
Self-powered NH3 synthesis by trifunctional Co2B-based high power density Zn–air batteries†
Divyani
Gupta
 a,
Alankar
Kafle
a,
Prajna Parimita
Mohanty
bc,
Tisita
Das
c,
Sudip
Chakraborty
c,
Rajeev
Ahuja
bd and
Tharamani C.
Nagaiah
*a
a,
Alankar
Kafle
a,
Prajna Parimita
Mohanty
bc,
Tisita
Das
c,
Sudip
Chakraborty
c,
Rajeev
Ahuja
bd and
Tharamani C.
Nagaiah
*a
aDepartment of Chemistry, Indian Institute of Technology Ropar, Rupnagar, Punjab 140001, India. E-mail: tharamani@iitrpr.ac.in
bDepartment of Physics, Indian Institute of Technology Ropar, Rupnagar, Punjab 140001, India
cMaterials Theory for Energy Scavenging (MATES) Lab, Harish-Chandra Research Institute (HRI) Allahabad, HBNI, Chhatnag Road, Jhunsi, Prayagraj, Allahabad 211019, India
dCondensed Matter Theory Group, Department of Physics and Astronomy, Uppsala University, Uppsala, SE-75120, Sweden
First published on 17th May 2023
Abstract
The electrochemical production of NH3 by Zn–air batteries is a viable and economical approach to realize sustainable and competent energy conversion. We report the environment friendly, cost-effective, and energy efficient sonochemical synthesis of amorphous Co2B nanosheets for trifunctional electrocatalysis. The catalyst exhibits a high NH3 yield rate (2.98 mg h−1 mgcat.−1), F.E (20.45%), and TOF of 0.74 h−1 at −0.3 V vs. RHE, thereby unveiling an outstanding performance for the artificial ammonia synthesis. The reliable and true NH3 production is premediated by following rigorous protocol that involves the purification of gas supplies, elimination of N-contaminants, and quantification of NH3 by different methods, UV-Vis spectroscopy and 15N2 isotope labelling experiments. More interestingly, DFT calculations on the Co2B catalyst surface shed light on the efficient NRR owing to the presence of Co active sites and possible HER suppression. The optimized Co2B catalyst shows outstanding oxygen bifunctional activity. When employed as an air-cathode for Zn–air batteries, it exhibited remarkable electrocatalytic activity delivering an open circuit potential of 1.45 V with a high power density of 500 mW cm−2 and an energy density of 1078 W h kg−1, which can perform NH3 generation with an overall NH3 production yield rate of 1.048 mg h−1 mgcat.−1.
Introduction
There are exigent demands to develop efficient and robust electrochemical energy conversion and storage devices to muddle through future energy crises and environmental pollution.1 The N2 cycle (N2/NH3, i.e. N2 fixation) is a crucial process for humans to lead a sustainable life on earth because the produced ammonia provides food to a huge percentage of the world's population, can store hydrogen indirectly, and act as a carbon free energy carrier.2 Amid different technologies for artificial N2 fixation, a century-old Haber–Bosch process3 is applied for the large scale production of ammonia, which is highly energy intensive (about 1–3% world's energy consumption), operates at high temperatures (400–500 °C) and pressure (100–200 atm), and consumes excessive natural gas (e.g., CH4 for H2 production; 3–5% world's natural gas production).4 Above all, it is accompanied by immense carbon emissions responsible for around 1.5% of greenhouse gas production averaging about 2.9 tons CO2 per ton of NH3 globally.5,6 Consequently, it is critical to deal with severe energy and environmental concerns raised due to the traditional Haber–Bosch process.7 The development of cost-effective and energy efficient alternative technologies with renewable energy sources is of great interest.8,9 In recent years, the conversion of abundant N2 to NH3via an electrochemical pathway has gained significant attention owing to the ease of operation under ambient conditions using water as a source of H2 and renewable energy source derived electric power supply subsequently without CO2 emissions.5,10,11In addition, the utilization of high-power density rechargeable metal–air batteries viz. Zn (ZAB) for electrochemical ammonia synthesis12 is a game-changing strategy because of the earth's abundant, economical and environment friendly metal anode and its high theoretical energy density that is three times higher than that of Li-ion batteries (1216 W h kg−1).13 More importantly, integrating ZAB in electrochemical NH3 production provides cumulative efficiency compared to its device.14,15 To accomplish these necessities, the principal task is to develop highly efficient and robust catalysts active towards nitrogen reduction reaction and oxygen bifunctional catalyst viz. oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) for ZAB.16 For Zn–air batteries, the degradation of air cathodes during consecutive cycling due to the sluggish multielectron process of OER and ORR remains the major disadvantage.17 Similarly, the adsorption and activation of strong N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N triple bond with high dissociation binding energy of 940.95 kJ mol−1, negative e− affinity of −1.8 eV, high ionization potential of 15.8 eV, high HOMO–LUMO gap of 10.82 eV and the permanent dipole is a prerequisite for the efficacious synthesis of NH3.18 To date, precious Pt/C is the state-of-art ORR catalysts but are not efficient towards OER/NRR. However, IrO2/RuO2 is the benchmark OER catalyst but is not efficient for ORR/NRR.19–21
N triple bond with high dissociation binding energy of 940.95 kJ mol−1, negative e− affinity of −1.8 eV, high ionization potential of 15.8 eV, high HOMO–LUMO gap of 10.82 eV and the permanent dipole is a prerequisite for the efficacious synthesis of NH3.18 To date, precious Pt/C is the state-of-art ORR catalysts but are not efficient towards OER/NRR. However, IrO2/RuO2 is the benchmark OER catalyst but is not efficient for ORR/NRR.19–21
Henceforth, the primary challenge for sustainable and renewable energy is the discovery and design of efficient electrocatalysts exhibiting superior trifunctional activity for NRR and rechargeable ZABs with improved stability during their operation.22 More specifically, the expensive, scarce nature of noble metal-based state-of-art catalysts along with their sluggish kinetics impede their real-time application. This limitation demands the development of a cost-effective non-noble metal-based catalyst for OER, ORR and NRR, which holds the potential to compete with or even surpass the activity of precious benchmark catalysts.23,24 Moreover, the designing of such a catalyst, which possesses an ample number of active sites and robustness towards three reactions with completely different fundamental mechanisms, is yet complicated but the need of the hour.25–28 However, so far, only three reports are available wherein ZABs have been used to power electrochemical NH3 synthesis.12,29,30 Several classes of catalysts, including noble-metal-based,31–33 non-noble metal-based34,35 and metal free-based catalysts,36–38 have been explored towards NRR in aqueous media, but the faradaic efficiency (F.E.) and NH3 production yield rate obstruct their attempt to be put into practice.39–41 Therefore, the development of suitable, cost-effective and active catalysts is vital to achieving good bifunctional activity.24 In the past decade, extensive efforts have been devoted to developing precious metal free oxygen bifunctional catalysts,42–46 including transition-metal oxides/hydroxides, sulphides, carbides, carbonaceous materials, molecular catalysts, nitrides, and phosphides.47–50 Transition metal borides (TMBs) have begun to appear as an important inorganic class of materials with wide-spread applications in various fields, such as energy storage,51,52 energy conversion,53 superconductivity, and thermoelectricity.54 TMBs, including cobalt and nickel, have been generally used in electrocatalysis because of their enhanced electrical conductivity, stability and hardness.55,56 TMBs exhibit superior oxygen evolution reaction (OER) performance in alkaline media compared with noble metal catalysts, metal oxides, and metal alloy counterparts.57 Very recently, Co2B was explored in a report by Jose et al.58 for the ORR in alkaline media to achieve higher activity than the benchmark catalyst. In addition, several theoretical studies have reported the potential of TMBs for the NRR.59–63 For now, there are very limited reports on TMBs for oxygen bifunctional electrocatalysis64–66 because of the complicated synthesis of TMBs relying on expensive methods such as physical/chemical vapor deposition, high-energy ball milling, and arc-plasma. These methods usually involve toxic gaseous precursors, and/or complex procedures as well as high-cost devices, incurring the problem of practical applicability.54,67 Therefore, in this study, we exploit a cost-effective, energy efficient, less time consuming, and facile sonochemical approach for the synthesis of TMBs.68,69 Moreover, this approach has the advantage of obstructing particle agglomeration under a chemical environment70 owing to ultrasound waves generated during sonochemical synthesis.
Herein, we report the synthesis of amorphous cobalt boride with porous nanosheet morphology as a trifunctional catalyst for NRR, ORR, and OER under ambient conditions. The optimized cobalt boride was equipped in a Zn–air battery as an air cathode and successfully powered a self-driven NH3 synthesis. To the best of our knowledge, this is the first report to explore metal borides as a trifunctional catalyst for battery-driven NH3 synthesis under ambient conditions. The catalyst shows good activity towards electrochemical NH3 synthesis with F.E. of 20.45% in alkaline media at −0.3 V vs. RHE. When employed as an air-cathode catalyst for Zn–air battery, Co2B exhibits remarkable electrocatalytic activity, delivering open circuit potential of 1.45 V with a high-power density of 500 mW cm−2 and an energy density of 1078 W h kg−1, which can carry out NH3 generation with an overall NH3 production yield rate of 1.048 mg h−1 mgcat.−1.
Results and discussion
The Co2B catalysts were prepared using a green and facile sonochemical approach with cobalt chloride and NaBH4 (Scheme 1) under an inert atmosphere. Briefly, the dropwise addition of aq. NaBH4 aqueous solution was carried out into aq. CoCl2 solution under continuous bath sonication and probe sonication at regular intervals (details are in ESI†) with a probe frequency of 20 Hz. To understand the effect of boron on the morphology, microstructure thence trifunctional activity, three variants of cobalt boride were synthesized by varying the ratio of cobalt chloride to NaBH4 designated as Co2B (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4/1:8/1:12).
:4/1:8/1:12).
