plane with an angle of 45°. The ORR overpotential on Fe as the active site with this model is calculated to be 0.72 V (
Fig. 7b). FePP on a single graphene sheet (Ph-ring out of plane) shows an ORR overpotential of 0.81 V (
Fig. 7c). Experimentally, upon electrophoretic exfoliation with graphene, the Ph ring of the FePP polymer network happened to reorient itself planarly with the underlying graphene making an almost 0° angle with adjacent pyrene and porphyrin units. Therefore, FePP with periodic graphene at the top and bottom was used. Hence, the atoms in the graphene sheet and flake are fixed, where the Ph ring is now completely in plane. The overpotential for this structure is calculated to be 0.64 V (
Fig. 7d) from the free energy plot. In both the cases the OOH* formation is the potential determining step (PDS). The d-band centre which is a well-known catalytic activity descriptor for metals is also calculated (see ESI, Fig. S27
†). There is also a small increase in the state near the Fermi level for the in-plane case (indicated by the arrow in Fig. S27
†). The change in the d-band centre is too small to explain the underlying reason for the enhanced catalytic activity. So, Bader charge distribution and charge density analyses have been performed. From the analyses, the Fe atom is naturally charge deficient in the FeN
4 environment. In FePP, the charge deficiency from the Fe atom is 1.11 |
e|, while that on FePP/G and FePP@G
30/3/7 is 1.15 |
e| and 1.05 |
e| respectively (positive represents charge deficiency). This matches well with the overpotential trend and our charge density distribution analyses. A comparative summary of all the electronic as well as electrochemical parameters of FePP@G
30/3/7 and FePP catalysts is given in ESI, Table T7.
† Overall, the graphene substrate helps in maintaining the Ph ring planarity which leads to improved ORR activity due to a better charge transfer effect.
 |
| | Fig. 7 (a) Top and side views of the Fe-porphyrin-pyrene (FePP) moiety with the Ph ring out of plane at 45°, FePP on a single graphene sheet (Ph-ring out of plane) and FePP@G30/3/7, and FePP between a fixed graphene sheet and flake, where the Ph ring is completely in-plane. Green, grey, blue and brown colored spheres represent hydrogen, carbon, nitrogen and iron atoms respectively. (b) Free energy plot for the ORR on Fe in FePP with the Ph ring out of plane at 45°. (c) Free energy plot for the ORR on Fe in FePP on a single graphene sheet (Ph-ring out of plane). (d) Free energy plot for the ORR on Fe in FePP between a fixed graphene sheet and flake with the Ph ring completely in plane. | |
Conclusions
In conclusion, a new noble-metal-free conjugated porous polymer (FePP) has been synthesized having a high surface area and superior electrochemical activity towards the ORR. Further we grafted the catalyst onto a graphene sheet via a one-step electrophoretic exfoliation technique under acidic conditions. A strong pi–pi interaction not only facilitates the exfoliation but also allows the phenyl ring flipping to attain an all-planner geometry, which increased the pi–electron mobility to the active center of the catalyst. This modification helps to achieve a symmetric alignment of the p-orbitals of all the constituent elements facilitating a free charge cloud movement, which improved the catalytic efficiency by decreasing the electrical resistivity of the covalent porous polymer. DFT study has shown that the graphene support facilitates the phenyl ring's in-plane orientation which aids in better charge accumulation on the active site, thereby improving the ORR efficiency and the 4-electron mechanistic pathway has been revealed and advocated by in situ Raman and FTIR studies. This eco-friendly, simple and cost-effective technique would certainly open a new avenue for improvisation of the less conductive polymer materials towards the ORR as well as different electrochemical applications in the near future.
Author contributions
RSD and SKD conceived the idea of this work. SKD and GK did synthesis and detailed physical characterization. GK and SKD did the electrochemical measurements. SKD and GK analysed data and wrote the manuscript with the aid of all authors. AB, SB and MD helped in data analysis, data presentation and draft editing. EES and RT did computational analysis and contributed to the manuscript writing. All the authors have checked the final draft of the manuscript and approved the submission.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
GK wishes to thank UGC, New Delhi, SKD, AB, SB and MD wish to thank INST, Mohali for a fellowship, and RSD acknowledges DST SERB (CRG/2020/005683) for financial support. RT thanks DST SERB, India (Grant No. CRG/2021/000620) and National Supercomputer Mission (NSM), India for financial support (Ref No. DST/NSM/R&D_HPC_Applications/2021/19). The authors gratefully acknowledge the RRCAT, Indore for XAS measurements.
