
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHarnessing ion–dipole interactions: a simple and effective approach to high-performance lithium receptors†
Chengkai
Xu
 ,
Quy
Tran
,
Lukasz
Wojtas
and
Wenqi
Liu
*
,
Quy
Tran
,
Lukasz
Wojtas
and
Wenqi
Liu
*
Department of Chemistry, University of South Florida, 4202 E. Fowler Ave, Tampa, FL 33620, USA. E-mail: wenqi@usf.edu
First published on 23rd May 2023
Abstract
The burgeoning demand for lithium across various sectors, most notably in lithium-ion batteries, necessitates the development of efficient extraction and purification methodologies. As a response to this imperative, the design of synthetic receptors exhibiting high selectivity and affinity for lithium ions has emerged as a crucial area of research. This investigation proposes a simple and effective approach to high-performance lithium receptors that capitalizes on ion–dipole interactions as the principal driving force for lithium binding. Our investigation encompasses the design, synthesis, and evaluation of five distinct ionophores characterized by varied ion–dipole interactions with lithium, culminating in significantly enhanced binding affinity and Li+/Na+ selectivity compared to conventional macrocyclic crown ether-based receptors. Moreover, we identify a new building block based on pyridine-N-oxide, which serves as an efficacious motif for developing receptors with augmented lithium-binding capacities. Additionally, our findings demonstrate a rapid and efficient solid–liquid extraction process for LiCl in the presence of a substantial excess of NaCl and KCl, employing the newly discovered ionophore. Collectively, this study contributes valuable insights into molecular design strategies for high-performance lithium receptors and advocates for continued exploration of sustainable molecular materials to enhance lithium recognition and extraction efficiencies.
 Chengkai Xu | Chengkai Xu is a PhD student in the Department of Chemistry at the University of South Florida. He received his BSc in Chemistry from Zhejiang University in 2020. His research interests lie in the development of molecular receptors for critical minerals and biomolecules. |
 Wenqi Liu | Dr Wenqi Liu is an Assistant Professor in the Department of Chemistry at the University of South Florida. He received his BSc in Chemistry from Shandong University in 2013 and his PhD in Supramolecular Organic Chemistry from the University of Notre Dame in 2018 under the supervision of Professor Bradley Smith. He then conducted postdoctoral research at Northwestern University from 2018 to 2021, working with Professor Fraser Stoddart. Dr Liu's research interests lie in host–guest chemistry, particularly in relation to critical minerals and bioactive molecules. He is interested in designing and synthesizing functional molecular materials to address challenges in clean energy and health. |
Introduction
Lithium (Li+) has emerged as a critical element due to its extensive applications across pharmaceutical, scientific, and technological sectors. As a mood stabilizer, Li+ serves a crucial role in treating depression and various mental health disorders.1,2 Moreover, it is utilized to enhance strength and thermal stability in the ceramics and glass industries and improve lubricants' viscosity.3,4 The most significant application of Li+ lies in its indispensable role as a key component in lithium-ion batteries.5–8 The ongoing global energy transition from fossil fuels to renewable sources has precipitated a Li+ supply crisis, highlighting the imperative to develop novel technologies for extracting and enriching Li+ from natural resources and anthropogenic waste.9–15 Consequently, the design and implementation of molecular receptors capable of effectively recognizing, sensing, extracting, and purifying this critical mineral have become paramount in addressing this pressing challenge.16–22The design and synthesis of high-performance receptors for Li+ have traditionally focused on developing macrocyclic receptors, featuring small binding cavities meticulously tailored19,20,23–30 to selectively bind Li+ while excluding competing ions such as sodium (Na+) and potassium (K+). A pioneering work in this domain was exemplified31–36 by Donald Cram's spherands, which displayed an extraordinary binding affinity exceeding 1016 M−1 for Li+ due to their rigid and preorganized binding cavities. In alignment with this foundational principle, numerous macrocyclic scaffolds have been devised for Li+ binding, employing molecular design strategies based on crown ether derivatives,21,27,28,37–43 ion pair receptors,16,25,25,44–48 mechanically interlocked molecules,26,49–52 and organometallic macrocycles.18,19,29,53 However, despite the success in achieving strong binding affinity for Li+, the majority of these macrocyclic receptors exhibit limited Li+/Na+ selectivity (ratio <1000) and necessitate high-dilution conditions during synthesis. These constraints present considerable challenges for large-scale production and the practical implementation of these receptors in Li+ extraction and separation processes.
In contrast to macrocyclic receptors, acyclic receptors54–57 offer a relatively straightforward synthesis process. However, their inherent flexibility complicates the achievement of effective selectivity without relying on size matching. We hypothesize that addressing this challenge is achievable by leveraging58 the high charge density inherent to Li+, enabling the establishment of selectivity predicated upon ion–dipole interactions within the context of acyclic receptor architectures. This study introduces a simple and effective approach to high-performance Li+ receptors, emphasizing ion–dipole interactions as the primary driving force for selective Li+ binding. Our research encompasses the design, synthesis, and evaluation of five ionophores that display strong ion–dipole interactions with Li+, resulting in superior binding affinity and Li+/Na+ selectivity compared to conventional macrocyclic crown ether-based receptors. Additionally, we uncover a new building block based upon pyridine-N-oxide, serving as an effective motif for designing receptors with augmented Li+ binding capabilities. Our results further illustrate a rapid and efficient solid–liquid extraction process for LiCl in the presence of a substantial excess of NaCl and KCl, employing the newly identified ionophore. This investigation provides valuable insights into molecular design strategies for high-performance Li+ receptors and paves the way for continued exploration of cutting-edge molecular architectures to achieve sustainable molecular materials with enhanced Li+ recognition and extraction efficiencies.
Experimental
Synthesis
1H NMR titration
1H NMR titrations in CD3CN were conducted at 298 K on a Varian Unity Inova 400 MHz system. Aliquots from a stock solution containing the corresponding salts were added sequentially to an NMR tube containing a solution of the ionophores (600 μL). The 1H NMR spectrum was acquired after each addition. The 1H NMR titration spectra were analyzed by MestReNova software. The NMR titration isotherms were fitted to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 host–guest binding model using Thordarson's equations59,60 at https://app.supramolecular.org/bindfit/. The data were then plotted using OriginLab software. The binding constants Ka were presented with standard deviations from the fitting outcomes.
:1 host–guest binding model using Thordarson's equations59,60 at https://app.supramolecular.org/bindfit/. The data were then plotted using OriginLab software. The binding constants Ka were presented with standard deviations from the fitting outcomes.