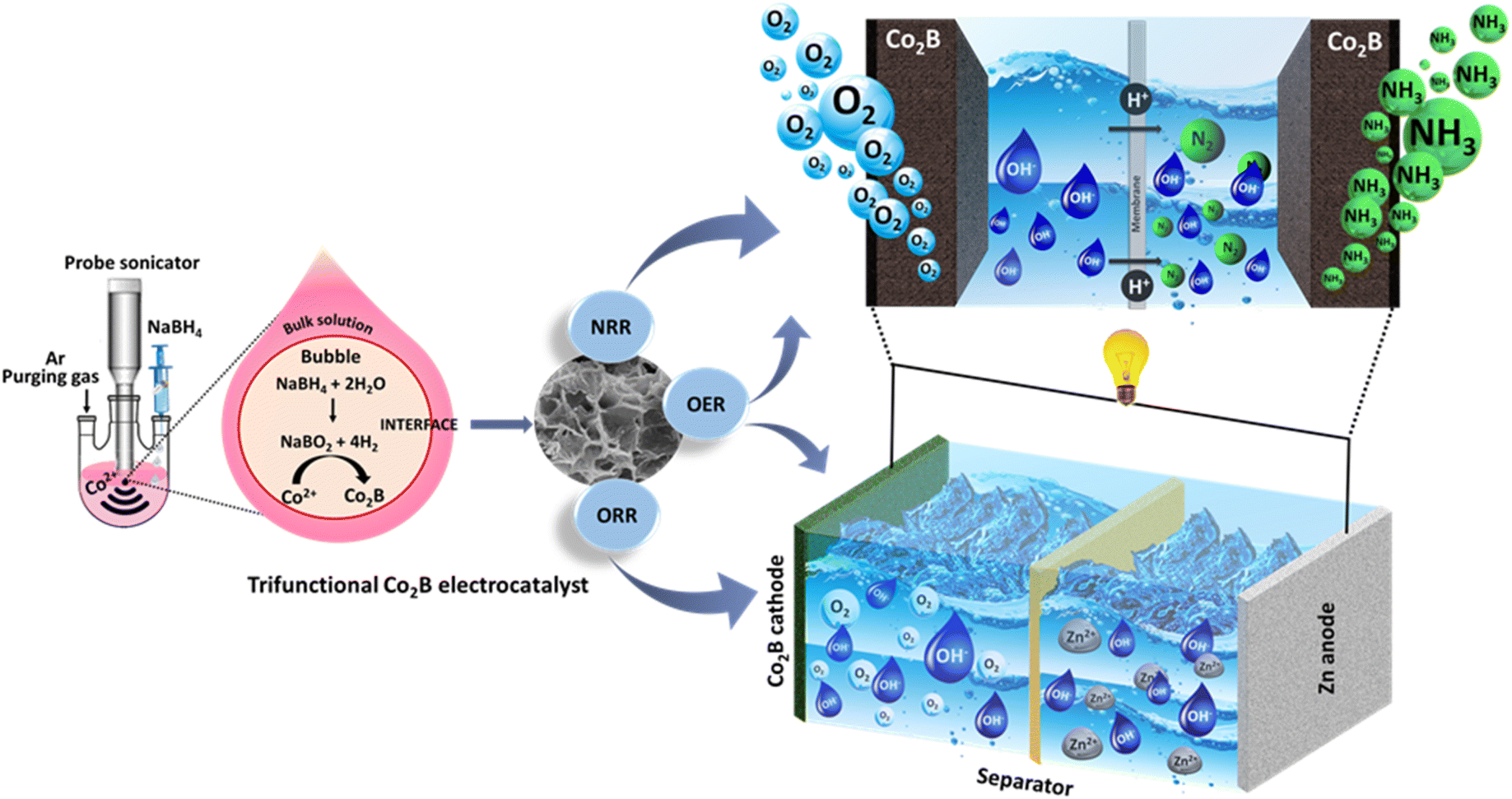 | ||
| Scheme 1 Schematic representation of the controlled sonochemical synthesis of cobalt boride catalysts signifying trifunctional activity towards NRR/ORR/OER for application in electrochemical ammonia production by as-assembled batteries. | ||
Electrochemical dinitrogen reduction (NRR)
To understand the potential efficacy of the synthesized catalyst towards electrocatalytic NRR, linear sweep voltammograms (LSV) were recorded in Ar and N2-saturated 0.1 M KOH electrolytes. Because of any impurities present in the Ar or N2 gas feed streams, we purified the gas with KMnO4 and H2SO4 trap prior to NRR measurements (Fig. S1, ESI†).71 LSV for Co2B catalysts exhibit a distinct increase in cathodic current density under an N2 atmosphere than in Ar-saturated electrolytes (Fig. 1a and S2, ESI†). Co2B (1:8) demonstrates a higher current density, which suggests pronounced activity of the same for NRR than other composites. Under the continuous N2 gas flow in the cathode compartment of the H-cell, N2 on the surface of Co2B catalysts react with H+ ions to form NH3 with potentiostatic reduction, wherein the chronoamperometry was performed for 2 h (Fig. S3, ESI†). The quantification of the NH3 formed during the reaction was performed by applying the indophenol blue method (Fig. S4 and S5A, ESI†).45 As shown in Fig. S4,† the highest faradaic efficiency (F.E.) of 20.45% and NH3 yield of 2980.22 μg h−1 mgcat.−1 were achieved for Co2B (1:8) variant at −0.3 V vs. RHE with a high TOF value of 0.74 h−1. When compared with the recently reported literature in alkaline media, the Co2B catalyst in this study outperforms in terms of F.E. and NH3 production yield rate (Table S1A, ESI†). The presence of a peak related to NH stretching (1455 cm−1) in the FT-IR spectra designates the formation of NH3 after NRR (Fig. S5B, ESI†).72 More interestingly, Nessler's test data acquired for NH3 standard solutions (Fig. S5C, ESI†) and electrolyte samples collected after NRR by Co2B (1:8) for 2 h, validate the NH3 production, in which both of the quantifications methods, i.e. indophenol blue and Nessler's reagent methods produced NH3 yields in the order of 2.98 and 2.35 mg h−1 mgcat.−1, respectively, as presented in Table S1B (ESI).† However, Co2B (1:4) and Co2B (1:12) showed F.E. of 9.03% and 5.86% with NH3 yield rates of 844.78 μg h−1 mgcat.−1 and 363.72 μg h−1 mgcat.−1, respectively (Fig. 1b). It is noteworthy to mention that no hydrazine was detected during NRR when tested by applying the Watt–Chrisp method (Fig. S6, ESI†), revealing the selectivity of the catalyst for NH3 production. The superior activity of Co2B (1:8) towards NRR could be attributed to the facile kinetics of the interfacial charge transfer process, which is in turn related to the electron transfer at the electrode–electrolyte interface caused by the change in the electronic structure of the catalyst material due to a strong M–B bond to enhance NRR (explained in detail in the subsequent section).59 This was evidenced by electrochemical impedance spectroscopic (EIS) studies. As expected, the Co2B (1:8) composite revealed the lowest charge transfer resistance (Rct) of 35.85 Ω shown by the semicircle behavior compared to the other two variants following the order Co2B (1:8) < Co2B (1:4) < Co2B (1:12) (Fig. 1c and Table S2A, ESI†). The lower Rct of Co2B (1:8) signifies the dominance of faster kinetics towards e-NRR due to higher conductivity and facilitated electron transport at the electrode–electrolyte interface. This facile kinetics could be due to the exposure of higher numbers of electroactive sites than the other variants viz., Co2B (1:4) and Co2B (1:12). This was supported by electrochemical surface area (ECSA) evaluated by double layer capacitance in the non-faradaic region in which Co2B (1:8) showed the highest Cdl of 420 μF and ECSA of 10.5 cm2, respectively, followed by Co2B (1:4) and Co2B (1:12) (Fig. S7 and Table S2B, ESI†). The higher surface area of Co2B (1:8) originates from both the inter-sheet space and intra-sheet holes, thus resulting in slit-shaped porosity. This is supported by field emission scanning electron microscopic (FE-SEM) images (Fig. 1d). However, Co2B (1:4) and Co2B (1:12) show variation in morphological features, in which spherical nanoparticles were observed for Co2B (1:12) with an increase in boron content, and stacks of assembled nano-particles were observed for Co2B (1:4) depicted in Fig. S8A;† further increase in boron content (Co2B (1:8)) resulted in surface nanostructuring, thus forming porous nanosheets. The elemental dot mappings of Co2B (1:8) porous nanosheets show a uniform distribution of the respective elements Co and B over the scanned area, as illustrated in Fig. 1e.
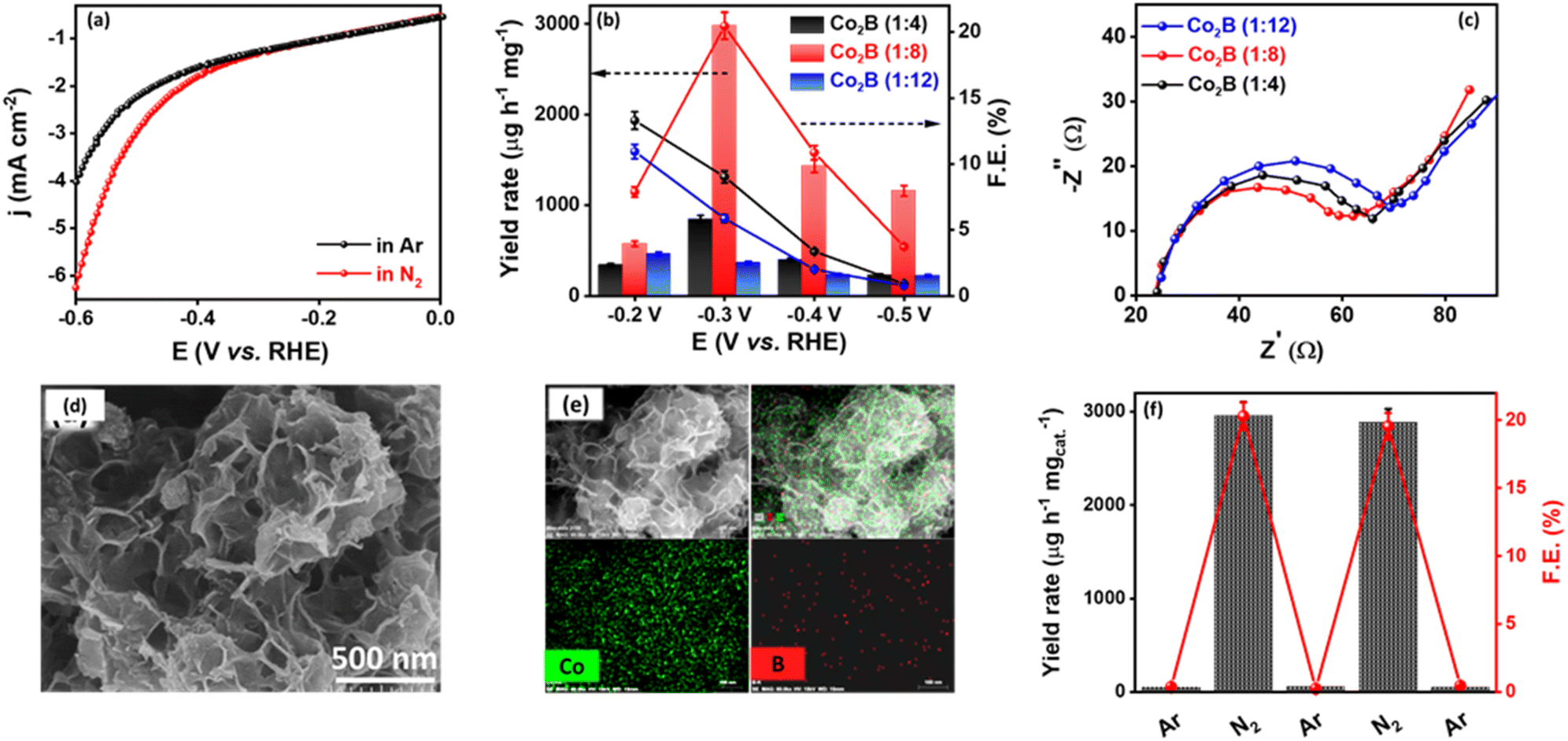 | ||
| Fig. 1 (a) Linear sweep voltammograms for Co2B (1:8) in Ar- and N2-saturated 0.1 M KOH, (b) comparison of yield rates and F.E. of Co2B catalysts at different applied potentials, (c) Nyquist plots, (d) FE-SEM image of Co2B (1:8) and (e) corresponding elemental dot mapping showing uniform distribution of Co and B elements over the scanned area. (f) F.E. and yield rates obtained during NRR under switching gas-feed (Ar/N2) environments. | ||
More interestingly, the BET surface area measurements were carried out for all the catalysts to determine the pore size and surface area values. The N2 adsorption–desorption isotherms of Co2B catalysts represent a type IV curve with a hysteresis loop, indicating the mesoporous nature of all the designed catalysts. Co2B (1:8) catalyst possesses a large surface area of 103 m2 g−1, which is higher than that of the BET surface area obtained for the Co2B (1:4) catalyst. On further increasing the NaBH4 concentration in the Co2B (1:12) catalyst, the overall surface area of the composite decreased. Thus, the active sites for reactant adsorption over the Co2B (1:8) catalyst were provided by its high surface area (Fig. S8B and Table S2C, ESI†).