References
- S. Mukherjee, A. Mukherjee, A. Bhagi-Damodaran, M. Mukherjee, Y. Lu and A. Dey, Nat. Commun., 2015, 6, 8467 CrossRef CAS PubMed.
- J. Vilhjálmsdóttir, R. B. Gennis and P. Brzezinski, Sci. Rep., 2018, 8, 7502 CrossRef PubMed.
- S. Chatterjee, K. Sengupta, S. Samanta, P. K. Das and A. Dey, Inorg. Chem., 2013, 52(17), 9897–9907 CrossRef CAS PubMed.
- J. Wang, X. Sun, H. Hu, T. Shen, G. Liu, Z. Li, D. Cao, L. He and Y.-F. Song, Chem. – Eur. J., 2022, 28, e202103601 CAS.
- A. Singha, A. Mondal, A. Nayek, S. G. Dey and A. Dey, J. Am. Chem. Soc., 2020, 142, 21810–21828 CrossRef CAS PubMed.
- Q. Hua, K. E. Madsen, A. M. Esposito, X. Chen, T. J. Woods, R. T. Haasch, S. Xiang, A. I. Frenkel, T. T. Fister and A. A. Gewirth, ACS Catal., 2022, 12, 1139–1149 CrossRef CAS.
- M. Hanana, H. Arcostanzo, P. K. Das, M. Bouget, S. Le Gac, H. Okuno, R. Cornut, B. Jousselme, V. Dorcet, B. Boitrel and S. Campidelli, New J. Chem., 2018, 42, 19749–19754 RSC.
-
F. Si, Y. Zhang, L. Yan, J. Zhu, M. Xiao, C. Liu, W. Xing and J. Zhang, Rotating Electrode Methods and Oxygen Reduction Electrocatalysts, ed. W. Xing, G. Yin and J. Zhang, Elsevier, Amsterdam, The Netherlands, 2014, pp. 133–170 Search PubMed.
- H. A. Hansen, V. Viswanathan and J. K. Nørskov, J. Phys. Chem. C, 2014, 118, 6706–6718 CrossRef CAS.
- K. Kusada, D. Wu, T. Yamamoto, T. Toriyama, S. Matsumura, W. Xie, M. Koyama, S. Kawaguchi, Y. Kubota and H. Kitagawa, Chem. Sci., 2019, 10, 652–656 RSC.
- M. Kiani, X. Q. Tian and W. Zhang, Coord. Chem. Rev., 2021, 441, 213954 CrossRef CAS.
- K. Iwase, S. Nakanishi, M. Miyayama and K. Kamiya, ACS Appl. Energy Mater., 2020, 3, 1644–1652 CrossRef CAS.
- V. Raut, B. Bera, M. Neergat and D. Das, J. Chem. Sci., 2021, 133, 34 CrossRef CAS.
- L. Xie, X. P. Zhang, B. Zhao, P. Li, J. Qi, X. Guo, B. Wang, H. Lei, W. Zhang, U. P. Apfel and R. Cao, Angew. Chem., Int. Ed., 2021, 60, 7576–7581 CrossRef CAS PubMed.
- A. Biswas, S. Sarkar, M. Das, N. Kamboj and R. S. Dey, Inorg. Chem., 2020, 59, 16385–16397 CrossRef CAS PubMed.
- K. Jayaramulu, M. Horn, A. Schneemann, H. Saini, A. Bakandritsos, V. Ranc, M. Petr, V. Stavila, C. Narayana, B. Scheibe, Š. Kment, M. Otyepka, N. Motta, D. Dubal, R. Zbořil and R. A. Fischer, Adv. Mater., 2021, 33, 2004560 CrossRef CAS PubMed.
- A. E. Baumann, D. A. Burns, B. Liu and V. S. Thoi, Commun. Chem., 2019, 2, 1–14 CrossRef.
- A. Ostadhossein, J. Guo, F. Simeski and M. Ihme, Commun. Chem., 2019, 2, 1–11 CrossRef.
- X. Y. Zhou, C. Xu, P. P. Guo, W. L. Sun, P. J. Wei and J. G. Liu, Chem. – Eur. J., 2021, 27, 9898–9904 CrossRef CAS PubMed.