Isothermal titration calorimetry
Isothermal titration was performed on the MicroCal ITC200 system at 25 °C. The experiments were conducted in the 200 μL working volume of the sample cell. The capacity of the injection syringe is 40 μL. The stirring speed was set at 750 rpm. Host and guest solutions were prepared in MeCN. A host solution was placed in the titration cell, and the guests were loaded into the syringe. In each case, 20–25 injections were performed. The heat of dilution was measured by titrating the guest into a blank solution. The heat of dilution was subtracted before analyzing with MicroCal ITC200 software using a 1:1 host–guest binding model and plotted by Origin Lab software.
Computational modeling
Structural Optimization and Binding Energy Analysis: the Cartesian coordinates for the calculations were directly created using the GaussView 6 program. All optimizations were performed with density functional theory (DFT) in the Orca program61 (version 4.2.1) using the Becke '88 exchange and Lee–Yang–Parr correlation (BLYP) functional,62 the Ahlrich's double zeta Def2-SVP basis sets63 with geometrical counterpoise (gCP) scheme,64 and Grimme's third-generation dispersion correction65 with Beck Johnson damping (D3BJ). In order to speed up the DFT optimizations, the Coulomb integral66 and numerical chain-of-sphere integration67 for the HF exchanges (RIJCOSX) method was applied with the Def2/J auxiliary basis (AuxJ).68 All optimizations were performed in an acetonitrile continuum with the Conductor-like Polarizable Continuum Model (CPCM) in Orca. Multiwfn 3.6 program69 was used to analyze the electrostatic potential map,70 partial charges,71 and noncovalent surfaces.72,73Solid–liquid extraction
A solution of receptor I5 (5 mM, 1 mL) in CDCl3 was mounted on top of an excess amount of solid LiCl, NaCl and KCl, or a mixture of the three salts. The mixture was sonicated for 5 min. The resulting solution was pipetted into an NMR tube and measured by 1H NMR spectroscopy. To release the captured LiCl, D2O (1 mL) was added to the extractant, and the solution mixture was vortexed for 1 min, after which the organic phase was separated and measure by 1H NMR.Result and discussion
In pursuit of potential molecular scaffolds capable of binding Li+ with strong affinity and high selectivity, we identified ionophore I1, initially reported54 in 1986 for Li+ sensing but with undetermined binding affinity and selectivity for ion binding. Intrigued by I1's simple structure and unusual Li+ selectivity, we opted to reexamine its binding properties. I1 was synthesized following the original literature procedures. Its binding with Li+ was initially assessed using 1H NMR titrations in CD3CN, employing LiBF4 as a soluble salt. The binding affinity (Ka > 106 M−1) between I1 and Li+ was found to be too high for reliable determination (Fig. S16 and S17†) by 1H NMR titration at mM concentrations. We were only able to establish a 1:1 binding stoichiometry using this method. Alternatively, the binding constant (Ka = 3.5 × 106 M−1) was determined (Fig. 2) through isothermal titration calorimetry (ITC) experiments at a micromolar (70 μM) concentration. The binding affinity between I1 and Na+ is weak and can be determined by both 1H NMR and ITC, which yielded a Ka in the order of 103 M−1, resulting in a remarkable Li+/Na+ selectivity of 2214. This selectivity ranks among one of the highest reported25,28,33,39,74–76 values for Li+ receptors.