The synthesized porous Co2B (1:8) nanosheets are highly hydrophobic compared to other variants due to the ‘lotus effect’ resulting in surface nanostructuring, which influences the electrocatalytic activity of the catalyst towards NRR.73 It is clear from the water contact angle measurements that Co2B (1:8) exhibits a higher surface hydrophobicity with a contact angle of 115°, which is higher than other variants viz. Co2B (1:4), Co2B (1:12) with a contact angle of 105° and 96.5° respectively (Fig. S8C, ESI†). Thus, the increased surface hydrophobicity of the Co2B (1:8) catalyst can effectively decrease the rate of accessibility of H2O molecules (H+ donor) to the catalyst, resulting in the obstruction of HER. This demonstrates the remarkable activity of Co2B (1:8) towards NRR owing to suppressed HER at the cathode. To substantiate HER suppression by the catalyst, we performed chronoamperometry under Ar and N2 saturated 0.1 M KOH subsequently by switching the gas feed from Ar to N2 and vice versa after every 2 h. A rapid increase in cathodic current density was observed when the gas feed is switched from Ar to N2, whereas a decrease was observed when the gas feed is switched back to Ar from N2 (Fig. S9, ESI†). This is evident in the catalyst's ability to suppress HER in the presence of N2 and the high stability of the catalyst during electrolysis. Upon the quantification of the samples collected after the experiment, a rapid increase in both yield and F.E. is observed when the Ar gas stream is changed to N2. However, on switching back to Ar, no NH3 production was observed, indicating inactivity towards NRR in the absence of an N2 atmosphere (Fig. 1f, S9 and S10, ESI†), which implies that the source of nitrogen in the NH3 production is externally purged N2 only. This was further supported by chronoamperometry measurements at open circuit potential (OCP), in which no NH3 production exists in the presence of Ar after chronoamperometry at −0.3 V, indicating that only the N2 purged into the electrolyte contributes to the NH3 production (Fig. S10, ESI†). Notably, any nitrate/nitrite species in the electrolyte, which may add to the false estimation of NH3 yield, are found to be absent as well (Fig. S11, ESI†).74
Another potential source of contamination during NRR is the presence of NO, NO2, and N2O in the gas supplies used and needs to be detected, quantified and subsequently eliminated to avoid any overestimation of NH3 yield. To serve this purpose, we followed a rigorous protocol in which NOx concentration in gas supplies was traced and quantified before NRR.75 First, the concentration of N2O if any in the gas supplies viz.14N2, 15N2 and Ar were quantified using GC-MS,76 from which a total of 0.06 ppm and 0.07 ppm N2O contamination was detected, while the N2O was below the detection limit (below 0.01 ppm) for Ar gas supply (Fig. S12A, ESI†). Other impurities, including trace NO/NO2 and NH4+, were quantified by employing the colorimetric method, which yielded a total of 0.09 and 0.08 ppm NH4+ in 14N2 and 15N2 gas-feed but no NH4+ in Ar gas-supply when trapped in acid solution. Note that 0.6 ppm, 1.1 ppm and 1.2 ppm of NOx were estimated for Ar, 14N2 and 15N2 gas supplies, respectively (Fig. S12B, ESI†). Even this small amount of contamination could impose a huge overestimation of NH3 yield because NOx may undergo a reduction reaction under identical experimental conditions and potential ranges. Therefore, after tracing all these possibilities of interference by NOx contamination during NRR, the same was then eliminated by carrying out gas purification by KMnO4 trap and H2SO4 solution (scrubbing solution) prior to every NRR measurement in this study. Additionally, to confirm the possible elimination of the NOx contaminants, we again quantified the same gaseous samples after purification by applying GC-MS and colorimetric methods (Fig. S13, ESI†). The absence of NOx and NH4+ impurities by both procedures is evident in the true estimation of NH3 yield in this study. For better understanding, a distinct comparison of NOx contamination in gas supplies before and after the purification is provided in Table S3 (ESI).† Later, the high NH3 yield rate achieved during NRR further prompted us to conduct 15N2 isotopic labelling experiments to confirm the true estimation of NH3. After bulk electrolysis for 2 h under a purified 15N2 gas supply, the formation of 15NH4+ was evaluated using 1H nuclear magnetic resonance (NMR) spectroscopy.
After potential electrolysis by Co2B (1:8) at −0.3 V (vs. RHE) for 2 h using Ar, 15N2 and 14N2 gas supply, the 1H NMR spectra for samples under N2 environment match well with standard couplings, revealing that the NH3 produced in this study is indeed from the electroreduction of supplied N2 rather than any possible contaminants (Fig. S14, ESI†). In addition to the qualitative tracing of true NH3 source, it is equally important to quantify the obtained yield rates under both 14N2 and 15N2 environments. To serve this purpose, we adapted two methods, i.e.1H-NMR spectroscopy and liquid chromatography-mass spectroscopy (LC-MS), respectively. The calibration curves were first extracted from the 1H-NMR spectrum of the standard aqueous 15NH4+ and 14NH4+ solutions with different concentrations from 1.0 to 5.0 ppm (Fig. S15A, ESI†). The NH3 yield was then quantified for NRR by Co2B catalyst at different time intervals (1 h and 2 h) performed under both 14N2 and 15N2 saturated electrolytic conditions (Fig. 2a and b). The NH3 yield rate was calculated to be 1.87 (1 h) and 2.94 mg h−1 mgcat.−1 (2 h) for 14NH4+ as well as 1.8 mg h−1 mgcat.−1 (1 h) and 2.92 mg h−1 mgcat.−1 (2 h) for 15NH4+ produced during NRR, respectively, which are analogous to each other. However, the NH3 yield rate obtained from the indophenol blue method for 14NH3 (2.94 mg h−1 mgcat.−1) and 15NH3 (2.93 mg h−1 mgcat.−1) also complements each other. In the end, LC-MS was employed to validate the NH3 yield after the isotope labelling experiment, which yields overall ammonia production yield rates of 2.98 and 2.95 mg h−1 mgcat.−1 for NRR performed under 14N2- and 15N2-saturated electrolyte conditions, respectively (Fig. S15B, ESI†). Fig. 2c and Table S4 (ESI)† demonstrate the comparison of NH3 yield obtained after isotope labelling experiments via various methods.
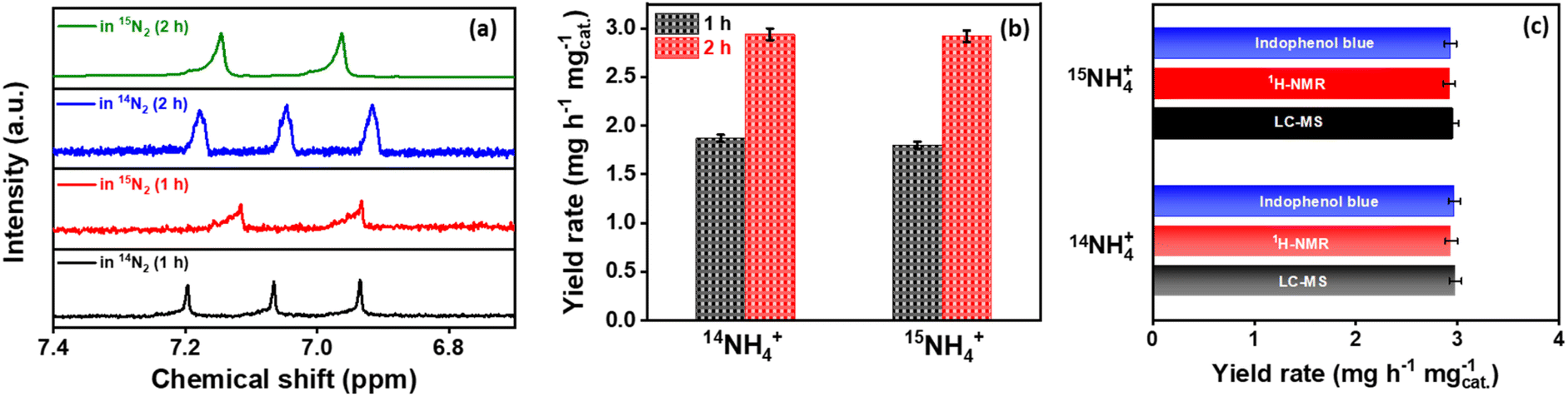 | ||
| Fig. 2 (a) 1H-NMR spectrum acquired after NRR isotope labelling experiments using Co2B (1:8) catalyst for different time intervals and (b) corresponding yield rates calculated from the same. (c) Bar diagram comparison of 14NH4+ and 15NH4+ yield rates obtained using different quantification methods. | ||
The disparity in the NRR activity of the catalysts with only a difference in metal:boron ratio during synthesis was then scrutinized by performing a series of ex situ characterizations. The P-XRD pattern of Co2B (1:8) in Fig. S16A (ESI)† reveals the formation of Co2B with a distinct peak at 2θ = 45° with no other peaks, indicating that the formation of pure amorphous Co2B phase agrees with that of the literature.77 The presence of additional peaks at 26° and 36.8° related to B2O3 (JCPDF card: 06-0297) and elemental B (JCPDF card: 12-0377) imply the amorphous boron-oxide layer over the catalyst surface in Co2B (1:12). However, we cannot deny the fact that the high intensity peak for Co2B can suppress the peaks for metallic cobalt and Co(OH)2 phase in XRD patterns. Therefore, to check the purity of the Co2B catalyst prepared, we further performed Raman spectroscopy analysis. As shown in Fig. S16B (ESI),† the Raman spectra for Co2B (1:4) and Co2B (1:8) do not reveal any peak related to metallic cobalt or Co(OH)2 phase, while the Raman spectra of Co2B (1:12) show the bands corresponding to B2O3 at 650 and 950 cm−1, respectively, along with a low intensity band at 465 cm−1 related to the O–Co–O bond in Co(OH)2. These results show that the bulk phase in the Co2B (1:8) catalyst is Co2B only, and upon further increasing the amount of boron, the catalyst forms a composite of Co2B/B2O3/Co(OH)2 with a major phase as Co2B only.