- F. P. Kinik, A. Ortega-Guerrero, D. Ongari, C. P. Ireland and B. Smit, Chem. Soc. Rev., 2021, 50, 3143–3177 RSC.
- C. Wang, F. Liu, J. Chen, Z. Yuan, C. Liu, X. Zhang, M. Xu, L. Wei and Y. Chen, Energy Storage Mater., 2020, 32, 448–457 CrossRef.
- B. Cui and G. Fu, Nanoscale, 2022, 14, 1679–1699 RSC.
- C. Guo, F. Duan, S. Zhang, L. He, M. Wang, J. Chen, J. Zhang, Q. Jia, Z. Zhang and M. Du, J. Mater. Chem. A, 2022, 10, 475–507 RSC.
- H. Jia, Z. Sun, D. Jiang, S. Yang and P. Du, Inorg. Chem. Front., 2016, 3, 821–827 RSC.
- P. M. Usov, B. Huffman, C. C. Epley, M. C. Kessinger, J. Zhu, W. A. Maza and A. J. Morris, ACS Appl. Mater. Interfaces, 2017, 9, 33539–33543 CrossRef CAS PubMed.
- Q. Hua, K. E. Madsen, A. M. Esposito, X. Chen, T. J. Woods, R. T. Haasch, S. Xiang, A. I. Frenkel, T. T. Fister and A. A. Gewirth, ACS Catal., 2022, 12, 1139–1149 CrossRef CAS.
- C. Wang, F. Liu, S. Yan, C. Liu, Z. Yu, J. Chen, R. Lyu, Z. Wang, M. Xu, S. Dai, Y. Chen and L. Wei, Carbon, 2022, 190, 412–421 CrossRef CAS.
- A. Biswas, S. Sarkar, M. Das, N. Kamboj and R. S. Dey, Inorg. Chem., 2020, 59, 16385–16397 CrossRef CAS PubMed.
- G. Mohmad, S. Sarkar, A. Biswas, K. Roy and R. S. Dey, Chem. – Eur. J., 2020, 26, 12664–12673 CrossRef CAS PubMed.
- K. S. Novoselov, V. I. Fal'Ko, L. Colombo, P. R. Gellert, M. G. Schwab and K. Kim, Nature, 2012, 490, 192–200 CrossRef CAS PubMed.
- S. K. Das, S. Shyamal, M. Das, S. Mondal, A. Chowdhury, D. Chakraborty, R. S. Dey and A. Bhaumik, Front. Chem., 2021, 9, 1094 Search PubMed.
- G. Luo, Y. Wang and Y. Li, Sci. Bull., 2017, 62, 1337–1343 CrossRef CAS PubMed.
- C. Zhang, H. Yang, D. Zhong, Y. Xu, Y. Wang, Q. Yuan, Z. Liang, B. Wang, W. Zhang, H. Zheng, T. Cheng and R. Cao, J. Mater. Chem. A, 2020, 8, 9536–9544 RSC.
- A. Ghatak, S. Bhunia and A. Dey, ACS Catal., 2020, 10, 13136–13148 CrossRef CAS.
- Z. Xiang, Y. Xue, D. Cao, L. Huang, J. F. Chen and L. Dai, Angew. Chem., Int. Ed., 2014, 53, 2433–2437 CrossRef CAS PubMed.
- S. Sarkar, A. Biswas, E. E. Siddharthan, R. Thapa and R. S. Dey, ACS Nano, 2022, 16, 7890–7903 CrossRef CAS PubMed.
- D. Dasler, R. A. Schäfer, M. B. Minameyer, J. F. Hitzenberger, F. Hauke, T. Drewello and A. Hirsch, J. Am. Chem. Soc., 2017, 139, 11760–11765 CrossRef CAS PubMed.
- J. Wang, X. Yang, T. Wei, J. Bao, Q. Zhu and Z. Dai, ACS Appl. Bio Mater., 2018, 1, 382–388 CrossRef CAS PubMed.
- N. Zhang, T. Zhou, M. Chen, H. Feng, R. Yuan, C. Zhong, W. Yan, Y. Tian, X. Wu, W. Chu, C. Wu and Y. Xie, Energy Environ. Sci., 2020, 13, 111–118 RSC.
- X. Liang, Z. Li, H. Xiao, T. Zhang, P. Xu, H. Zhang, Q. Gao and L. Zheng, Chem. Mater., 2021, 33, 5542–5554 CrossRef CAS.