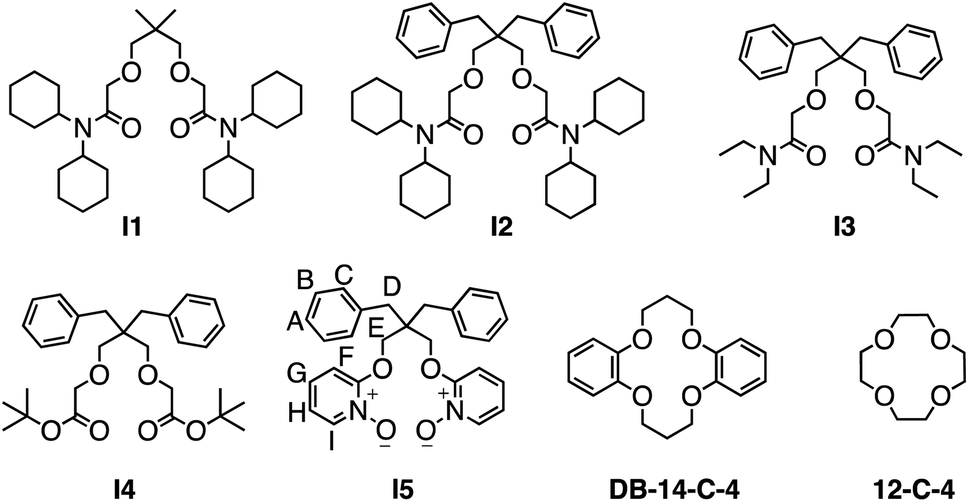 | ||
| Fig. 1 Structural formula of the investigated Li+ ionophores. | ||
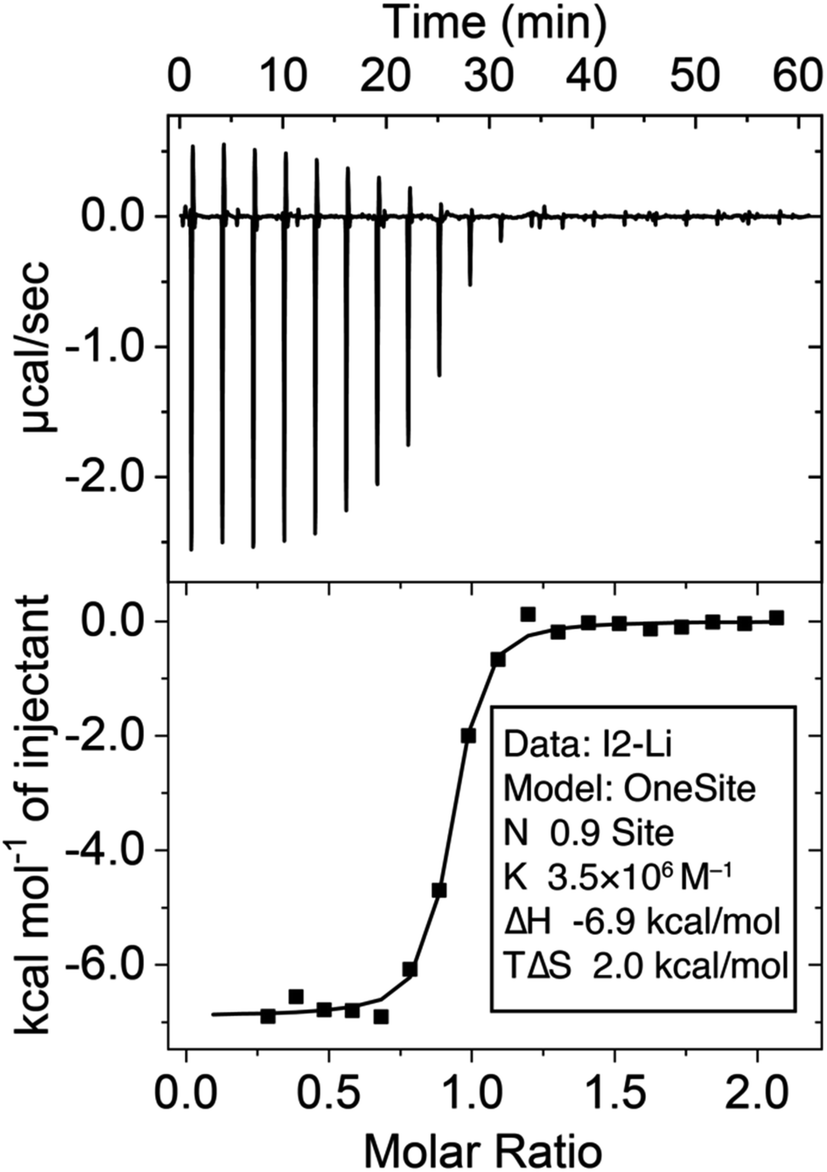 | ||
| Fig. 2 Isothermal titration calorimetry of the binding between I2 (0.1 mM in the cell) and LiBF4 (1 mM in the syringe) in MeCN at 25 °C. | ||
To provide a comparative assessment, we evaluated the binding affinities of two widely employed ionophores for Li+ binding and capture, 12-crown-4 and dibenzo-14-crown-6. The 12-crown-4 exhibited a low binding affinity in the order of 103 M−1 for both Li+ and Na+. In fact, this macrocycle displays a slightly stronger affinity for Na+ than Li+, resulting in a Li+/Na+ selectivity of only 0.5. Consequently, the use of this macrocycle as a Li+ ionophore is not recommended for selective Li+ binding and capture.77–80 In contrast, dibenzo-14-crown-4 demonstrated a higher Li+ binding affinity in the order of 104 M−1 and a more favorable Li+/Na+ selectivity of 25, making it a popular choice for selective Li+ binding. However, compared to I1, 12-crown-4 and dibenzo-14-crown-6 appear less attractive as ideal ionophores for Li+ binding. Furthermore, the synthesis of I1 is significantly simpler, as it does not involve a macrocyclization step, rendering I1 a more practical option for serving as an ionophore for Li+.
Motivated to elucidate the structural features responsible for the high affinity and selectivity for Li+ associated with I1, we synthesized a library of its analogs, I2–I5. Initially, we incorporated two benzyl groups in I2, as evidence suggests81 their potential to enhance Li+ binding selectivity. ITC binding studies revealed that I2 exhibited a binding affinity for Li+ identical to that of I1. However, I2 demonstrated a lower binding affinity for Na+, resulting in an improved Li+/Na+ selectivity of 5645, which is over 200 times better than commonly used crown ether derivatives. ITC further indicated that the Li+ binding of I1 and I2 is driven by favorable enthalpies, supported by a modest increase in entropy. The binding enthalpy of I2 is slightly lower than that of I1, which is also evidenced by the less negative charges (Fig. S60 and S62†) associated with the oxygen atoms in I2 compared to I1. This slight decrease in enthalpy is counterbalanced by a more favorable binding entropy due to the enhanced structural rigidity of I2.
A single crystal structure of I2 (Fig. 3) was obtained via the slow evaporation of its solution in CHCl3. The benzyl groups and N,N-dicyclohexylamide groups present in I2 contribute to steric hindrances, restricting flexibility and supporting the hypothesis that the benzyl groups enhance the receptor I2's rigidity for Li+ binding. Our attempts to obtain a single crystal structure of I2 in conjunction with Li+ were unsuccessful. Instead, we derived an optimized structure (Fig. 3b) using density functional theory (DFT) calculations based on the reported crystal structure of the Li+ complex with I1. Upon Li+ binding, I2 can reorganize its conformations to resemble the binding cavity of 14-crown-4, providing an ideal cavity size for Li+. The Li+ is positioned at the center of the binding cavity, stabilized by four [Li+⋯O] ion–dipole interactions. One of the benzyl groups is situated below the Li+ ion. The electrostatic potential (ESP) map (Fig. 3c) indicates that the benzyl group generates a negatively charged surface area, potentially promoting Li+ binding through favorable electrostatic attractions. The 14-crown-4-like binding cavity associated with I1 and I2 offers not only the appropriate size for Li+ but also stronger ion–dipole interactions, rendering it superior for Li+ binding.