The nanosheet structure of Co2B (1:8) is supported by transmission electron microscopy (TEM) images (Fig. 3a and b). The high resolution-TEM (HR-TEM) images in Fig. 3c show the edge site of entangled nanosheets; these images do not show any clear lattice fringes but a very short-range ordered atomic arrangement at a few locations with an interlayer d spacing of 0.2 nm corresponding to the (210) plane of Co2B (Fig. 3d and e), and these arrangements could be embedded within the diffused amorphous phase,78,79 which are consistent with XRD data.77 The energy dispersive spectroscopic (EDS) elemental dot mapping reveals the co-existence of Co, B, and O elements, which are uniformly distributed in the scanned area, and their compositions are presented in Table S4 (Fig. S17, ESI).† The ratio of Co:B in Co2B (1:8) was estimated from the atomic percentage obtained from EDS (Table S5, ESI†) and was found to be 1.83:1 with a product comprising B (26.07 at%) and Co (47.76 at%) and the others mostly oxygen, yielding an approximate atomic Co:B ratio of 2:1. From XPS, the ratio of Co:B was observed to be 2.3:1, yielding the final composition of Co2.3B1.0 ≈ Co2B. Moreover, the MP-AES analysis produces a total of 79.1% cobalt and 10.5% boron in the catalyst, which describes the presence of Co2B as a major phase in the proposed catalyst with minute impurities.
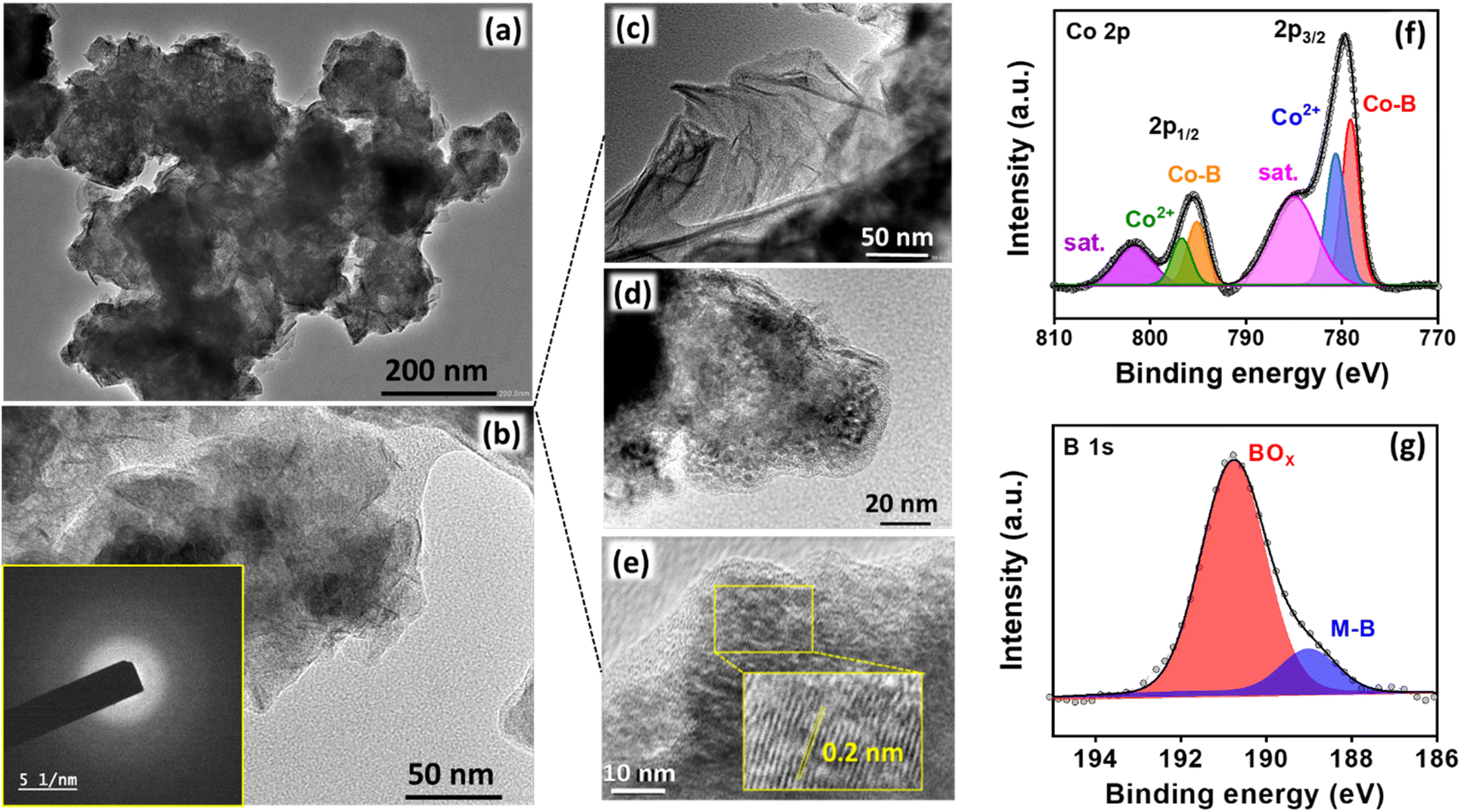 | ||
| Fig. 3 (a–e) HR-TEM images of Co2B (1:8) at different magnifications (inset of (b) shows SAED pattern for Co2B (1:8) revealing an amorphous nature and inset of (e) shows d-spacing corresponding to Co2B with short range atomic arrangement at few locations). (f) Deconvoluted XP spectra of Co 2p and (g) XP spectra of B 1s for Co2B (1:8) catalyst. | ||
Subsequently, to understand the surface properties of Co and B in the Co2B (1:8) catalyst, X-ray photoelectron spectroscopy (XPS) analysis was performed. The XP survey spectrum indicates the presence of both Co 2p and B 1s peaks (Fig. S18, ESI†) with no additional peaks due to impurity, which reinforces the fact that the synthesized catalyst is highly pure.
The deconvoluted Co 2p XP spectra in Fig. 3f display two distinct peaks at binding energies of 779.07 and 795.4 eV with additional satellite peaks at 785.1 eV and 801.7 eV corresponding to Co 2p3/2 and Co 2p1/2, respectively. The presence of a peak at 778.5 and 795 eV is attributed to the formation of metal-boride.80 However, B 1s XP spectra were deconvoluted into two peaks in which the peak at 188.6 eV corresponds to the interaction of boron with cobalt, which is shifted towards higher energies by 1.6 eV w.r.t elementary boron (187 eV) most probably due to electron transfer from B to vacant d-orbital of Co metal.81 Moreover, an additional dominant peak at 191.5 is due to boron-oxo species (Fig. 3g), which is characteristic of amorphous metal-borides upon exposure to the atmosphere. The reason behind the large B–O peak in the B 1s XP spectrum is the presence of oxidized boron species at a binding energy value of 191.5 eV. This indicates that the Co2B nanoparticles were oxidized to a certain degree, while the bulk phase remains in the Co2B state. The formation of boron oxide was quite expected as the work-up (filtering and washing) of the prepared catalyst was performed in the air. This BO2− species might have appeared because of the self-hydrolysis of the non-stabilized NaBH4. The commercial bulk CoxB sample also shows the peaks related to boride and borate/oxidized boron.64,80 Moreover, the activity and stability of the catalyst during NRR are crucial, for which a stability test was conducted by applying chronoamperometry at −0.3 V for 10 h. Fig. S19 (ESI)† illustrates a stable current density response, and the quantification of NH3 after every 2 h reveals a negligible change in both faradaic efficiency and NH3 yield rate (Fig. 4a), assisting the high stability of electrocatalyst. Moreover, to observe the structural stability of the catalyst after NRR, we examined its physicochemical properties by P-XRD, FE-SEM and EDS dot mapping. The morphology was retained after the 10 h of NRR stability test, with well-distributed boron and cobalt over the entire scanned area in EDS dot mapping analysis, as shown in Fig. S20 (ESI).† All these findings demonstrate the high durability of Co2B for electrochemical NH3 production.