- D. Deng, X. Chen, L. Yu, X. Wu, Q. Liu, Y. Liu, H. Yang, H. Tian, Y. Hu, P. Du, R. Si, J. Wang, X. Cui, H. Li, J. Xiao, T. Xu, J. Deng, F. Yang, P. N. Duchesne, P. Zhang, J. Zhou, L. Sun, J. Li, X. Pan and X. Bao, Sci. Adv., 2022, 1, e1500462 CrossRef PubMed.
- L. Yang, D. Cheng, H. Xu, X. Zeng, X. Wan, J. Shui, Z. Xiang and D. Cao, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 6626–6631 CrossRef CAS PubMed.
- S. Lim, W. Lee, H. Choo and C. Lee, Constr. Build. Mater., 2017, 157, 42–50 CrossRef CAS.
- G. Das, B. P. Biswal, S. Kandambeth, V. Venkatesh, G. Kaur, M. Addicoat, T. Heine, S. Verma and R. Banerjee, Chem. Sci., 2015, 6, 3931–3939 RSC.
- E. v. Basiuk, M. Martínez-Herrera, E. Álvarez-Zauco, L. V. Henao-Holguín, I. Puente-Lee and V. A. Basiuk, Dalton Trans., 2014, 43, 7413–7428 RSC.
- A. H. Al-Bagawi, A. M. Bayoumy and M. A. Ibrahim, Heliyon, 2020, 6, e04456 CrossRef PubMed.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- S. Liu, H. Ou, Y. Li, H. Zhang, J. Liu, X. Lu, R. T. K. Kwok, J. W. Y. Lam, D. Ding and B. Z. Tang, J. Am. Chem. Soc., 2020, 142, 15146–15156 CrossRef CAS PubMed.
- C. C. L. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem.
Soc., 2015, 137, 4347–4357 CrossRef CAS PubMed.
- Y. Mun, S. Lee, K. Kim, S. Kim, S. Lee, J. W. Han and J. Lee, J. Am. Chem. Soc., 2019, 141, 6254–6262 CrossRef CAS PubMed.
- T. Shinagawa, A. T. Garcia-Esparza and K. Takanabe, Sci. Rep., 2015, 5, 13801 CrossRef PubMed.
- S. Kanti Das, S. Chatterjee, A. Banerjee, G. Kumar, A. Kumar Patra, R. Sundar Dey, A. J. Pal and A. Bhaumik, Chem. Eng. J., 2023, 451, 138766 CrossRef CAS.
- T. Boruah, S. K. Das, G. Kumar, S. Mondal and R. S. Dey, Chem. Commun., 2022, 58, 5506–5509 RSC.
- M. Das, A. Biswas and R. S. Dey, Chem. Commun., 2022, 58, 1934–1937 RSC.
- J. Wei, D. Xia, Y. Wei, X. Zhu, J. Li and L. Gan, ACS Catal., 2022, 12, 7811–7820 CrossRef CAS.
- X. Li, C. S. Cao, S. F. Hung, Y. R. Lu, W. Cai, A. I. Rykov, S. Miao, S. Xi, H. Yang, Z. Hu, J. Wang, J. Zhao, E. E. Alp, W. Xu, T. S. Chan, H. Chen, Q. Xiong, H. Xiao, Y. Huang, J. Li, T. Zhang and B. Liu, Chem, 2020, 6, 3440–3454 CAS.
- S. Mondal, D. Bagchi, M. Riyaz, S. Sarkar, A. K. Singh, C. P. Vinod and S. C. Peter, J. Am. Chem. Soc., 2022, 144, 11859–11869 CrossRef CAS PubMed.
- J. P. Vivek, N. G. Berry, J. Zou, R. J. Nichols and L. J. Hardwick, J. Phys. Chem. C, 2017, 121, 19657–19667 CrossRef CAS.
- W. Cheng, X. Zhao, H. Su, F. Tang, W. Che, H. Zhang and Q. Liu, Nat. Energy, 2019, 4, 115–122 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, XPS, BET, EXAFS, additional electrochemical data regarding the ORR, DFT calculation and tables. See DOI: https://doi.org/10.1039/d3ta03055e |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Sakshi
Bhardwaj
a,
Manisha
Das
a,
Ranjit
Thapa
b and
Ramendra Sundar
Dey
a,
Sakshi
Bhardwaj
a,
Manisha
Das
a,
Ranjit
Thapa
b and
Ramendra Sundar
Dey