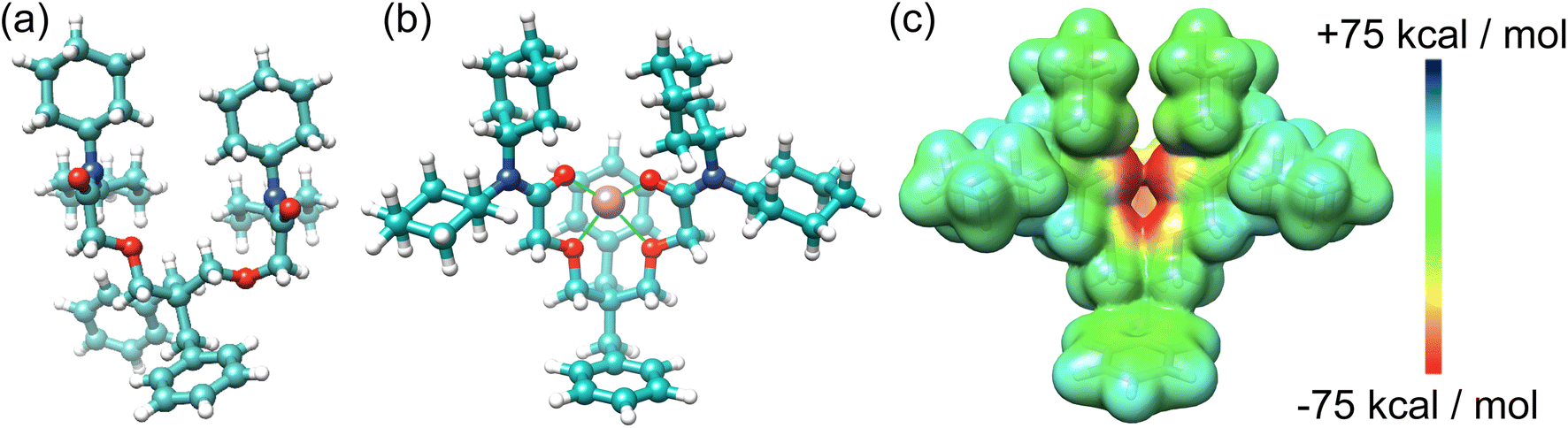 | ||
| Fig. 3 (a) X-ray single crystal structure of I2 and (b) an optimized structure of I2 complexed with Li+. (c) ESP of I2 with reorganized conformation to fit the binding of Li+. | ||
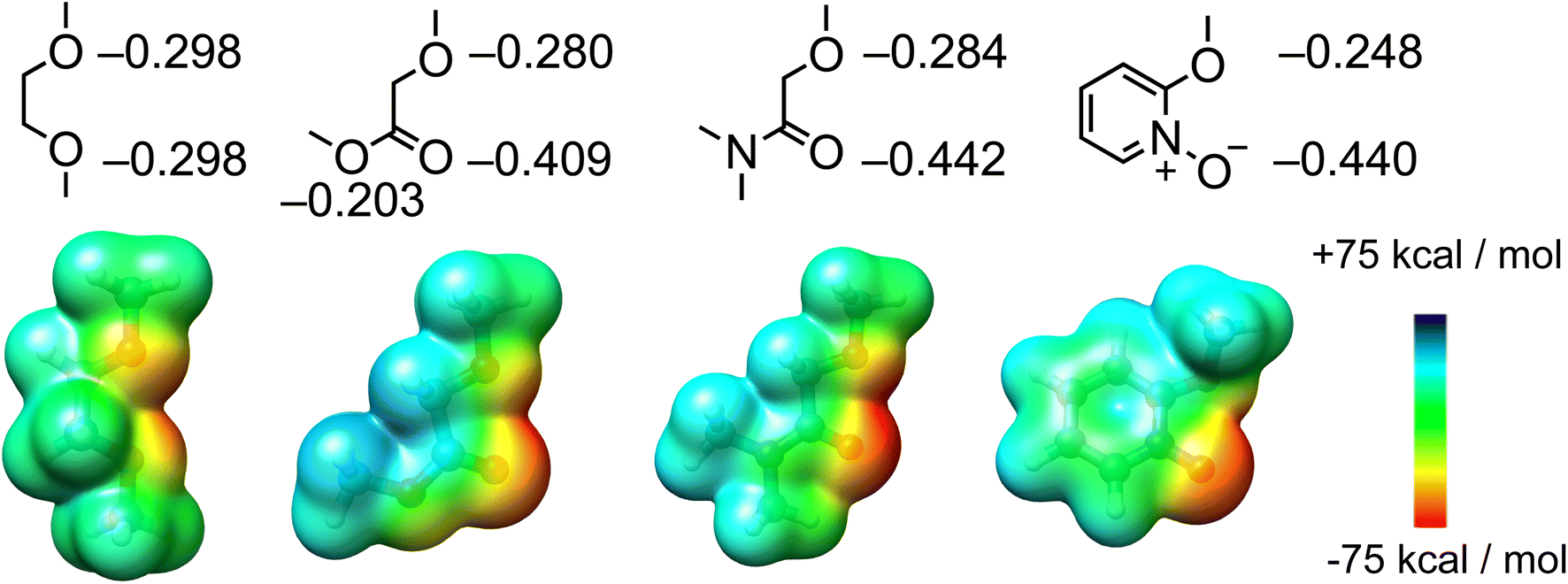 | ||
| Fig. 4 Structural formula of the building blocks to construct Li+ ionophores and calculated partial charges and ESP. | ||
I1 and I2 exhibit a significantly lower binding affinity for Na+ than Li+. The primary reason is the larger ionic radius and, consequently, the lower charge density of Na+, which results in weaker ion–dipole interactions. This diminished ion–dipole interaction with Na+ is evidenced (Table 1 entries 2 and 4) by lower binding enthalpies. The presence of dibenzyl groups in I2 further disfavors Na+ binding by imposing an entropy penalty of −0.7 kcal mol−1, suggesting a higher reorganization energy barrier required to accommodate the larger Na+ ion within the 14-crown-4-like binding cavity.
| Entry | Complex | K a ( M−1) NMR | ITC | ΔG (kcal mol−1) | ΔH (kcal mol−1) | TΔS (kcal mol−1) | Li+/Na+ selectivity |
|---|---|---|---|---|---|---|---|
| a Standard errors are presented in the ESI. b The binding affinity to beyond the lower limit of ITC. c The low affinity and potential low binding enthalpy prevent ITC measurement. d The selectivity is based on ITC measurements. e The selectivity is based on NMR measurements. | |||||||
| 1 | I1-Li+ | >106 | 3.1 × 106 | −8.6 | −7.5 | 1.4 | 2214d |
| 2 | I1-Na+ | 2.4 × 103 | 1.4 × 103 | −4.3 | −3.8 | 0.5 | NA |
| 3 | I2-Li+ | >106 | 3.5 × 106 | −8.9 | −6.9 | 2.0 | 5645d |
| 4 | I2-Na+ | 2.5 × 102 | 6.2 × 102 | −3.8 | −3.1 | −0.7 | NA |
| 5 | I3-Li+ | >106 | 1.4 × 106 | −8.4 | −8.2 | 0.18 | 1167d |
| 6 | I3-Na+ | 2.9 × 102 | 1.2 × 103 | −4.2 | −2.5 | 1.7 | NA |
| 7 | I4-Li+ | 1.8 × 102 | 4.2 × 101 | −2.2 | −3.3 | −1.1 | NA |
| 8 | I4-Na+ | <5 | NAb | NA | NA | NA | NA |
| 9 | I5-Li+ | 1.2 × 105 | 8.0 × 104 | −6.7 | −5.5 | 1.2 | 1154e |
| 10 | I5-Na+ | 1.0 × 102 | NAc | NAb | NAc | NAc | NA |
| 11 | DB-14-C-4-Li+ | 3.0 × 104 | 3.3 × 104 | −6.2 | −5.7 | 0.5 | 25d |
| 12 | DB-14-C-4-Na+ | 6.1 × 102 | 1.3 × 103 | −4.2 | −2.4 | 1.8 | NA |
| 13 | 12-C-4-Li+ | 2.5 × 103 | 3.5 × 103 | −4.8 | −1.2 | 3.6 | 0.5d |
| 14 | 12-C-4-Na+ | 1.4 × 104 | 6.4 × 103 | −5.2 | −7.0 | −1.8 | NA |
Considering the clumsiness of N,N-dicyclohexyl substitutes, we aim to achieve better atomic efficiency for Li+ capture using a smaller-sized ionophore I3 with N,N-diethyl substituents. I3 exhibited a comparable order of binding affinity and Li+/Na+ selectivity as I1 and I2, though its overall performance decreased by a factor of two. I3 displayed a stronger binding enthalpy in comparison to I1 and I2. Owing to the smaller N,N-diethyl substituents, the size reduction increased I3's structural flexibility, resulting in a less entropically favored Li+ binding. The entropy–enthalpy compensation led to minimal changes in I3's overall performance for Li+ binding, suggesting that N,N-dicyclohexyl substituents are not essential for Li+ binding. The binding between I3 and Na+ was slightly greater than that of I2, attributable to I3's increased structural flexibility, which better accommodates Na+. This interpretation is supported by a favorable entropy of 1.7 kcal mol−1 observed in ITC experiments.