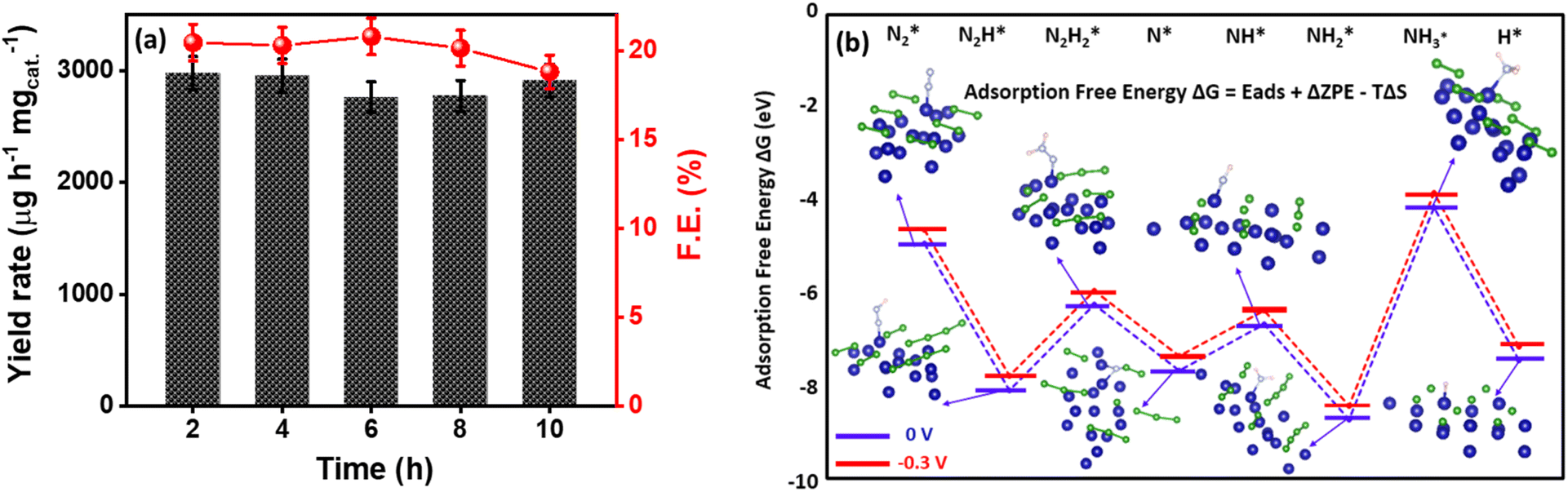 | ||
| Fig. 4 (a) F.E. and NH3 yield obtained after chronoamperometry tests performed for 10 h under N2-saturated 0.1 M KOH for Co2B (1:8). (b) Free energy diagrams of the NRR distal pathway on Co2B surface (Co active site) at applied potentials of 0 and −0.3 V and H+ adsorption at identical potentials. | ||
Theoretical investigations
To gain a deep insight into the NRR mechanism over the Co2B surface, systematic electronic structure calculations along with the framework of density functional theory (DFT) formalism were performed to anticipate the nitrogen reduction to ammonia on the experimentally synthesized Co2B catalyst surface. Because no hydrazine formation was witnessed during the quantification of products after NRR, we determined the adsorption free energies for seven intermediates (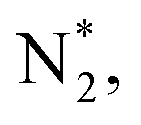 N2H*,
N2H*, 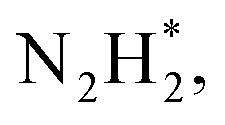 N*, NH*,
N*, NH*, 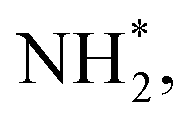 and
and 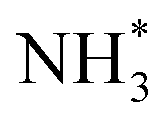 ) corresponding to both the Co (Fig. 4b) and B sites (Fig. S21, ESI†) of the catalytic Co2B surface. It seems that the Co site is more favorable for the NRR. The greater electron affinity of Co could be a possible reason for the favorable NRR mechanism. We also calculated the adsorption free energy of hydrogen (H*) to check the possibility of a hydrogen evolution reaction. In the plot reaction, coordinates were generated from Gibbs adsorption free energy (ΔG) corresponding to different electrode potentials of 0 V and −0.3 V (Table S5B and C, ESI,† where the maximum NH3 F.E. is achieved). Here, the adsorption energy is defined as follows:
) corresponding to both the Co (Fig. 4b) and B sites (Fig. S21, ESI†) of the catalytic Co2B surface. It seems that the Co site is more favorable for the NRR. The greater electron affinity of Co could be a possible reason for the favorable NRR mechanism. We also calculated the adsorption free energy of hydrogen (H*) to check the possibility of a hydrogen evolution reaction. In the plot reaction, coordinates were generated from Gibbs adsorption free energy (ΔG) corresponding to different electrode potentials of 0 V and −0.3 V (Table S5B and C, ESI,† where the maximum NH3 F.E. is achieved). Here, the adsorption energy is defined as follows:| Ead = Esub+ads − Esub − Eads, |
| ΔG = Ead + ΔZPE − TΔS + ΔGU, |
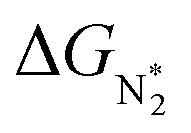 value over the Co2B surface is −4.93 eV at 0 V of the applied potential, which supports the high activity of the catalyst at −0.3 V (with a
value over the Co2B surface is −4.93 eV at 0 V of the applied potential, which supports the high activity of the catalyst at −0.3 V (with a 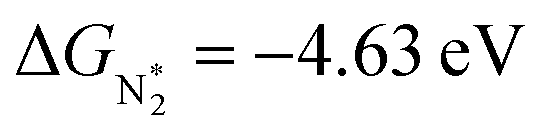 ). Further NRR mechanism analysis was performed by the free energy diagram, in which the first step proceeds via end-on adsorption of N2 molecule over the Co active site of the Co2B surface with an adsorption energy of −4.63 eV at −0.3 V (vs. RHE). Thereafter, the adsorbed N2
). Further NRR mechanism analysis was performed by the free energy diagram, in which the first step proceeds via end-on adsorption of N2 molecule over the Co active site of the Co2B surface with an adsorption energy of −4.63 eV at −0.3 V (vs. RHE). Thereafter, the adsorbed N2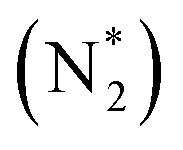 species undergoes the first hydrogenation step (H+/e−) to form N2H* with a downhill in the corresponding free energy profile by −7.74 eV. In the second hydrogenation step, the N2H* intermediate captures another H+ + e− pair to form
species undergoes the first hydrogenation step (H+/e−) to form N2H* with a downhill in the corresponding free energy profile by −7.74 eV. In the second hydrogenation step, the N2H* intermediate captures another H+ + e− pair to form 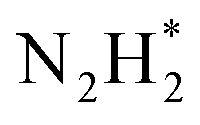 with an uphill Gibbs free energy change of 1.81 eV (rate-limiting step). The third hydrogenation step proceeds with the formation of N* + NH3, leading to the release of the first NH3 molecule by following an exothermic pathway with a free energy change of −1.39 eV. Successful hydrogenation of the remaining adsorbed N (N*) occurs with corresponding free energy changes of 0.98, −2.03 and 4.51 eV to form NH*,
with an uphill Gibbs free energy change of 1.81 eV (rate-limiting step). The third hydrogenation step proceeds with the formation of N* + NH3, leading to the release of the first NH3 molecule by following an exothermic pathway with a free energy change of −1.39 eV. Successful hydrogenation of the remaining adsorbed N (N*) occurs with corresponding free energy changes of 0.98, −2.03 and 4.51 eV to form NH*,  and
and 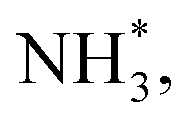 respectively, followed by the desorption of the second NH3 molecule.
respectively, followed by the desorption of the second NH3 molecule.
In addition to the activity of the catalyst for the adsorption and activation of the NN bond, selectivity over H+ adsorption to suppress competing HER is an imperative aspect. Under thermoneutral conditions, the optimal catalysts should not attain an adsorption free energy close to zero for H+ adsorption. The value of adsorption free energy corresponding to N2 is calculated to be −4.63 eV at −0.3 V, while it is −7.12 eV for H+ adsorption. This supports the fact that the Co2B catalyst surface provides optimum active sites for NRR than HER at an identical potential.
Oxygen bifunctional activity for the air-cathode
We further investigated the potential efficacy of Co2B variants in oxygen bifunctional activity viz. the oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) to understand its ability to serve as an air cathode in Zn–air batteries. Initially, the catalytic activity of the Co2B catalyst towards ORR was examined in a 0.1 M KOH electrolyte. The cyclic voltammetry (CV) curves of Co2B (1:8) exhibit the most positive reduction peak potential at 0.97 V vs. RHE (Fig. 5a and S22, ESI†), indicating its superior ORR activity compared with those of Co2B (1:4) and Co2B (1:12). As observed from voltammetric curves in Fig. 5a, the Co2B (1:8) shows an obvious increase in cathodic current at a potential of 0.97 V vs. RHE in an O2 saturated 0.1 M KOH electrolyte, and no such behaviour was observed in an oxygen free electrolyte (N2 saturated). The obtained reduction potential is more positive compared to those of the other two variants. These results can be attributed to a reduction in oxygen. To understand the kinetics of the ORR, rotating ring disc electrode measurements (RRDE) were performed at various rotating rates under hydrodynamic conditions in 0.1 M KOH electrolyte (Fig. S23, ESI†). The RRDE curves depicted in Fig. 5b disclose an exponential current increment for Co2B (1:8) with an increase in rotation rate and reached up to the diffusion-limited current density of −4.9 mA cm−2 at 1300 rpm with an onset potential (Eonset) of 0.98 V vs. RHE (Table S6A, ESI†). The comparative analysis of disc and ring current densities depicted in Fig. 5c indicates that Co2B (1:8) has activity superior activity compared to other variants. Notably, the ring current of Co2B (1:8) is lower than those of the other variants. The half wave potentials can be a descriptor of ORR activity in the mixed kinetic/diffusion-controlled region, wherein Co2B (1:8) exhibits a half-wave potential (E1/2) of 0.83 V, which is 170 and 140 mV higher than that of Co2B (1:12) and Co2B (1:4) respectively, suggesting superior ORR activity. More interestingly, compared with the benchmark catalyst, i.e. Pt/C, a comparable activity is observed in terms of onset potential and diffusion limiting current density and specific mass activity at a similar potential (Fig. 5c, d and S24, ESI†). Hence, apart from the remarkable NRR activity, Co2B (1:8) exhibits outstanding ORR activity. The kinetic parameters were evaluated using Koutecky–Levich (K–L) plots derived from LSV curves to gain deeper insights into the mechanism governing the ORR reaction. The K–L plots represent a linear behavior between j−1vs. ω−1/2 attributed to first-order reaction kinetics with respect to the dissolved O2 (Fig. S25, ESI†).
