
Modulating the microenvironment of AuPd nanoparticles using metal–organic frameworks for selective methane oxidation†

Abstract
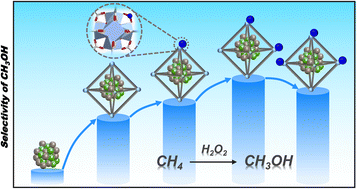
Direct and selective oxidation of methane (CH4) into methanol (CH3OH) under ambient conditions remains a grand challenge because of the high energy barrier of CH4 activation and the complicated processes involved. Herein, by incorporating AuPd alloy nanoparticles (NPs) into a series of Cu2+-doped metal–organic frameworks (MOFs), namely AuPd@Cu-UiO-66x, efficient and direct catalytic conversion of CH4 to CH3OH can be achieved at mild temperature (70 °C) with H2O2 as an oxidant. The Cu-UiO-66 serving as the microenvironment parameter not only regulates the electronic state of AuPd NPs to improve the CH4 adsorption and activation, but also affects the generation of hydroxyl radicals (˙OH) in H2O2 reaction pathways, which consequently results in a volcano-type dependency of the CH3OH selectivity on the Cu contents. This work represents the first finding on achieving direct catalytic oxidation of CH4 to CH3OH under mild conditions by modulating the microenvironment of catalytic centers based on a MOF platform.
- This article is part of the themed collection: Functional Framework Materials
 Please wait while we load your content...
Please wait while we load your content...