Replacing the amide with esters in I4 significantly diminished ion binding performance (Table 1, entries 7 and 8). The low affinities are because the carbonyl oxygen in esters is less electronegative than amide oxygen. Consequently, ester oxygen is a weaker electron donor for establishing ion–dipole interactions. As a result, I4 demonstrated a significant decrease in binding enthalpy and affinity for Li+ and Na+.
By examining structural analogs of I1, we discovered that establishing strong ion–dipole interactions is crucial for achieving strong Li+ binding affinity and high Li+/Na+ selectivity. To further validate this hypothesis, we designed receptor I5 based on a new building block, pyridine-N-oxide, which exhibits a formal charge on the oxygen atom and should facilitate strong ion–dipole interactions. I5 was designed with a binding pocket akin to that of 14-crown-4 and equipped with functionalities capable of establishing stronger ion–dipole interactions. I5 can be readily synthesized (Fig. 5) from the diol precursor in two steps. A nucleophilic aromatic substitution reaction was employed to link two pyridine residues to the diol, followed by oxidation of the pyridine to pyridine-N-oxide, yielding the target ionophore I5 in high efficiency.
 | ||
| Fig. 5 Synthesis of new Li+ ionophore I5 based on pyridine-N-oxide. | ||
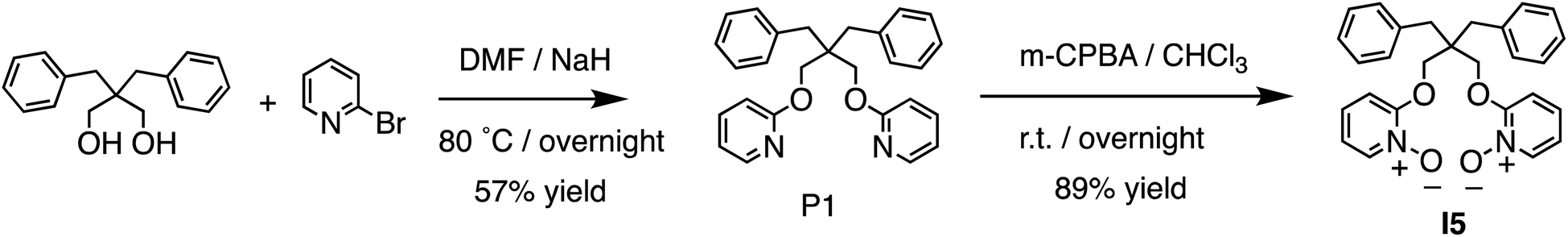
The binding of I5 with Li+ was investigated through 1H NMR titration experiments. Upon the addition of LiBF4 to a solution of I5 in CD3CN, notable downfield shifts were observed for several proton signals (Fig. 6a). The most significant shifts originated from protons E, G, and I (proton labels depicted in Fig. 1), which are all in proximity to the binding pocket. The shifts ceased (Fig. 6c) after adding approximately one molar equivalent of LiBF4, suggesting a 1:1 binding stoichiometry. By fitting the downfield shift of proton E, we determined (Fig. 6b) a binding affinity of 1.2 × 105 M−1 using a 1:1 binding model. The Li+ binding by I5 was independently corroborated by ITC, which exhibited a comparable binding constant in the order of 105 M−1. ITC further revealed a high binding enthalpy (−5.5 kcal mol−1) coupled with a favorable entropy (1.2 kcal mol−1). The high enthalpy can be ascribed to the anticipated strong ion–dipole interactions between the pyridine-N-oxide residues and Li+, while the favorable entropy can be attributed to the relative rigidity of the molecular scaffold. The binding of I5 with Na+ was directly determined using 1H NMR titrations, yielding a low Ka of 104 M−1, which resulted in an outstanding Li+/Na+ selectivity of 1167. The experimental observation of strong Li+ binding affinity and high Li+/Na+ selectivity facilitated by ion–dipole interactions was further corroborated by DFT calculations. Both amide and pyridine-N-oxide demonstrated larger binding energies (Table S2†) and stronger noncovalent interactions (Fig. S60–S69†) in comparison to ester. These findings validate our hypothesis that potent ion–dipole interactions play a critical role in attaining substantial binding affinity and exceptional Li+/Na+ selectivity.
 | ||
| Fig. 6 (a) 1H NMR (400 MHz, CD3CN) spectrum of I5 titrated with LiBF4. (b) Nonlinear fitting for the chemical shift of proton E using a 1:1 binding model. (c) The molar ratio of I5 and its Li+ complex upon titrating LiBF4. | ||
The binding affinity of I5 for Li+ is an order of magnitude lower than that of amide-based ionophores, such as I1–I3. ITC experiments suggest that this reduced binding affinity results from a lower binding enthalpy, indicating a weaker ion–dipole interaction for I5. This observation is further supported by the partial charges and ESP comparison (Fig. 4) between pyridine-N-oxide and etheryl amide building blocks. Although the pyridine-N-oxide oxygen exhibits a comparable electronegativity to the amide oxygen, the oxygen atom attached to the ortho position of the pyridine ring is less electronegative due to its conjugation with the aromatic ring. Consequently, the overall ion–dipole interaction associated with I5 is weaker than that of the amide building block. However, considering the straightforward and cost-effective synthesis, coupled with its high binding affinity and Li+/Na+ selectivity, receptor I5 represents an accessible ionophore for engineering Li+ binding materials that outperform traditional crown ethers.