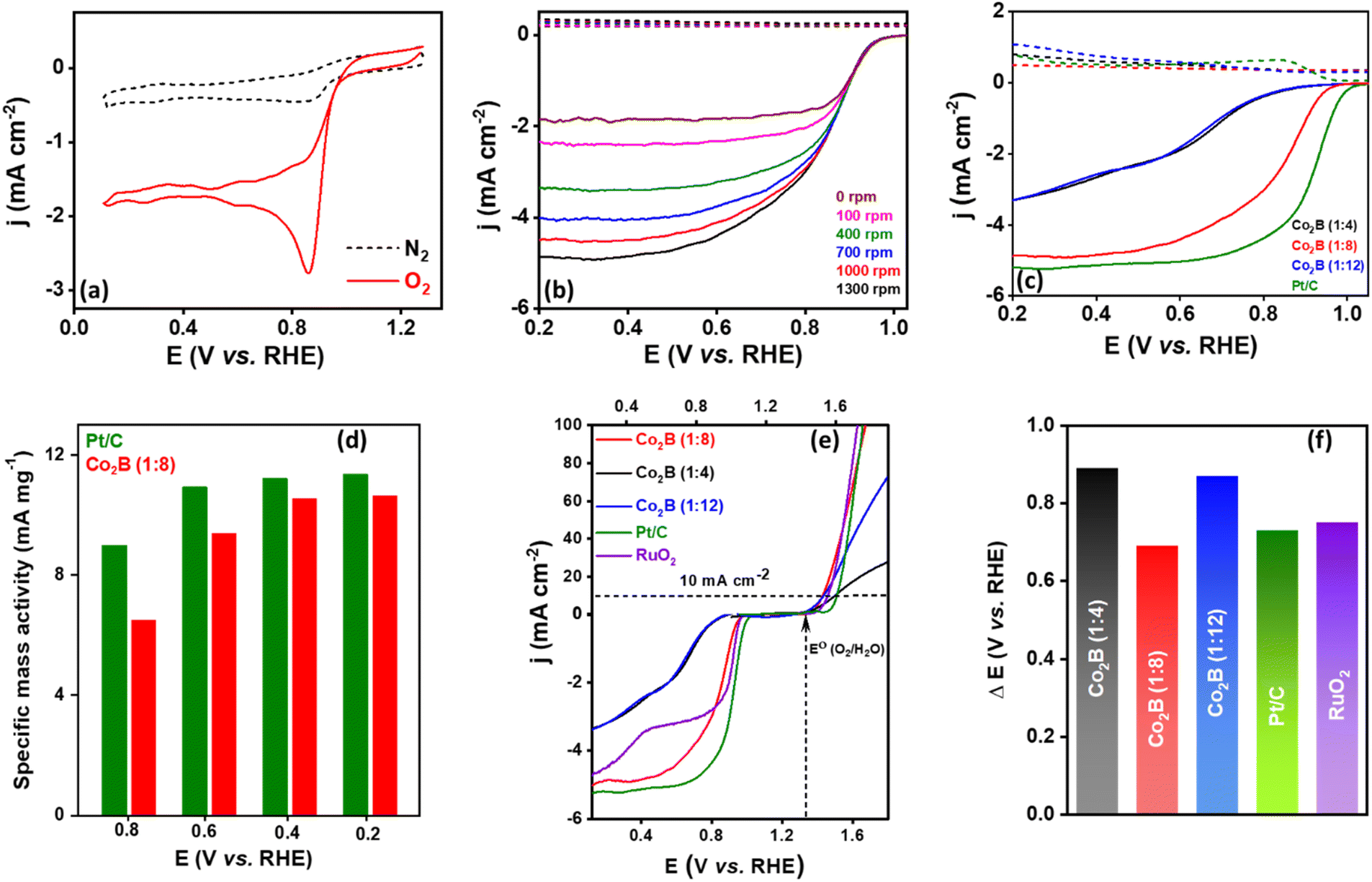 | ||
| Fig. 5 (a) Cyclic voltammogram of Co2B (1:8) for oxygen reduction activity in 0.1 M KOH at 25 mV s−1. (b) RRDE polarization curves of Co2B (1:8) in the presence of O2 at varying rotation rates and at a scan rate of 5 mV s−1. (c) Comparison of RRDE curves of Co2B catalysts with state-of-art Pt/C catalyst. (d) Comparison of specific mass activity for Pt/C and Co2B (1:8) at different potentials, (e) oxygen bifunctional activity of Co2B catalysts and commercial benchmark catalysts and (f) comparison of a potential gap for Zn–air battery. | ||
Further, an apparent 4e− pathway, followed by Co2B (1:8) during oxygen reduction, was confirmed by calculating the number of e− transfer (n) from RRDE curves, and the quantitative estimation of peroxide (Fig. S26 and Table S6B, ESI†) produced during ORR was obtained using the following equations:
| n = 4Id/[Id + (Ir/N)], | (1) |
| % H2O2 = 200 × (Ir/N)/[Id + (Ir/N)]. | (2) |
:8) impersonates a complete four electron transfer (n ≈ 4), revealing its ability to conduct the direct 4e− pathway during O2 reduction to H2O as a final product. Additionally, the yield of H2O2 generated was found to be very low (less than 9%) for Co2B (1:8), which is highly anticipated, because H2O2 production could adversely affect the cell components.82 Therefore, chronoamperometry, performed at an onset potential of ORR for a time duration of 1 day in O2-saturated 0.1 M KOH, showed a stable current response even after a prolonged stability study (Fig. S28, ESI†). These outcomes clearly validate the capability of Co2B (1:8) to reduce O2 with enhanced activity and stability, which is very essential for practical applications. To determine the electrocatalytic ability of the Co2B catalysts towards the OER, linear sweep voltammetry (LSV) experiments were performed in 0.1 KOH at a scan rate of 5 mV s−1. Analogous to the activity obtained during ORR, Co2B (1:8) displays significantly enhanced OER activity than Co2B (1:4) and Co2B (1:12) under identical conditions. It is evident from Fig. S29 (ESI)† that oxygen gas evolution was observed at 1.5 V on the surface of Co2B (1:8) and became sturdy as the potential scan progressed towards anodic potentials, reaching 100 mA cm−2 at 1.6 V. The Co2B (1:8) achieves a 10 mA cm−2 current density at an overpotential of (η) 290 mV, which is lower than that of Co2B (1:4) and Co2B (1:12) with an η value of 360 and 300 mV, respectively (Table S7, ESI†).
Hence, apart from the superior ORR activity, Co2B (1:8) also shows remarkable OER activity. The higher OER activity could be attributed to the strain originating in Co2B due to the chemical hybridization of B 2p and Co vacant 3d orbitals, which may presumably reduce the activation energy barrier for Co oxidation.57 This strong hybridization is supported by the presence of the M–B bond revealed by XPS studies (Fig. 3g), which could facilitate the OOH* intermediate formation and in turn OER (M–OH/M–OOH species being an active site). The Tafel slope of the Co2B (1:8) supports facilitated OER, which was found to be the least when compared with other catalysts, indicating the faster reaction rate during OER (Fig. S30, ESI†). The chronopotentiometric stability test of Co2B (1:8) shows a negligible change in the overpotential required to deliver 10 mA cm−2 even over 24 h (Fig. S31a, ESI†). In addition, the stability was further examined under different current densities from 10 to 50 mA cm−2, in which a continuous increase in cell potential was observed as the current density increased but underwent rapid stabilization (Fig. S31b, ESI†). The cell potential was again restored when the applied current density returns to 10 from 50 mA cm−2, indicating superior stability.
Metal borides are usually susceptible to undergo in situ electrochemical oxidation during OER to generate metal oxyhydroxides and metal borates.83 The post-OER XPS analysis acquired (Fig. S32, ESI†) after 24 h stability test reveals the formation of Co–OOH species at 779.1 eV in Co 2p XP spectra. The deconvoluted B 1s XP spectra show a peak related to boron-oxo species (B–O), but the diminished intensity of the B peak related to boride could be attributed to the shielding of the catalyst surface by the oxyhydroxide layer after the OER. This B–O species may either stick onto the catalyst surface or dissolve in the electrolyte and further create pores to increase active sites during OER.57 During OER in alkaline electrolytes, either Co–OOH or B–O species could serve as an active site. In one case, Co–OOH may serve as the major active site, while borate may modulate the adsorption energies of the reaction intermediates. The other case could be the improved electron transfer between partially oxidized metal oxyhydroxide and the current collector due to active non-oxidized metal boride centers.84 Therefore, the overall significant electrocatalytic activity of Co2B (1:8) indicates that it is an extremely capable bifunctional oxygen catalyst. This is further validated by the oxygen polarization curves representing ORR and OER bifunctional activity (Fig. 5e). An exclusive factor to investigate the ability of different catalysts towards oxygen bifunctional activity is the potential gap (ΔE). In this study, the ΔE (ΔE = E@10 mA cm−2 − E1/2@ORR) value was calculated using the potential values amid OER (10 mA cm−2) and ORR (half-wave potential). The lower ΔE value of 0.69 V for Co2B (1:8) considerably demonstrates the superior electrocatalytic performance for ORR/OER than 0.89 V for (1:4) and 0.87 V for the (1:12) ratio of Co2B, 0.73 V for Pt/C and 0.75 V for RuO2 (Fig. 5f and Table S8, ESI†). A Zn–air battery equipped with a Co2B (1:8) air cathode was used. To investigate the potential application of the outstanding oxygen bifunctional Co2B (1:8) catalyst towards energy conversion and storage devices, a homemade Zn–air battery was assembled by employing Co2B (1:8) air-cathode catalyst with Zn foil anode using a 6 M KOH + 0.2 M zinc acetate electrolyte. The assembled battery exhibits a high open circuit voltage of 1.45 V (Fig. 6a), which is close to that of Pt/C + RuO2 air cathode (1.48 V) with identical mass loading (2 mg cm−2). The polarization curves during discharge and the corresponding power density plots are presented in Fig. 6b and c, respectively. By employing Co2B (1:8) as an air cathode, the peak power density of the battery was found to be 500 mW cm−2, which is superior to that of Pt/C + RuO2 (200 mW cm−2, Fig. 6c) and those reported in the literature. The high-power density of the battery mainly depends on the discharge efficacy of the catalyst at the cathode, i.e. to carry out ORR with high activity in which the high O2 diffusion rate at the cathode is responsible for high discharge current density and thus the high power density. Therefore, in this case, the high peak power density can be related to the following properties.
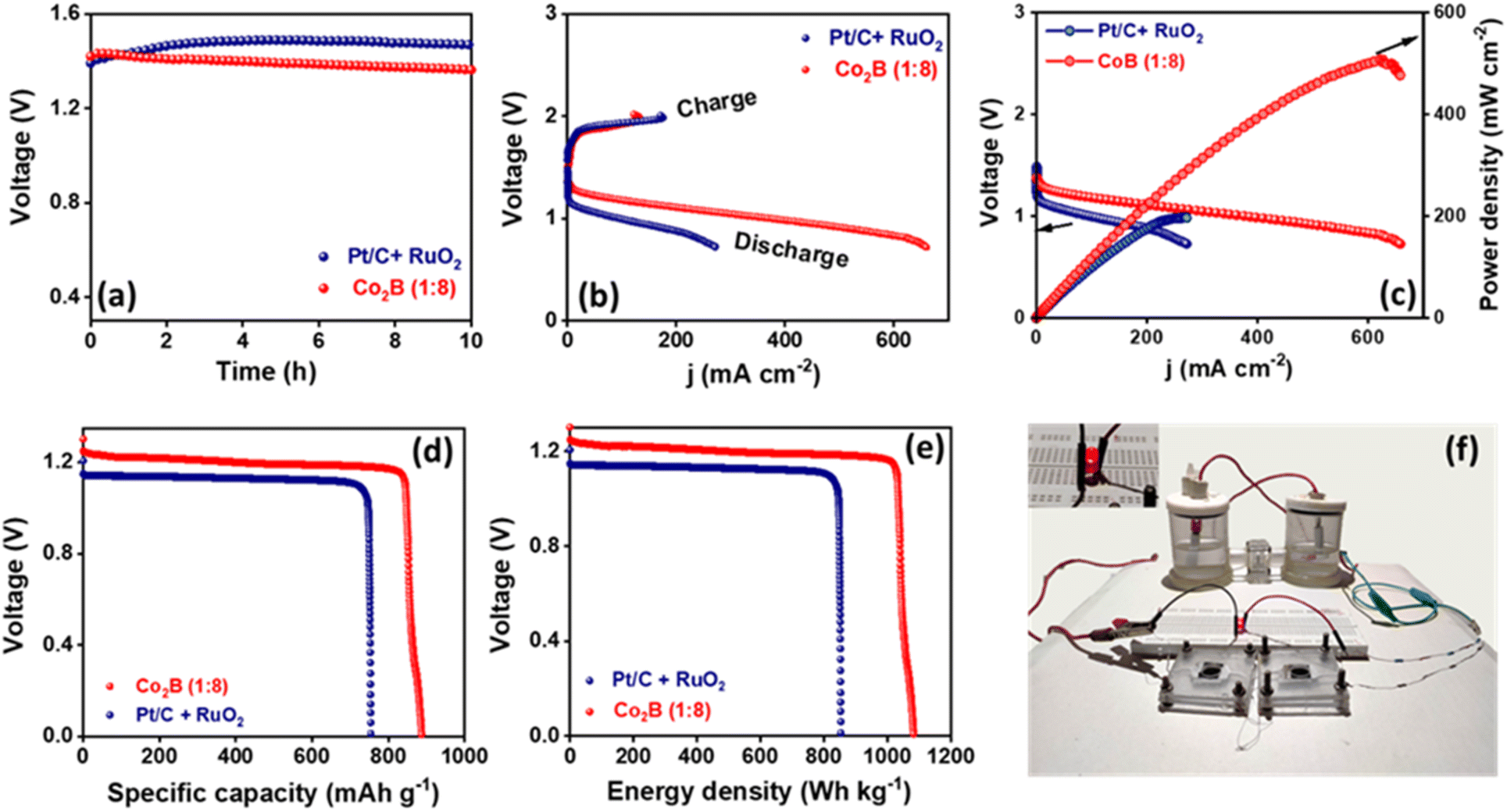 | ||
| Fig. 6 (a) Open circuit voltage, (b) charge–discharge polarization curves, (c) power density plots extracted from discharge polarization curve for Zn–air battery assembled using Co2B (1:8) and Pt/C as an air cathode. (d) Corresponding specific capacity and (e) energy density curves obtained at 10 mA cm−2. (f) Photograph of Zn–air battery-powered NRR full cell setup (air cathode in battery: Co2B (1:8); cathode and anode in NRR full cell: Co2B (1:8)). | ||
More importantly, the Co2B (1:8) air cathode shows a small charge–discharge voltage gap of 0.61 V, which is less than that of Pt/C + RuO2, demonstrating a promising future as an air-cathode alternative to noble metal-based electrodes in Zn–air batteries. Co2B (1:8) air cathode further demonstrates a stable response at 10 mA cm−2 with an overall cell voltage of 1.27 V (Fig. 6d and e), resulting in a high specific capacity of 889 mA h g−1 and an energy density of 1078 W h kg−1, which are higher than those of Pt/C + RuO2 (748 mA h g−1 and 860 W h kg−1, normalized w.r.t the mass of lost zinc). The durability of the proposed Co2B (1:8) air cathode by galvanostatic-cycling performance of the battery at 10 mA cm−2 demonstrates that an initial overall potential gap of 0.61 V is retained to 0.75 V for 17 h, with each cycle having 10 min of discharging and 10 min of charging (Fig. S33A, ESI†). However, Pt/C + RuO2 suffers severe degradation in the potential gap (from 0.63 V to 1.6 V) within 45 cycles (15 h) most probably due to Ostwald ripening and carbon corrosion, leading to the aggregation of noble metals. Further, to validate the practicability of the designed battery, it was connected in series and applied to power an LED. As shown in Fig. S33B (ESI),† two Zn–air batteries equipped with the catalyst provide adequate voltage to power a red LED (2.0 V).