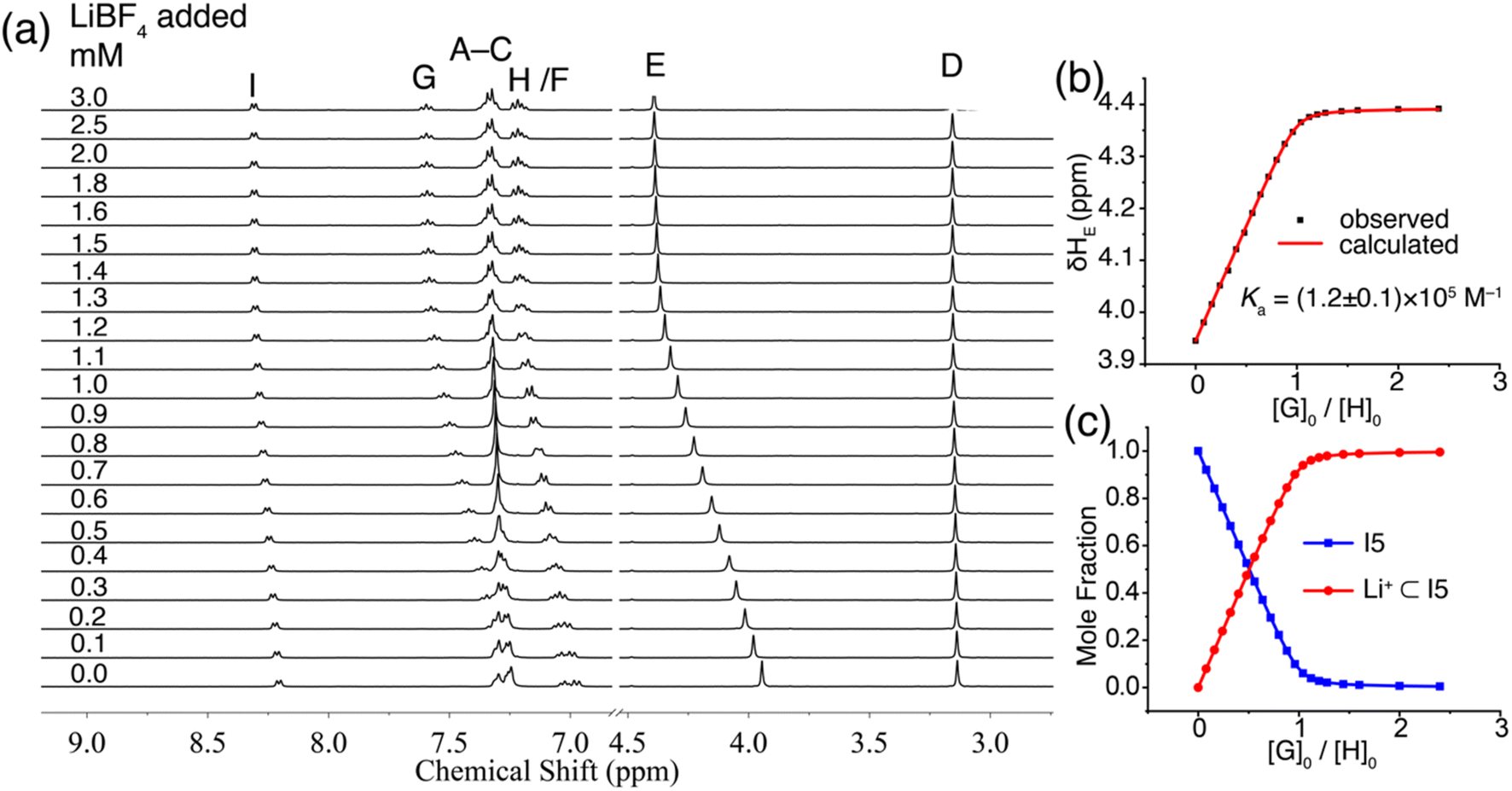
To demonstrate the potential application of ionophore I5 in Li+ separation, we conducted a solid–liquid extraction experiment. A solution of I5 in CDCl3 was placed atop an excess amount of LiCl or NaCl. The solution was sonicated for 5 minutes, and the extractant was monitored using 1H NMR spectroscopy. The 1H NMR spectrum of I5 after NaCl extraction remained (Fig. 7) identical to that of the free I5, indicating no NaCl was extracted into the organic phase. In contrast, the 1H NMR spectrum of I5 after LiCl extraction displayed a distinct downfield shift of proton E, suggesting that I5 can rapidly achieve complexation with LiCl and extracting it into the organic phase. This process involves the extraction of LiCl from its crystal lattice, overcoming the associated electrostatic attractions. Ion pair receptors, which possess a binding motif for cations and a binding motif for anions, are typically required for such solid–liquid extractions, and the process is generally slow, taking days to complete.25,44 The high affinity and selectivity of I5 enable rapid extraction of LiCl into the organic phase within 5 minutes, demonstrating its exceptional performance in Li+ extraction. The extracted Li+ can be easily released from the organic phase to an aqueous solution by a simple wash with D2O, after which the 1H NMR spectrum of I5 resumes (Fig. 7a) back to its uncomplex state. We performed (Fig. 7b) four cycles of LiCl extraction followed by its release into D2O without observing a significant loss of its extraction capacity.
 | ||
| Fig. 7 (a) 1H NMR (400 MHz, CDCl3) spectra of I5 to track the solid–liquid extraction of LiCl and NaCl. (b) Changes in the chemical shift of proton E during four cycles of LiCl extraction using CDCl3 and release in D2O using I5. | ||
To further demonstrate the applicability of receptor I5 for Li+ separation, we conducted a solid–liquid extraction experiment from a sample containing only 0.2% LiCl in the presence of a significant excess of NaCl and KCl, two common salts that coexist in large excess in real-world scenarios. The 1H NMR spectrum of I5 after extraction (see Fig. S70 and S71†) showed the same chemical shift as the one saturated with LiCl, suggesting the high Li+ selectivity associated with I5.
Conclusion
In summary, we have demonstrated the binding of Li+ using five ionophores and compared their performances with traditional Li+ receptors based on crown ethers. These ionophores demonstrated a much better binding affinity and Li+/Na+ selectivity when associated with functional groups that show strong ion–dipole interactions. In addition, a new building block based on pyridine-N-oxide was identified as an effective motif for the design of receptors to bind Li+. We also demonstrated a rapid and efficient process for solid–liquid extraction of LiCl in the presence of large access of NaCl and KCl to organic solvent using ionophore I5. Overall, these studies are expected to advance our understanding of molecular design strategies for producing high-performance receptors targeted for the recognition and extraction of the critical mineral lithium. These receptors are expected to facilitate lithium extraction and separation when grafted onto a polymeric backbone as an immobilized extraction agent. We are currently exploring other molecular skeletons integrated with strong ion–dipole interactions to achieve better affinities and selectivities for Li+.Author contributions
C. X. and Q. T. performed the experiments. L. W. solved the crystal structure. W. L. and C. X. designed the experiment, analyzed the results, and wrote the manuscript. All authors discussed the results and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for this work was provided by the University of South Florida start-up funding. This work was supported, in part, by the University of South Florida Research & Innovation Internal Awards Program under Grant No. 0154360. This research made use of the X-RAY and CPAS Core facilities at the University of South Florida. The research was supported in part by the computational resources provided by the CIRCE research cluster facility at the University of South Florida.Notes and references
- S. K. Sarai, H. M. Mekala and S. Lippmann, Innov. Clin. Neurosci., 2018, 15, 30–32 Search PubMed.