Zn–air battery-powered NRR
To demonstrate the ability of the assembled Zn–air battery to power the two-electrode NRR cell, we made a setup of two batteries connected in series to supply power to drive ammonia synthesis (Fig. 6f). During the integration of Zn–air batteries (2.9 V) with the full cell NH3 synthesis device, we utilised a resistor (300 Ω) to limit the excessive current supply to the cell, thus maintaining the efficiency of the device to achieve the maximum NH3 production yield rate. A two-electrode NRR cell was constructed by employing Co2B (1:8) as a bifunctional catalyst at both the anode and cathode. The bifunctional ability of the catalyst to carry out NRR at the cathode and OER at the anode is supported using LSV acquired under full cell conditions in a potential ranging from 0 to 2.5 V (Fig. S34, ESI†). The steep increase in current density at 1.6 V describes the onset potential for overall NH3 synthesis. Additionally, LSV was evaluated in Ar and N2 saturated electrolytes to check the ammonia production in full cells using a potentiostat. An increase in current density (cathodic) in the presence of N2 gives a picture of successful nitrogen reduction to ammonia (Fig. S35, ESI†). On further performing 2 h of electrolysis, the home-built NRR cell setup powered by assembled Zn–air batteries could deliver an overall NH3 yield rate of 1048.2 μg h−1 mgcat.−1 (Fig. S35, ESI†). These results indicate the ability of the catalyst to be applied for practical applications. To date, very few self-powered electrochemical NH3 synthesis setups have been reported, amongst which our study reveals the maximum yield rate within 2 h (Table S9, ESI†). The outstanding trifunctional activity towards NRR/OER/ORR could lead to a new pathway towards the development of cost-effective catalysts for Zn–air battery and NH3 synthesis and their implementation towards powering the ammonia production by sustainable and high-power density rechargeable Zn–air batteries.
Conclusions
In summary, we report the synthesis of cobalt boride (Co2B) catalyst via an environment friendly, economical and less-time-consuming sonochemical route using non-expensive precursors. The catalyst demonstrates remarkable activity with a high faradaic efficiency and yield rate for NRR. This high activity is attributed to the strong M–B bond and the nanosheet morphology, providing an ample number of active channels for mass and charge transfer. Systematic DFT investigations are carried out to estimate NRR activity over the Co2B catalyst surface, in which the Co acts as an active site to adsorb and activate strong NN and produce NH3via an associative distal pathway. The undesired products viz. H2 owing to the competition between NRR and HER can be effectively suppressed at the catalyst active sites, indicating the high selectivity of Co2B towards NH3 production. When employed as a cathode catalyst for Zn–air battery, Co2B (1:8) unveils substantial electrocatalytic performance, yielding a reduced potential gap, improved rate of reaction, and high durability. Interestingly, the assembled Zn–air battery can power NH3 synthesis with an appreciable NH3 production yield of 1048.2 μg h−1 mgcat.−1 after two continuous hours of electrolysis. Being a non-noble, simple and facile synthesis strategy, the high trifunctional activity of Co2B holds great promise for energy conversion and storage devices.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. Gupta thanks DST for DST-INSPIRE fellowship (DST/INSPIRE/03/2017/000005), A. Kafle thanks IIT Ropar for fellowship. T. C. Nagaiah thanks Science and Engineering Research Board (SPG/2021/002415). T. D. and S. C. would like to acknowledge HRI Allahabad and DST-SERB Funding (SRG/2020/001707) for the infrastructure and funding. Computational work for this study was carried out at the cluster computing facility in the Harish-Chandra Research Institute (https://www.hri.res.in/cluster). P. P. M. would like to acknowledge INSPIRE PhD Fellowship and IIT Ropar for infrastructure. R. A. also thanks the Swedish Research Council (VR-2016-06014 and VR-2020-04410) for financial support. We are thankful to Sophisticated Analytical Instrumentation Facility (SAIF) and Central Instrumentation Facility (CIL), Panjab University, Chandigarh, India, for providing FE-SEM and HRT-TEM facilities. We also thank LC-MS facility at IIT Ropar procured through FIST-DST grant (SR/FST/CS-I/2018/55) for quantitative product analysis. We acknowledge Prof. Sanjay Mandal and his group at IISER Mohali, Punjab, India, for providing the BET measurements used in this work.References
- D. Bogdanov, A. Gulagi, M. Fasihi and C. Breyer, Appl. Energy, 2021, 283, 116273 CrossRef.
- V. Rosca, M. Duca, M. T. de Groot and M. T. Koper, Chem. Rev., 2009, 109, 2209–2244 CrossRef CAS PubMed.
- G. Leigh, in Catalysts for Nitrogen Fixation, Springer, 2004, pp. 33–54 Search PubMed.
- G. Soloveichik, Nat. Catal., 2019, 2, 377–380 CrossRef CAS.
- V. Kyriakou, I. Garagounis, A. Vourros, E. Vasileiou and M. Stoukides, Joule, 2020, 4, 142–158 CrossRef CAS.
- M. Wang, M. A. Khan, I. Mohsin, J. Wicks, A. H. Ip, K. Z. Sumon, C.-T. Dinh, E. H. Sargent, I. D. Gates and M. G. Kibria, Energy Environ. Sci., 2021, 14, 2535–2548 RSC.
- C. Smith, A. K. Hill and L. Torrente-Murciano, Energy Environ. Sci., 2020, 13, 331–344 RSC.
- C. A. Fernandez and M. C. Hatzell, J. Electrochem. Soc., 2020, 167, 143504 CrossRef CAS.
- S. Ghavam, M. Vahdati, I. Wilson and P. Styring, Front. Energy Res., 2021, 9, 34 Search PubMed.
- U. B. Shahid, K. Siddharth and M. Shao, Curr. Opin. Electrochem., 2021, 30, 100790 CrossRef CAS.
- H. Xu, K. Ithisuphalap, Y. Li, S. Mukherjee, J. Lattimer, G. Soloveichik and G. Wu, Nano Energy, 2020, 69, 104469 CrossRef CAS.
- C. Zhao, S. Zhang, M. Han, X. Zhang, Y. Liu, W. Li, C. Chen, G. Wang, H. Zhang and H. Zhao, ACS Energy Lett., 2019, 4, 377–383 CrossRef CAS.
- Q. Liu, Z. Pan, E. Wang, L. An and G. Sun, Energy Storage Mater., 2020, 27, 478–505 CrossRef.
- M. Kumar and T. C. Nagaiah, J. Mater. Chem. A, 2022, 10, 4720–4730 Search PubMed.
- X. Meng, J. Han, L. Lu, G. Qiu, Z. L. Wang and C. Sun, Small, 2019, 15, 1902551 CrossRef CAS PubMed.
- X. Yang, K. Li, D. Cheng, W.-L. Pang, J. Lv, X. Chen, H.-Y. Zang, X.-L. Wu, H.-Q. Tan and Y.-H. Wang, J. Mater. Chem. A, 2018, 6, 7762–7769 RSC.
- Z. Zhao, X. Fan, J. Ding, W. Hu, C. Zhong and J. Lu, ACS Energy Lett., 2019, 4, 2259–2270 CrossRef CAS.
- G. Qing, R. Ghazfar, S. T. Jackowski, F. Habibzadeh, M. M. Ashtiani, C.-P. Chen, M. R. Smith III and T. W. Hamann, Chem. Rev., 2020, 12, 5437–5516 CrossRef PubMed.
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- Z.-L. Wang, D. Xu, J.-J. Xu and X.-B. Zhang, Chem. Soc. Rev., 2014, 43, 7746–7786 RSC.
- D. U. Lee, P. Xu, Z. P. Cano, A. G. Kashkooli, M. G. Park and Z. Chen, J. Mater. Chem. A, 2016, 4, 7107–7134 RSC.
- Y. Kong, Y. Li, B. Yang, Z. Li, Y. Yao, J. Lu, L. Lei, Z. Wen, M. Shao and Y. Hou, J. Mater. Chem. A, 2019, 7, 26272–26278 RSC.
- G. Fenya, L. Hongwei, Z. Mengzhe, X. Zhengqi, Z. Yueqing and L. Tingting, Prog. Chem., 2020, 32, 33 Search PubMed.
- K. Zeng, X. Zheng, C. Li, J. Yan, J. H. Tian, C. Jin, P. Strasser and R. Yang, Adv. Funct. Mater., 2020, 30, 2000503 CrossRef CAS.
- Z. Lu, J. Wang, S. Huang, Y. Hou, Y. Li, Y. Zhao, S. Mu, J. Zhang and Y. Zhao, Nano Energy, 2017, 42, 334–340 CrossRef CAS.
- S. Ghosh and R. N. Basu, Nanoscale, 2018, 10, 11241–11280 RSC.
- T. Meng, J. Qin, S. Wang, D. Zhao, B. Mao and M. Cao, J. Mater. Chem. A, 2017, 5, 7001–7014 RSC.
- J. Yang, X. Wang, B. Li, L. Ma, L. Shi, Y. Xiong and H. Xu, Adv. Funct. Mater., 2017, 27, 1606497 Search PubMed.
- Q. Zhang, F. Luo, Y. Ling, S. Xiao, M. Li, K. Qu, Y. Wang, J. Xu, W. Cai and Z. Yang, J. Mater. Chem. A, 2020, 8, 8430–8439 RSC.
- A. Kafle, D. Gupta, A. Bordoloi and T. C. Nagaiah, Nanoscale, 2022, 14, 16590–16601 RSC.
- J. Li, F. Wei, C. Dong, Z. Wang, Z. Xiu and X. Han, Mater. Today Energy, 2021, 100766 CrossRef CAS.
- D. Gupta, A. Kafle, S. Kaur, P. P. Mohanty, T. Das, S. Chakraborty, R. Ahuja and T. C. Nagaiah, J. Mater. Chem. A, 2022, 10, 20616–20625 RSC.
- D. Gupta, A. Kafle and T. C. Nagaiah, Faraday Discuss., 2023 10.1039/D2FD00150K.
- Z. Xiang, L. Li, Y. Wang and Y. Song, Chem.–Asian J., 2020, 15, 1791–1807 CrossRef CAS PubMed.
- D. Gupta, A. Kafle and T. C. Nagaiah, Small, 2023, 2208272 CrossRef PubMed.
- S. Zhao, X. Lu, L. Wang, J. Gale and R. Amal, Adv. Mater., 2019, 31, 1805367 CrossRef PubMed.
- L.-H. Zhang, F. Yu and N. R. Shiju, ACS Sustain. Chem. Eng., 2021, 9, 7687–7703 CrossRef CAS.
- D. Gupta, A. Kafle, S. Kaur, T. S Thomas, D. Mandal and T. C. Nagaiah, ACS Appl. Mater. Interfaces, 2023, 15, 4033–4043 CrossRef CAS PubMed.
- D. Liu, M. Chen, X. Du, H. Ai, K. H. Lo, S. Wang, S. Chen, G. Xing, X. Wang and H. Pan, Adv. Funct. Mater., 2021, 31, 2008983 CrossRef CAS.
- S. L. Foster, S. I. P. Bakovic, R. D. Duda, S. Maheshwari, R. D. Milton, S. D. Minteer, M. J. Janik, J. N. Renner and L. F. Greenlee, Nat. Catal., 2018, 1, 490–500 CrossRef.
- X. Guo, H. Du, F. Qu and J. Li, J. Mater. Chem. A, 2019, 7, 3531–3543 RSC.
- Y. Sun, X. Liu, Y. Jiang, J. Li, J. Ding, W. Hu and C. Zhong, J. Mater. Chem. A, 2019, 7, 18183–18208 RSC.
- Y. Gao, T. Zhang, Y. Mao, J. Wang and C. Sun, J. Electroanal. Chem., 2023, 935, 117315 CrossRef CAS.
- J. Bian, X. Cheng, X. Meng, J. Wang, J. Zhou, S. Li, Y. Zhang and C. Sun, ACS Appl. Energy Mater., 2019, 2, 2296–2304 CrossRef CAS.
- E. Marini, L. Jörissen and S. Brimaud, J. Power Sources, 2021, 482, 228900 CrossRef CAS.
- T. C. Nagaiah, D. Gupta, S. D. Adhikary, A. Kafle and D. Mandal, J. Mater. Chem. A, 2021, 9, 9228–9237 RSC.
- S. Haller, V. Gridin, K. Hofmann, R. W. Stark, B. Albert and U. I. Kramm, Energy Technol., 2021, 9, 2001106 CrossRef CAS.
- J. Han, H. Bao, J.-Q. Wang, L. Zheng, S. Sun, Z. L. Wang and C. Sun, Appl. Catal., B, 2021, 280, 119411 CrossRef CAS.
- J. Han, X. Meng, L. Lu, Z. L. Wang and C. Sun, Nano Energy, 2020, 72, 104669 CrossRef CAS.
- N. Thakur, M. Kumar, D. Mandal and T. C. Nagaiah, ACS Appl. Mater. Interfaces, 2021, 13, 52487–52497 CrossRef CAS PubMed.
- Z. Pu, T. Liu, G. Zhang, X. Liu, M. A. Gauthier, Z. Chen and S. Sun, Small Methods, 2021, 5, 2100699 CrossRef CAS PubMed.
- L. Yi, Z. Zeng, Y. Liao, Y. Tang, X. Li, N. Lv, Y. Xu, H. Li and Y. Wang, Energy Fuels, 2021, 12, 10315–10321 CrossRef.
- R. K. Tripathy, A. K. Samantara and J. Behera, Sustain. Energy Fuels, 2021, 5, 1184–1193 RSC.
- S. Carenco, D. Portehault, C. Boissiere, N. Mezailles and C. Sanchez, Chem. Rev., 2013, 113, 7981–8065 CrossRef CAS PubMed.
- X. Ma, K. Zhao, Y. Sun, Y. Wang, F. Yan, X. Zhang and Y. Chen, Catal. Sci. Technol., 2020, 10, 2165–2172 RSC.
- N. Xu, G. Cao, Z. Chen, Q. Kang, H. Dai and P. Wang, J. Mater. Chem. A, 2017, 5, 12379–12384 RSC.
- S. Gupta, M. K. Patel, A. Miotello and N. Patel, Adv. Funct. Mater., 2020, 30, 1906481 CrossRef CAS.
- V. Jose, J. M. V. Nsanzimana, H. Hu, J. Choi, X. Wang and J. M. Lee, Adv. Energy Mater., 2021, 11, 2100157 CrossRef CAS.
- Y. Xiao, C. Shen and T. Long, Chem. Mater., 2021, 33, 4023–4034 CrossRef CAS.
- X. Guo, S. Lin, J. Gu, S. Zhang, Z. Chen and S. Huang, Adv. Funct. Mater., 2021, 31, 2008056 CrossRef CAS.
- G. Qin, Q. Cui, A. Du, W. Wang and Q. Sun, ChemCatChem, 2019, 11, 2624–2633 CrossRef CAS.
- S. Qi, Y. Fan, L. Zhao, W. Li and M. Zhao, Appl. Surf. Sci., 2021, 536, 147742 CrossRef CAS.
- H. Zhang, S. Wang, H. Wang, B. Huang, S. Dong, Y. Dai and W. Wei, Nanoscale, 2021, 13, 17331–17339 RSC.
- K. Elumeeva, J. Masa, D. Medina, E. Ventosa, S. Seisel, Y. U. Kayran, A. Genç, T. Bobrowski, P. Weide and J. Arbiol, J. Mater. Chem. A, 2017, 5, 21122–21129 RSC.
- X. Liu, Y. Wang, L. Chen, P. Chen, S. Jia, Y. Zhang, S. Zhou and J. Zang, ACS Appl. Mater. Interfaces, 2018, 10, 37067–37078 CrossRef CAS PubMed.
- J.-l. Ma, N. Li, Q. Zhang, X.-b. Zhang, J. Wang, K. Li, X.-f. Hao and J.-m. Yan, Energy Environ. Sci., 2018, 11, 2833–2838 RSC.
- H. Park, A. Encinas, J. P. Scheifers, Y. Zhang and B. P. Fokwa, Angew. Chem., Int. Ed., 2017, 56, 5575–5578 CrossRef CAS PubMed.
- H. Xu, B. W. Zeiger and K. S. Suslick, Chem. Soc. Rev., 2013, 42, 2555–2567 RSC.
- B. Coşkuner, A. K. Figen and S. Pişkin, J. Chem., 2014, 2014, 185957 Search PubMed.
- Z. Li, T. Zhuang, J. Dong, L. Wang, J. Xia, H. Wang, X. Cui and Z. Wang, Ultrason. Sonochem., 2021, 71, 105384 CrossRef CAS PubMed.
- U. B. Shahid, Y. Chen, S. Gu, W. Li and M. Shao, Trends Chem., 2021, 4, 142–156 CrossRef.
- J.-T. Ren, L. Chen, Y. Liu and Z.-Y. Yuan, J. Mater. Chem. A, 2021, 9, 11370–11380 RSC.
- W. Lei, D. Portehault, D. Liu, S. Qin and Y. Chen, Nat. Commun., 2013, 4, 1–7 Search PubMed.
- Y. Zhao, R. Shi, X. Bian, C. Zhou, Y. Zhao, S. Zhang, F. Wu, G. I. Waterhouse, L. Z. Wu and C. H. Tung, Adv. Sci., 2019, 6, 1802109 CrossRef PubMed.
- J. Choi, B. H. Suryanto, D. Wang, H.-L. Du, R. Y. Hodgetts, F. M. F. Vallana, D. R. MacFarlane and A. N. Simonov, Nat. Commun., 2020, 11, 1–10 CrossRef PubMed.
- D. Ekeberg, G. Ogner, M. Fongen, E. J. Joner and T. Wickstrøm, J. Environ. Monit., 2004, 6, 621–623 RSC.
- J. Masa, P. Weide, D. Peeters, I. Sinev, W. Xia, Z. Sun, C. Somsen, M. Muhler and W. Schuhmann, Adv. Energy Mater., 2016, 6, 1502313 CrossRef.
- Y. Tsarfati, S. Rosenne, H. Weissman, L. J. Shimon, D. Gur, B. A. Palmer and B. Rybtchinski, ACS Cent. Sci., 2018, 4, 1031–1036 CrossRef CAS PubMed.
- B. Jiang, H. Song, Y. Kang, S. Wang, Q. Wang, X. Zhou, K. Kani, Y. Guo, J. Ye and H. Li, Chem. Sci., 2020, 11, 791–796 RSC.
- G. Arzac, T. C. Rojas and A. Fernández, ChemCatChem, 2011, 3, 1305–1313 CrossRef CAS.
- B. Guan, L. Fan, X. Wu, P. Wang, Y. Qiu, M. Wang, Z. Guo, N. Zhang and K. Sun, J. Mater. Chem. A, 2018, 6, 24045–24049 RSC.
- V. A. Sethuraman, J. W. Weidner, A. T. Haug, M. Pemberton and L. V. Protsailo, Electrochim. Acta, 2009, 54, 5571–5582 CrossRef CAS.
- J. M. V. Nsanzimana, Y. Peng, Y. Y. Xu, L. Thia, C. Wang, B. Y. Xia and X. Wang, Adv. Energy Mater., 2018, 8, 1701475 Search PubMed.
- L. Cui, W. Zhang, R. Zheng and J. Liu, Chem.–Eur. J., 2020, 26, 11661–11672 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta02178e |
| This journal is © The Royal Society of Chemistry 2023 |