- Z. M. Kamal, S. Dutta, S. Rahman, A. Etando, E. Hasan, S. N. Nahar, W. F. S. Wan Ahmad Fakuradzi, S. Sinha, M. Haque and R. Ahmad, Cureus, 2022, 14, e29332 Search PubMed.
- C. Venkateswaran, H. Sreemoolanadhan and R. Vaish, Int. Mater. Rev., 2022, 67, 620–657 CrossRef CAS.
- N. Kumar, V. Saini and J. Bijwe, Tribol. Lett., 2020, 68, 124 CrossRef CAS.
- S. Yang, F. Zhang, H. Ding, P. He and H. Zhou, Joule, 2018, 2, 1648–1651 CrossRef.
- E. A. Olivetti, G. Ceder, G. G. Gaustad and X. Fu, Joule, 2017, 1, 229–243 CrossRef.
- X. Sun, M. Ouyang and H. Hao, Joule, 2022, 6, 1738–1742 CrossRef.
- M. Armand and J.-M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- S. Kim, H. Joo, T. Moon, S. H. Kim and J. Yoon, Environ. Sci.: Processes Impacts, 2019, 21, 667–676 RSC.
- C. Liu, Y. Li, D. Lin, P.-C. Hsu, B. Liu, G. Yan, T. Wu, Y. Cui and S. Chu, Joule, 2020, 4, 1459–1469 CrossRef CAS.
- Z. Li, C. Li, X. Liu, L. Cao, P. Li, R. Wei, X. Li, D. Guo, K. W. Huang and Z. Lai, Energy Environ. Sci., 2021, 14, 3152–3159 RSC.
- A. Alessia, B. Alessandro, V. G. Maria, V. A. Carlos and B. Francesca, J. Cleaner Prod., 2021, 300, 126954 CrossRef CAS.
- X. Luo, B. Guo, J. Luo, F. Deng, S. Zhang, S. Luo and J. Crittenden, ACS Sustainable Chem. Eng., 2015, 3, 460–467 CrossRef CAS.
- P. Xu, J. Hong, X. Qian, Z. Xu, H. Xia, X. Tao, Z. Xu and Q. Q. Ni, J. Mater. Sci., 2021, 56, 16–63 CrossRef CAS.
- J. F. Song, L. D. Nghiem, X. M. Li and T. He, Environ. Sci.: Water Res. Technol., 2017, 3, 593–597 RSC.
- K. I. Hong, H. Kim, Y. Kim, M. G. Choi and W. D. Jang, Chem. Commun., 2020, 56, 10541–10544 RSC.
- S. Tsuchiya, Y. Nakatani, R. Ibrahim and S. Ogawa, J. Am. Chem. Soc., 2002, 124, 4936–4937 CrossRef CAS PubMed.
- H. Piotrowski, K. Polborn, G. Hilt and K. Severin, J. Am. Chem. Soc., 2001, 123, 2699–2700 CrossRef CAS PubMed.
- Z. Grote, M. L. Lehaire, R. Scopelliti and K. Severin, J. Am. Chem. Soc., 2003, 125, 13638–13639 CrossRef CAS PubMed.
- Q. He, Z. Zhang, J. T. Brewster, V. M. Lynch, S. K. Kim and J. L. Sessler, J. Am. Chem. Soc., 2016, 138, 9779–9782 CrossRef CAS PubMed.
- H. Gohil, S. Chatterjee, S. Yadav, E. Suresh and A. R. Paital, Inorg. Chem., 2019, 58, 7209–7219 CrossRef CAS PubMed.
- X. Guo, Y. Yang, Z. Peng, Y. Cai, W. Feng and L. Yuan, Org. Chem. Front., 2019, 6, 2654–2661 RSC.
- K. Severin, Coord. Chem. Rev., 2003, 245, 3–10 CrossRef CAS.
- Q. He, G. I. Vargas-Zúñiga, S. H. Kim, S. K. Kim and J. L. Sessler, Chem. Rev., 2019, 119, 9753–9835 CrossRef CAS PubMed.
- Q. He, N. J. Williams, J. H. Oh, V. M. Lynch, S. K. Kim, B. A. Moyer and J. L. Sessler, Angew. Chem., Int. Ed., 2018, 57, 11924–11928 CrossRef CAS PubMed.
- V. K. Munasinghe, J. Pancholi, D. Manawadu, Z. Zhang and P. D. Beer, Chem.–Eur. J., 2022, 28, 1–8 CrossRef PubMed.
- Y. Luo, N. Marets and T. Kato, Chem. Sci., 2018, 9, 608–616 RSC.
- M. Kamenica, R. Kothur, A. Willows, B. Patel and P. Cragg, Sensors, 2017, 17, 2430 CrossRef PubMed.
- Z. Grote, R. Scopelliti and K. Severin, J. Am. Chem. Soc., 2004, 126, 16959–16972 CrossRef CAS PubMed.
- Z. Chen, O. F. Schall, M. Alcalá, Y. Li, G. W. Gokel and L. Echegoyen, J. Am. Chem. Soc., 1992, 114, 444–451 CrossRef CAS.
- D. J. Cram, T. Kaneda, G. M. Lein and R. C. Helgeson, J. Chem. Soc., Chem. Commun., 1979, 21, 948–950 RSC.
- D. J. Cram, S. P. Ho, C. B. Knobler, E. Maverick and K. N. Trueblood, J. Am. Chem. Soc., 1986, 108, 2989–2998 CrossRef.
- D. J. Cram and G. M. Lein, J. Am. Chem. Soc., 1985, 107, 3657–3668 CrossRef CAS.
- P. A. Kollman, G. Wipff and U. C. Singh, J. Am. Chem. Soc., 1985, 107, 2212–2219 CrossRef CAS.
- D. J. Cram, T. Kaneda, R. C. Helgeson, S. B. Brown, C. B. Knobler, E. Maverick and K. N. Trueblood, J. Am. Chem. Soc., 1985, 107, 3645–3657 CrossRef CAS.
- D. J. Cram and I. B. Dicker, J. Chem. Soc., Chem. Commun., 1982, 1219–1221 RSC.
- O. Shyshov, R. C. Brachvogel, T. Bachmann, R. Srikantharajah, D. Segets, F. Hampel, R. Puchta and M. von Delius, Angew. Chem., Int. Ed., 2017, 56, 776–781 CrossRef CAS PubMed.
- R. C. Brachvogel, F. Hampel and M. Von Delius, Nat. Commun., 2015, 6, 1–7 Search PubMed.
- H. Löw, E. Mena-Osteritz and M. Von Delius, Chem. Sci., 2018, 9, 4785–4793 RSC.
- S. Kitazawa, K. Kimura, H. Yano and T. Shono, J. Am. Chem. Soc., 1984, 106, 6978–6983 CrossRef CAS.
- B. P. Czech, D. A. Babb, B. Son and R. A. Bartsch, J. Org. Chem., 1984, 49, 4805–4810 CrossRef CAS.
- I. Oral and V. Abetz, Macromol. Rapid Commun., 2021, 42, 2000746 CrossRef CAS PubMed.
- K. Kobiro, Coord. Chem. Rev., 1996, 148, 135–149 CrossRef CAS.
- J. M. Mahoney, A. M. Beatty and B. D. Smith, Inorg. Chem., 2004, 43, 7617–7621 CrossRef CAS PubMed.
- H. Wang, L. O. Jones, I. Hwang, M. J. Allen, D. Tao, V. M. Lynch, B. D. Freeman, N. M. Khashab, G. C. Schatz, Z. A. Page and J. L. Sessler, J. Am. Chem. Soc., 2021, 143, 20403–20410 CrossRef CAS PubMed.
- Q. He, N. J. Williams, J. H. Oh, V. M. Lynch, S. K. Kim, B. A. Moyer and J. L. Sessler, Angew. Chem., Int. Ed., 2018, 57, 11924–11928 CrossRef CAS PubMed.
- Q. He, Z. Zhang, J. T. Brewster, V. M. Lynch, S. K. Kim and J. L. Sessler, J. Am. Chem. Soc., 2016, 138, 9779–9782 CrossRef CAS PubMed.
- C. Ren, V. Maurizot, H. Zhao, J. Shen, F. Zhou, W. Q. Ong, Z. Du, K. Zhang, H. Su and H. Zeng, J. Am. Chem. Soc., 2011, 133, 13930–13933 CrossRef CAS PubMed.
- M. Sheikh, M. Qassem, I. F. Triantis and P. A. Kyriacou, Sensors, 2022, 22, 736 CrossRef CAS PubMed.
- S. I. Pascu, T. Jarrosson, C. Naumann, S. Otto, G. Kaiser and J. K. M. Sanders, New J. Chem., 2005, 29, 80–89 RSC.
- G. Kaiser, T. Jarrosson, S. Otto, Y. F. Ng, A. D. Bond and J. K. M. Sanders, Angew. Chem., Int. Ed., 2004, 43, 1959–1962 CrossRef CAS PubMed.
- X. Ruan, C. Zhang, Y. Zhu, F. Cai, Y. Yang, J. Feng, X. Ma, Y. Zheng, H. Li, Y. Yuan and G. Zhu, Angew. Chem., Int. Ed., 2023, 62, e202216549 CrossRef CAS PubMed.
- M. L. Lehaire, R. Scopelliti, H. Piotrowski and K. Severin, Angew. Chem., Int. Ed., 2002, 41, 1419–1422 CrossRef CAS PubMed.
- E. Metzger, R. Aeschimann, M. Egli, G. Suter, R. Dohner, D. Ammann, M. Dobler and W. Simon, Helv. Chim. Acta, 1986, 69, 1821–1828 CrossRef CAS.
- M. Bocheńska and M. Gdaniec, J. Incl. Phenom. Mol. Recognit. Chem., 1994, 20, 53–71 CrossRef.
- A. R. De Sànchez, J. R. Anacona and V. E. Màrquez, Supramol. Chem., 1994, 4, 9–12 CrossRef.
- J. V. Gavette, J. Lara, L. L. Reling, M. M. Haley and D. W. Johnson, Chem. Sci., 2013, 4, 585–590 RSC.
- U. Olsher, R. M. Izatt, J. S. Bradshaw and N. K. Dalley, Chem. Rev., 1991, 91, 137–164 CrossRef CAS.
- D. Brynn Hibbert and P. Thordarson, Chem. Commun., 2016, 52, 12792–12805 RSC.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- H. Kruse and S. Grimme, J. Chem. Phys., 2012, 136, 154101 CrossRef PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- R. Izsák and F. Neese, J. Chem. Phys., 2011, 135, 144105 CrossRef PubMed.
- G. L. Stoychev, A. A. Auer and F. Neese, J. Chem. Theory Comput., 2017, 13, 554–562 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- J. Zhang and T. Lu, Phys. Chem. Chem. Phys., 2021, 23, 20323–20328 RSC.
- T. LU and F. CHEN, J. Theor. Comput. Chem., 2012, 11, 163–183 CrossRef CAS.
- T. Lu and Q. Chen, J. Comput. Chem., 2022, 43, 539–555 CrossRef CAS PubMed.
- C. Lefebvre, G. Rubez, H. Khartabil, J. C. Boisson, J. Contreras-García and E. Hénon, Phys. Chem. Chem. Phys., 2017, 19, 17928–17936 RSC.
- J. M. Lehn and J. P. Sauvage, J. Am. Chem. Soc., 1975, 97, 6700–6707 CrossRef CAS.
- D. J. Cram, T. Kaneda, R. C. Helgeson, S. B. Brown, C. B. Knobler, E. Maverick and K. N. Trueblood, J. Am. Chem. Soc., 1985, 107, 3645–3657 CrossRef CAS.
- K. Roy, C. Wang, M. D. Smith, P. J. Pellechia and L. S. Shimizu, J. Org. Chem., 2010, 75, 5453–5460 CrossRef CAS PubMed.
- S. J. Warnock, R. Sujanani, E. S. Zofchak, S. Zhao, T. J. Dilenschneider, K. G. Hanson, S. Mukherjee, V. Ganesan, B. D. Freeman, M. M. Abu-Omar and C. M. Bates, Proc. Natl. Acad. Sci., 2021, 118, e2022197118 CrossRef CAS PubMed.
- S. D. Alexandratos and C. L. Stine, React. Funct. Polym., 2004, 60, 3–16 CrossRef CAS.
- P. K. Mohapatra, D. S. Lakshmi, A. Bhattacharyya and V. K. Manchanda, J. Hazard. Mater., 2009, 169, 472–479 CrossRef CAS PubMed.
- M. Kazemabad, A. Verliefde, E. R. Cornelissen and A. D'Haese, J. Membr. Sci., 2020, 595, 117432 CrossRef.
- K. Kimura, H. Yano, S. Kitazawa and T. Shono, J. Chem. Soc., Perkin Trans. 2, 1986, 4, 1945–1951 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2252048. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ta01831h |
| This journal is © The Royal Society of Chemistry 2023 |